
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spin transition in a ferrous chloride complex supported by a pentapyridine ligand†
Manuel
Boniolo
 a,
Sergii I.
Shylin
a,
Petko
Chernev
a,
Mun Hon
Cheah
a,
Philipp A.
Heizmann
a,
Ping
Huang
a,
Nessima
Salhi
a,
Kamal
Hossain
a,
Anders
Thapper
a,
Marcus
Lundberg
a and
Johannes
Messinger
*ab
a,
Sergii I.
Shylin
a,
Petko
Chernev
a,
Mun Hon
Cheah
a,
Philipp A.
Heizmann
a,
Ping
Huang
a,
Nessima
Salhi
a,
Kamal
Hossain
a,
Anders
Thapper
a,
Marcus
Lundberg
a and
Johannes
Messinger
*ab
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, PO Box 523, 75120 Uppsala, Sweden. E-mail: johannes.messinger@kemi.uu.se
bDepartment of Chemistry, Chemical Biological Centre, Umeå University, 90187 Umeå, Sweden
First published on 5th February 2020
Abstract
Ferrous chloride complexes [FeIILxCl] commonly attain a high-spin state independently of the supporting ligand(s) and temperature. Herein, we present the first report of a complete spin crossover with T1/2 = 80 K in [FeII(Py5OH)Cl]+ (Py5OH = pyridine-2,6-diylbis[di(pyridin-2-yl)methanol]). Both spin forms of the complex are analyzed by X-ray spectroscopy and DFT calculations.
Spin-crossover (SCO) systems are excellent examples of bistable materials in which magnetic and optical properties,1 structure,2 and chemical reactivity3 can be switched between two distinct states. This crossover process is known for certain families of 3d4–3d7 metal complexes. The re-configuration of their electronic structures is induced by changes in temperature, pressure, light irradiation or absorption of guest molecules.4 In the case of ferrous complexes, the spin conversion occurs between the diamagnetic low-spin (S = 0, LS) state, in which all d-electrons are paired, and the paramagnetic high-spin (S = 2, HS) state, in which four d-electrons are unpaired. Because of the difference in the occupation of the anti-bonding molecular orbitals, metal-to-ligand distances in the HS state are significantly larger than in the LS state. Consequently, SCO occurs when the enthalpy of the stronger iron-to-ligand bonds in the LS state is overcome by the configurational and vibrational entropy of the HS form.5 Thus, the rational choice of ligands with a favorable ligand field strength is a key factor to design complexes with switchable behavior. There are a few families of known SCO-active iron(II) complexes in the literature, including molecular complexes,6–8 1D coordination polymers,9,10 2D layered structures and 3D metal organic frameworks.11 Within the realm of discrete ferrous complexes with SCO, those with 2,6-bis(pyrazolyl)pyridine6 and scorpionate ligands,7 as well as bis(thiocyanate) complexes supported by polypyridine ligands [FeLx(NCS)2],8 are among the best studied switchable materials. Polymeric 1,2,4-triazole-based iron(II) complexes have been a focus in both early studies of the SCO phenomenon and the most recent developments of functional materials.9 They have been used, for example, to design prototypes of thermochromic materials, microthermometers, chemical sensors, actuators and chiral switches.10 Heterobimetallic 2D and 3D coordination frameworks, known in literature as Hofmann-like clathrates, are excellent examples of the SCO systems due to their simplicity, universal design approach and porous structure.11 They are constructed of iron(II) ions coordinated by cyanometallic anions [M(CN)x]y− (where M = Ni, Pd, Pt, Cu, Ag, Au) and N-donor heterocyclic ligands. Notably, all these complexes feature the FeIIN6 chromophore that is frequently considered as a prerequisite for the existence of the SCO. However, a handful of SCO complexes with FeIIN4O2,12 FeIIN5O,13 FeIIN4S2,14 FeIIN5S,15 FeIIN4C2,16 FeIIN3C2O217 and even FeC3N18 coordination spheres have been also reported.
