
Isotopic effect on electrochemical CO2 reduction activity and selectivity in H2O- and D2O-based electrolytes over palladium†
Ji Hoon
Lee
 a,
Brian M.
Tackett
a,
Zhenhua
Xie
b,
Sooyeon
Hwang
c and
Jingguang G.
Chen
*ab
a,
Brian M.
Tackett
a,
Zhenhua
Xie
b,
Sooyeon
Hwang
c and
Jingguang G.
Chen
*ab
aDepartment of Chemical Engineering, Columbia University, New York, NY 10027, USA. E-mail: jgchen@columbia.edu
bChemistry Division, Brookhaven National Laboratory, Upton, NY 11973, USA
cCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 21st November 2019
Abstract
The isotopic effect on the electrochemical CO2 reduction reaction (CO2RR) is investigated in this study. A higher CO2RR selectivity over its competing hydrogen evolution reaction was observed in D2O-based electrolytes compared with the H2O-based counterparts, which can be attributed to the lower [D+] concentration than [H+].
The increasing demand for effectively mitigating the growing atmospheric CO2 level promotes the electrochemical CO2 reduction reaction (CO2RR) in aqueous electrolytes as one of the viable options.1–3 This is because the CO2RR, coupled with renewable energy resources, can produce value-added chemicals as a resource for downstream thermochemical reactions, thus potentially enabling the net reduction of CO2.3 Extensive studies have shown that the conversion and selectivity (i.e., total current density and faradaic efficiency, respectively) of catalysts can be controlled by alloy formation,4–6 phase transition,7,8 additives,9 surface functional groups,10–13 and different salts14,15 in the electrolyte. However, rarely has the isotopic effect of the aqueous media been explored.
The current study investigates the effect of deuterium oxide (D2O) on the CO2RR over carbon-supported Pd (Pd/C, see Fig. S1 for X-ray diffraction pattern in the ESI†) with four different 0.1 M alkali-ion bicarbonate electrolytes (0.1 M AHCO3 or 0.1 M ADCO3, A = Na+ or K+, see the ESI† for the experimental details). Interestingly, deuterium-based electrolytes exhibited markedly lowered D2 evolution reaction (DER) activity compared with the H2 evolution reaction (HER) activity in H2O-based electrolytes, which is ascribed to lower deuteron concentration ([D+]) than proton concentration ([H+]) in the corresponding electrolytes. The different [D+] and [H+] concentrations originated from the different dissociation constants (pK) of D2O and H2O.16 In contrast to the suppressed DER activity, the CO2RR activity was enhanced in D2O-based electrolytes, implying that the isotopic effect can allow for different product distribution in the synthesis gas (i.e., CO/D2 and CO/H2 ratios).
It is well-known that H+ is readily absorbed into the Pd lattice to form Pd hydride (PdH) at potentials for the CO2RR.6–8 This phase transformation thereby enables co-production of CO and H2; otherwise, metallic Pd would be poisoned by CO, resulting in a negligible yield of CO production. In order to investigate if Pd forms Pd deuteride (PdD) like PdH formation, in situ X-ray absorption fine structure (XAFS) analysis17 was conducted (see the ESI†) from 0.3 V to −0.8 V versus reversible hydrogen electrode (RHE). Note that all of the potentials (V) in this study are versus RHE. With potentials being applied more negatively, X-ray absorption near edge structure (XANES) spectra show the gradual shift toward lower energy, reflecting the progressive PdD and PdH formation under CO2RR conditions in 0.1 M NaDCO3 and NaHCO3 electrolytes, respectively (Fig. 1A and B). This result implies that the PdD formation likely occurs in the same manner as PdH formation, also evidenced by the interatomic distance profile of Pd and its first neighbor Pd (RPd–Pd) obtained from in situ extended XAFS (EXAFS) analysis (Fig. 1C, D and Fig. S2, Table S1, ESI†). In each electrolyte, D+(H+) diffusion into Pd starts around 0 V and saturates around −0.6 V to form PdD(PdH) with an increase in RPd–Pd by ∼2.4%, which is also consistent with the changes in the XANES peak positions.7 Meanwhile, the coordination number of Pd remains similar, implying that the particle size is unchanged during the CO2RR (Fig. S3 in the ESI†).