Ferrous complexes bearing at least one chloride ligand exist in the HS state due to the relatively large anionic radius of the chloride anion and its associated weak ligand field strength.19 It was, therefore, not surprising when the title complex [FeII(Py5OH)Cl](PF6) (Py5OH = pyridine-2,6-diylbis[di(pyridin-2-yl)methanol]) was reported as HS, based on its single crystal X-ray diffraction analysis at 100 K.20 The complex was employed as a catalyst in photo-induced water oxidation to dioxygen.
In an effort to trap and identify catalytically relevant intermediates, we observed the herein reported spin transition in [FeII(Py5OH)Cl](PF6) with a critical temperature of 80 K. This is confirmed by temperature dependent SQUID magnetic susceptibility measurements and synchrotron X-ray absorption studies, analyzing both the X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). The conclusions are supported by density functional theory (DFT) calculations of the title complex and the related HS analog [FeII(Py5OMe)Cl]+.13
Compound Py5OH was synthesized starting from 2,6-dibromopyridine and di(2-pyridyl)ketone (see ESI† for details). The [FeII(Py5OH)Cl](PF6) complex was obtained as microcrystalline yellow powder. Its powder X-ray diffraction (PXRD) profile shows a good match with the simulation that is based on the single crystal XRD structure reported previously (Fig. S1, ESI†).20 All iron ions in the structure, which are crystallographically identical, are coordinated by the five nitrogen atoms from Py5OH and by one chloride anion, which results in a distorted octahedral FeIIN5Cl environment.
The magnetic properties of the complex recorded in the cooling and warming regimes are shown in Fig. 1. The product of the molar magnetic susceptibility with temperature, χMT, as a function of temperature, reveals a complete one-step spin transition for this compound. At room temperature, χMT is 3.4 cm3 K mol−1, indicating that all iron(II) sites exist in the HS state. Upon cooling to ca. 140 K, χMT remains constant, as expected on the basis of Curie's law for the paramagnetic compound with S = 2. However, further cooling reveals a gradual decrease in χMT down to 0.3 cm3 K mol−1, which is due to a HS to LS transition of the iron centers. The transition is virtually complete at ∼40 K, and the T1/2 (the temperature at which half of the complex is in the LS state) is 80 K. Upon warming of the sample in the magnetic field, its susceptibility behavior was completely reversible, with only a narrow hysteresis at low temperatures, caused by kinetic effects and not cooperativity of the lattice (Fig. S4, ESI†).4d Due to a pronounced thermochromism of the spin transition, the yellow powder of [FeII(Py5OH)Cl](PF6) turns green upon cooling.
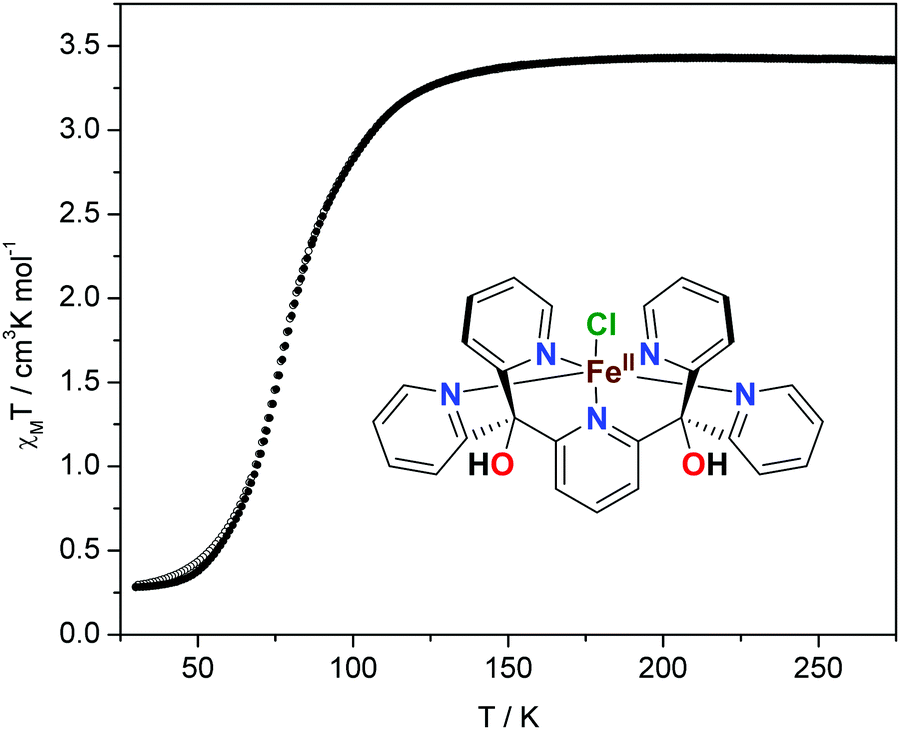 | ||
| Fig. 1 Magnetic properties of [FeII(Py5OH)Cl](PF6) powder in the form of χMT vs. T recorded in cooling (open circles) and warming (filled circles) modes with a scan rate of 2 K min−1. The molecular structure of the complex is shown as an inset. | ||
Compared to the common SCO systems, the transition temperature in the title complex is unusually low—the continuous spin conversion is observed down to ca. 40 K on cooling.6–11 In general, at temperatures below ca. 100 K, kBT is smaller than the energy difference between HS and LS states, which usually leads to a trapping of the HS state upon fast cooling.4c Thus, for iron(II) complexes with expected T1/2 below this point, the SCO is usually incomplete or is not observed at all.21 One of the recent examples is the molecular FeIIN6 complex with T1/2 = 95 K reported by Bao et al.; relaxation of its quenched HS state at 80 K occurs over the period of several hours.21a In contrast, [FeII(Py5OH)Cl](PF6) shows a complete transition even at lower temperature with a scan rate of 2 K min−1, indicating a weak cooperativity between the SCO centers in the compound.4,5 A similar SCO behavior with T1/2 = 81 K was reported for an organometallic four-coordinate complex with a pseudotetrahedral FeC3N ligation.18
X-ray absorption spectroscopy is a well-established technique for measuring metal–ligand distances in transition metal complexes (EXAFS) and for monitoring their oxidation and spin states (XANES). The XANES spectra of [FeII(Py5OH)Cl](PF6) acquired at different temperatures using synchrotron radiation are shown in Fig. 2. The spectra obtained at 150 K and 293 K are virtually identical. The position and shape of the Fe K-edge are consistent with those expected for iron(II) in the HS state,22 which corroborates our magnetic susceptibility measurements. In the XANES spectrum recorded at 20 K, the shift of the edge to higher energy reflects the completed HS to LS conversion. The inflection that occurs at about half-height (7123 eV) in the XANES spectrum of the LS complex (arrow B in Fig. 2) is as previously identified as a structure-sensitive