 | ||
| Fig. 1 In situ XAFS analysis measured at the Pd K-edge in different electrolytes. (A and B) In situ XANES profiles from 0.3 V (bottom) to −0.8 V (top) in 0.1 M (A) NaDCO3 and (B) NaHCO3 electrolytes. The dotted yellow lines are vertical lines to visualize the peak shifts. (C and D) In situ EXAFS profiles under the same conditions. | ||
Fig. S4 in the ESI† shows the linear scanning voltammetry (LSV) curves of Pd/C in 0.1 M NaHCO3 and NaDCO3 electrolytes saturated with CO2 or Ar. Under CO2-saturated conditions (Fig. S4A, ESI†), similar LSV profiles are observed in both electrolytes. In contrast, it can be seen that, under Ar-saturated conditions (Fig. S4B, ESI†), the current density in 0.1 M NaHCO3 electrolyte is higher than that in a 0.1 M NaDCO3 electrolyte, indicating that a simple replacement of H2O with D2O can significantly reduce the DER activity, as will be discussed later in the product analysis. To investigate the isotope effect of D2O on CO2RR behavior, the activity of Pd/C was evaluated by using chronoamperometry (CA) techniques from −0.6 to −1.0 V with a potential interval of 0.1 V in 0.1 M AHCO3 or ADCO3 electrolytes as shown in Fig. S5A–D (ESI†). The CA current densities in all of the electrolytes are stable. The gaseous products were analyzed by using gas chromatography, and the faradaic efficiency (FE) of each product was calculated (see the ESI†). As displayed in Fig. 2, CO and H2 (or D2) are the main products in H2O-based (or D2O-based) electrolytes, with the sum of FE(CO) and FE(H2/D2) being near or over 90%. The only exception is at −0.6 V in 0.1 M NaHCO3, which might be attributed to the formation of oxygenated product (i.e., formate), although the product yield is too low (∼10−5 M) even after a prolonged CO2RR period to be detected in the 1H-NMR analysis (see the ESI†). In addition, the long-term stability tests up to 8 h at −0.8 V in D2O-based electrolytes suggest that D2O is an effective media for the electrochemical CO2RR (Fig. S5E in the ESI†). After 8 h electrolysis, the FE(CO) values decreased by ∼7 and ∼12% in 0.1 M KDCO3 and NaDCO3 electrolytes (Fig. S6 in the ESI†), respectively, which seems to originate from catalyst agglomeration. This is also supported by transmission electron microscopy images taken before and after the long-term electrolysis, which show the well-dispersed Pd particles over C with a small increase in size (Fig. S7 in the ESI†).
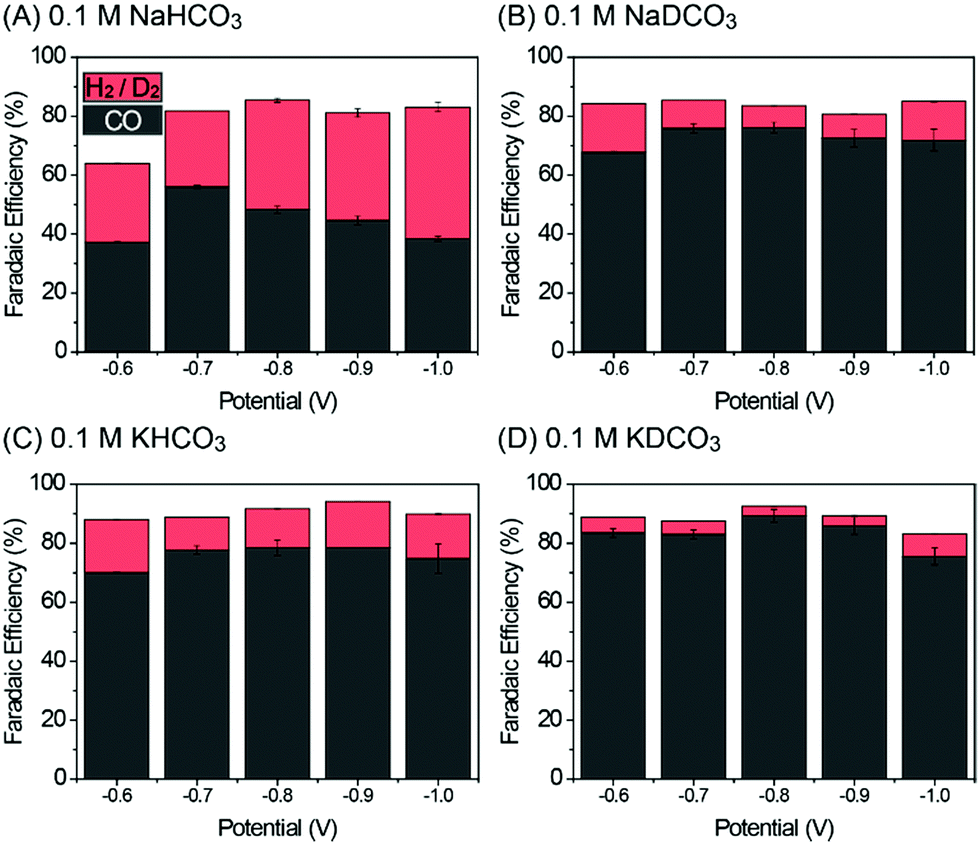 | ||