above-ionization multiple-scattering resonance.22b On the basis of the well-established correlation between edge absorption intensity and bond distance, this feature can be assigned to the presence of short Fe–N bonds in the LS state.22c In addition, the intensity of the pre-edge peak at 7112 eV (arrow A in Fig. 2), corresponding to the 1s → 3d excitation, increases upon transition to the LS state, similar to what has been observed for other iron SCO complexes.22d,e
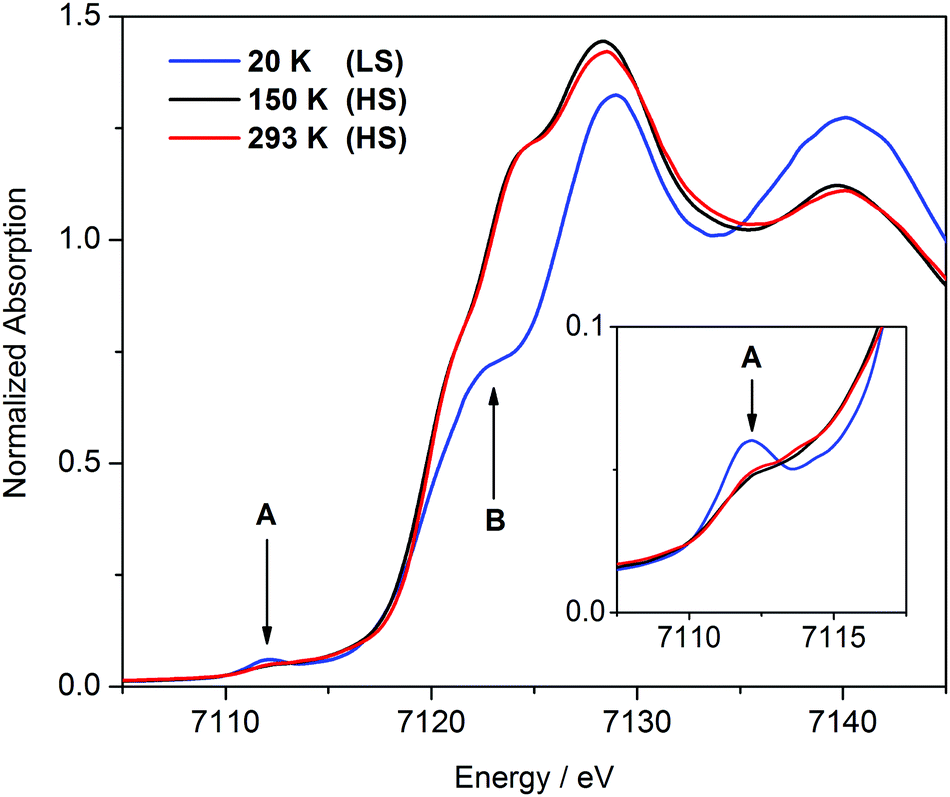 | ||
| Fig. 2 XANES spectra of [FeII(Py5OH)Cl](PF6) powder recorded at different temperatures. The inset shows enlargement of the pre-edge area. | ||
The EXAFS spectra and Fourier transform of EXAFS for the title complex are shown in Fig. S5 (ESI†) and Fig. 3. At 150 K and 293 K, the EXAFS of the title compound can be simulated well with Fe–N and Fe–Cl distances of 2.18 Å and 2.38 Å, respectively. These values are in good agreement with the X-ray diffraction data reported previously (〈Fe–N〉 = 2.177 Å; Fe–Cl = 2.417 Å at 100 K)20 and indicate the HS state of the divalent iron. When the sample is cooled below the SCO temperature, a clear contraction of the nitrogen shell can be observed in the Fourier transform of EXAFS. The spectrum at 20 K can be simulated with Fe–N and Fe–Cl bond distances of 1.99 Å and 2.38 Å, respectively, without notable contribution from possible HS residues. Remarkably, the Fe–Cl bond is not affected by the spin transition, while shortening of the Fe–N bonds by ca. 0.2 Å is as well documented for SCO complexes with the FeN6 chromophore.5 The essential characteristics of the title complex in both spin states derived from the XANES and EXAFS are summarized in Table 1. Full details of the simulation can be found in Table S1 (ESI†).
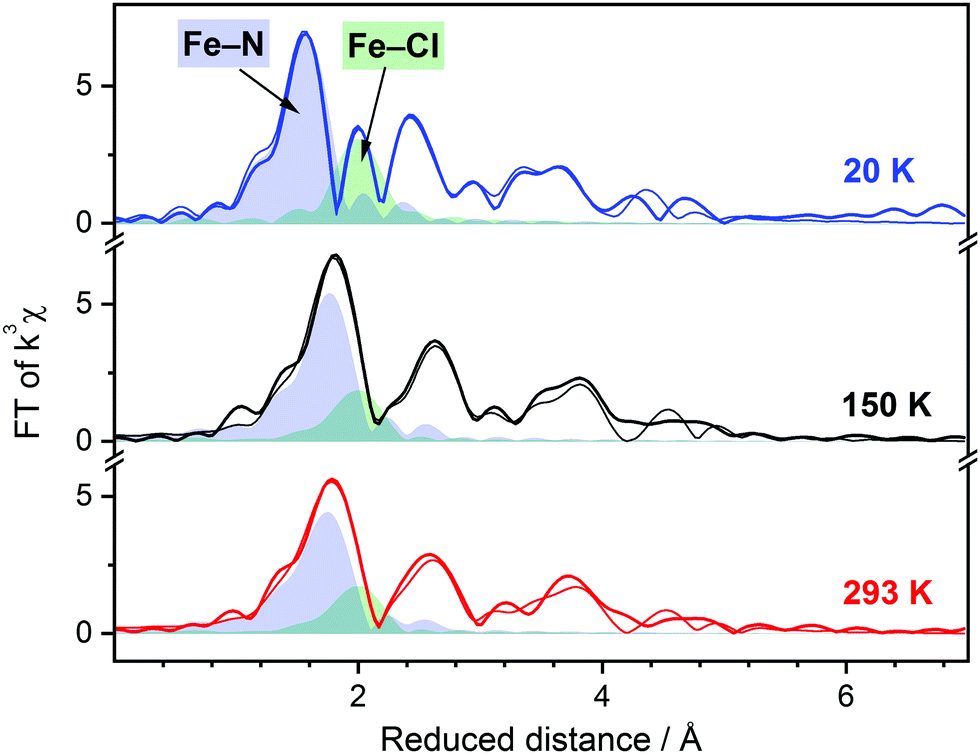 | ||
| Fig. 3 Fourier transform (FT) of k3-weighted EXAFS spectra of [FeII(Py5OH)Cl](PF6) powder recorded at different temperatures (EXAFS is shown in Fig. S5, ESI†). Simulations of the experimental data based on the molecular structure of the complex are shown as thin lines. The filled areas represent FT of the simulated single metal-to-ligand shells, Fe–N5, blue; Fe–Cl, green. The sum of the single shells in the FT is not equal to the shown final simulation due to destructive interference between neighboring shells. Temperature color code: 20 K, blue; 150 K, black; 293 K, red. | ||
| Temperature | 20 K | 150 K | 293 K |
|---|---|---|---|
| Spin state | LS | HS | HS |
| K-edge | 7120.5 | 7119.7 | 7119.7 |
| 〈Fe–N〉 | 1.993(3) | 2.178(6) | 2.169(7) |
| 〈Fe–Cl〉 | 2.378(6) | 2.38(2) | 2.38(2) |
In contrast to the title compound, the ferrous chloride complex with a pentapyridine ligand, which differs from Py5OH only by methylation of its hydroxy groups, remains HS down to 4 K.13 We performed DFT calculations to rationalize the contrasting spin state behavior between [FeII(Py5OH)Cl]+ and [FeII(Py5OMe)Cl]+. Optimization with the B3LYP functional reproduces the structural changes seen in EXAFS upon transition to the LS state, with Fe–N bonds shortening by 0.19 Å while the Fe–Cl bond length changes only marginally (by 0.03 Å). To reproduce the transition temperature of 80 K for the title complex we used energies from the B3LYP* functional (see ESI† for further details).23 At 0 K, the LS state of the title complex is stable by 0.6 kcal mol−1 (Fig. 4). As the temperature increases, the entropy contributions favor the HS state, leading to the spin transition at 80 K. The largest factor driving the transition to the HS state with temperature is the vibrational entropy contribution. This effect comes from the less stiff metal–ligand bonds in the quintet state with two electrons in the anti-bonding eg orbitals.
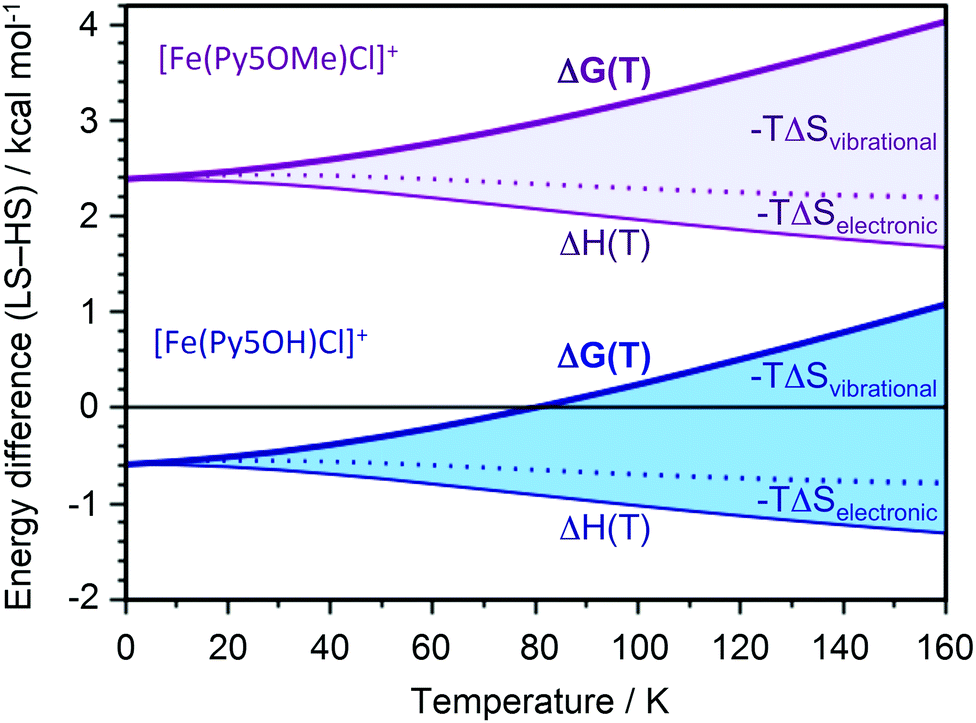 | ||
| Fig. 4 Calculated free energy of the LS state, relative to the HS state, as a function of temperature for both the title complex with hydroxo groups (thick blue line) and the reference complex with methoxy groups (thick purple line). The thin lines for each complex show the enthalpy, ΔH(T), while the shaded areas represent the electronic and vibrational entropy contributions. The effects of rotations are too small to be visible, and those from translations are zero. | ||
For the complex with methoxy groups, the HS state is stable by 2.3 kcal mol−1 at 0 K. Consequently, there is no SCO for this complex in agreement with experimental data.13 Methylating the ligand thus favors the quintet relative to the singlet by 2.9 kcal mol−1. The significant effect of methylating a position far from the potential SCO center comes from steric rather than electronic effects, with significant distortions of the axial pyridine ring. In [FeII(Py5OH)Cl]+, the axial pyridine plane is parallel to the z-axis, while in [FeII(Py5OMe)Cl]+ it intersects at 19° and 30° degrees for singlet and quintet, respectively. The distortion weakens the iron-to-ligand bond, which contributes to stabilizing the HS state.24 Similar calculations show that substitution of the coordinated chloride by bromide in the title complex favors the HS state down to 0 K (Table S2, ESI†).
In conclusion, we report a temperature-induced spin transition behavior of a mononuclear iron(II) complex with a coordination environment FeIIN5Cl. To the best of our knowledge, this is the first observation of spin transition in a ferrous chloride complex. Despite an unusually low critical temperature, the compound shows a complete transition to the LS state as confirmed experimentally by magnetic susceptibility measurements, XANES and EXAFS spectroscopy. DFT calculations show that both spin states can be stabilized in the title complex depending on temperature. Our study demonstrates that SCO can be found in a priori HS complexes, and the design principles revealed a new route towards the search for new switchable materials. As complexes with the pentapyridine ligands, including the title complex, are well-known for their catalytic activity,20,25 tuning the SCO temperature via modifications to the ligand molecule may be promising for the design of multifunctional materials.
This work was supported by the Swedish Energy Agency through the grant no. 45421-1. The authors thank the Helmholtz-Zentrum Berlin (HZB) for allocation of synchrotron radiation beamtime at the KMC-3 beamline (BESSY II synchrotron, Berlin Adlershof). Calculations were performed on resources provided by SNIC through the National Supercomputer Centre at Linköping University (Tetralith) under project snic2018-3-575. SIS thanks the ÅForsk Foundation for support (grant no. 19-349). AT and KH thank the Olle Engkvist Foundation for support (grant number 198-0369).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. B. Gaspar, V. Ksenofontov, M. Seredyuk and P. Gütlich, Coord. Chem. Rev., 2005, 249, 2661–2676 CrossRef CAS; (b) M. A. Halcrow, Spin-Crossover Materials, John Wiley & Sons Ltd, Chichester, 2013 CrossRef.
- (a) P. Guionneau, M. Marchivie, G. Bravic, J. F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97–128 CrossRef CAS; (b) I. A. Gural'skiy, B. O. Golub, S. I. Shylin, V. Ksenofontov, H. J. Shepherd, P. R. Raithby, W. Tremel and I. O. Fritsky, Eur. J. Inorg. Chem., 2016, 3191–3195 CrossRef; (c) M. Weselski, M. Książek, P. Mess, J. Kusz and R. Bronisz, Chem. Commun., 2019, 55, 7033–7036 RSC.
- I. A. Gural'skiy, S. I. Shylin, V. Ksenofontov and W. Tremel, Eur. J. Inorg. Chem., 2017, 3125–3131 CrossRef.
- For selected reviews on spin crossover, see: (a) P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef PubMed; (b) A. Bousseksou, G. Molnar, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC; (c) J. A. Real, A. B. Gaspar and M. C. Muñoz, Dalton Trans., 2005, 2062–2069 RSC; (d) S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- (a) P. Gütlich and H. Goodwin, Top. Curr. Chem., 2004, 233, 1–47 CrossRef; (b) W. Nicolazzi and A. Bousseksou, C. R. Chim., 2018, 21, 1060–1074 CrossRef CAS.
- (a) M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493–2514 CrossRef CAS; (b) M. A. Halcrow, New J. Chem., 2014, 38, 1868–1882 RSC; (c) L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2–12 CrossRef CAS.
- (a) G. J. Long, F. Grandjean and D. L. Reger, Top. Curr. Chem., 2004, 233, 91–122 CrossRef CAS; (b) O. Iasco, M.-L. Boillot, A. Bellec, R. Guillot, E. Rivière, S. Mazerat, S. Nowak, D. Morineau, A. Brosseau, F. Miserque, V. Repain and T. Mallah, J. Mater. Chem. C, 2017, 5, 11067–11075 RSC.
- (a) P. Guionneau, M. Marchivie, G. Bravic, J. F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97–128 CrossRef CAS; (b) J.-J. Lee, H. Sheu, C.-R. Lee, J.-M. Chen, J.-F. Lee, C.-C. Wang, C.-H. Huang and Y. Wang, J. Am. Chem. Soc., 2000, 122, 5742–5747 CrossRef CAS; (c) F. J. Valverde-Muñoz, A. B. Gaspar, S. I. Shylin, V. Ksenofontov and J. A. Real, Inorg. Chem., 2015, 54, 7906–7914 CrossRef PubMed.
- L. G. Lavrenova and O. G. Shakirova, Eur. J. Inorg. Chem., 2013, 670–682 CrossRef CAS.
- (a) G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 17003862 CrossRef PubMed; (b) S. Rat, M. Piedrahita-Bello, L. Salmon, G. Molnár, P. Demont and A. Bousseksou, Adv. Mater., 2018, 30, 1705275 CrossRef PubMed; (c) M. Piedrahita-Bello, K. Ridier, M. Mikolasek, G. Molnár, W. Nicolazzi, L. Salmon and A. Bousseksou, Chem. Commun., 2019, 55, 4769–4772 RSC; (d) I. A. Gural'skiy, V. A. Reshetnikov, A. Szebesczyk, E. Gumienna-Kontecka, A. I. Marynin, S. I. Shylin, V. Ksenofontov and I. O. Fritsky, J. Mater. Chem. C, 2015, 3, 4737–4741 RSC; (e) I. A. Gural'skiy, O. I. Kucheriv, S. I. Shylin, V. Ksenofontov, R. A. Polunin and I. O. Fritsky, Chem. – Eur. J., 2015, 21, 18076–18079 CrossRef PubMed.
- (a) M. C. Muñoz and J. A. Real, Coord. Chem. Rev., 2011, 255, 2068–2093 CrossRef; (b) N. F. Sciortino, K. R. Scherl-Gruenwald, G. Chastanet and C. J. Kepert, Angew. Chem., Int. Ed., 2012, 51, 10154–10158 CrossRef CAS PubMed; (c) O. I. Kucheriv, S. I. Shylin, V. Ksenofontov, S. Dechert, M. Haukka, I. O. Fritsky and I. A. Gural'skiy, Inorg. Chem., 2016, 55, 4906–4914 CrossRef CAS PubMed; (d) I. A. Gural'skiy, S. I. Shylin, B. O. Golub, V. Ksenofontov, I. O. Fritsky and W. Tremel, New J. Chem., 2016, 40, 9012–9016 RSC; (e) V. M. Hiiuk, S. Shova, A. Rotaru, V. Ksenofontov, I. O. Fritsky and I. A. Gural'skiy, Chem. Commun., 2019, 55, 3359–3362 RSC.
- B. Weber, Coord. Chem. Rev., 2009, 253, 2432–2449 CrossRef CAS , and ref. therein.
- D. J. Rudd, C. R. Goldsmith, A. P. Cole, T. D. P. Stack, K. O. Hodgson and B. Hedman, Inorg. Chem., 2005, 44, 1221–1229 CrossRef CAS PubMed.
- V. A. Grillo, L. R. Gahan, G. R. Hanson, R. Stranger, T. W. Hambley, K. S. Murray, B. Moubaraki and J. D. Cashion, J. Chem. Soc., Dalton Trans., 1998, 2341–2348 RSC.
- M. Yamasaki and T. Ishida, Chem. Lett., 2015, 44, 920–921 CrossRef CAS.
- J. S. Costa, C. Balde, C. Carbonera, D. Denux, A. Wattiaux, C. Desplanches, J. P. Ader, P. Gütlich and J. F. Létard, Inorg. Chem., 2007, 46, 4114–4119 CrossRef PubMed.
- P. Guionneau, F. Le Gac, A. Kaiba, J. Sanchez Costa, D. Chasseau and J. F. Letard, Chem. Commun., 2007, 3723–3725 RSC.
- J. J. Scepaniak, T. D. Harris, C. S. Vogel, J. Sutter, K. Meyer and J. M. Smith, J. Am. Chem. Soc., 2011, 133, 3824–3827 CrossRef CAS PubMed.
- M. A. Halcrow, Crystals, 2016, 6, 58 CrossRef.
- B. Das, A. Orthaber, S. Ott and A. Thapper, ChemSusChem, 2016, 9, 1178–1186 CrossRef CAS PubMed.
- (a) Y. S. Ye, X. Q. Chen, Y. D. Cai, B. Fei, P. Dechambenoit, M. Rouzières, C. Mathonière, R. Clérac and X. Bao, Angew. Chem., Int. Ed., 2019, 131, 19064–19067 CrossRef; (b) H. J. Shepherd, C. Bartual-Murgui, G. Molnár, J. A. Real, M. C. Muñoz, L. Salmon and A. Bousseksou, New J. Chem., 2011, 35, 1205–1210 RSC; (c) N. Moliner, M. C. Muñoz, S. Létard, X. Solans, N. Menéndez, A. Goujon, F. Varret and J. A. Real, Inorg. Chem., 2000, 39, 5390–5393 CrossRef CAS PubMed.
- (a) T. Westre, P. Kennepohl, J. DeWitt, B. Hedman, K. Hodgson and E. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS; (b) V. Briois, P. Sainctavit, G. J. Long and F. Grandjean, Inorg. Chem., 2001, 40, 912–918 CrossRef CAS; (c) H. Dau, P. Liebisch and M. Haumann, Anal. Bioanal. Chem., 2003, 376, 562–583 CrossRef CAS PubMed; (d) S. Mebs, N. Braun, R. Kositzki, C. Limberg and M. Haumann, Inorg. Chem., 2015, 54, 11606–11624 CrossRef CAS PubMed; (e) E. Collet and P. Guionneau, C. R. Chim., 2018, 21, 1133–1151 CrossRef CAS.
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- (a) N. Kroll, I. Speckmann, M. Schoknecht, J. Gülzow, M. Diekmann, J. Pfrommer, A. Stritt, M. Schlangen, A. Grohmann and G. Hörner, Angew. Chem., Int. Ed., 2019, 58, 13472–13478 CrossRef CAS PubMed; (b) A. E. King, Y. Surendranath, N. A. Piro, J. P. Bigi, J. R. Long and C. J. Chang, Chem. Sci., 2013, 4, 1578–1587 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, PXRD, IR, ESI-MS spectra, fit of the magnetic susceptibility data. See DOI: 10.1039/c9cc09630b |
| This journal is © The Royal Society of Chemistry 2020 |