| Fig. 2 Faradaic efficiency of CO and H2 in different electrolytes. (A) 0.1 M NaHCO3, (B) 0.1 M NaDCO3, (C) 0.1 M KHCO3, and (D) 0.1 M KDCO3. | ||
However, the resultant product distribution, CO/H2 or CO/D2, can be significantly affected by changing H2O with D2O over the entire potential range. For example, Pd/C in 0.1 M NaDCO3 shows a FE(CO) of 76.1% at 0.8 V, which is 1.58 times higher than that (48.2%) at the same potential in 0.1 M NaHCO3 (Fig. 2A and B). As the pH values of all electrolytes are near ∼6.8, one can exclude the possibility that the CO2RR performance is influenced by the pH effect. Therefore, such an enhancement can be explained by the different [D+] and [H+] concentrations in those near neutral-pH electrolytes. When H2O is replaced with D2O, [D+] is roughly 33% of [H+], owing to the higher pK value (14.491) of D2O than that (13.995) of H2O.16 This leads to impaired DER activity in comparison with HER activity. As a consequence, more catalytic sites would be available for the CO2RR rather than DER, in turn enhancing FE(CO) in 0.1 M NaDCO3 over 0.1 M NaHCO3. Such an isotopic effect of D2O is also observed in K-based electrolytes (Fig. 2C and D). The enhancement in FE(CO) in 0.1 M KHCO3 is attributed to the different hydrolysis capability of Na and K ions (the so-called, cation effect),14,15 which is now well-established. Based on the observed FE trend in Fig. 2, one can conclude that, regardless of the salt choice, D2O plays a role in facilitating CO2RR activity and decreasing DER activity, mainly due to the lower [D+] concentration.
It has been proposed that CO production primarily takes place via a carboxylic intermediate (*HOCO) while HER occurs via a hydrogen intermediate (*H) as shown in Fig. S8 (ESI†).18,19 DER is expected to undergo a similar intermediate (*D) as the HER. The proposed mechanisms imply that the protonation (deuteration) step is involved in both CO2RR and HER (DER),18 which in turn suggests that the total current density (J(total)) would be influenced by the solvent choice. As shown in Fig. 3A, in the case that the same cation salt is selected, H2O-based electrolytes exhibit slightly higher J(total) values than those in D2O-based electrolytes, confirming that D+/H+ are involved in the CO2RR as well as DER/HER. On the other hand, regardless of the isotopic effect, K-based electrolytes show higher J(total) values than those in Na-based electrolytes, which is attributed to the cation effect described above.
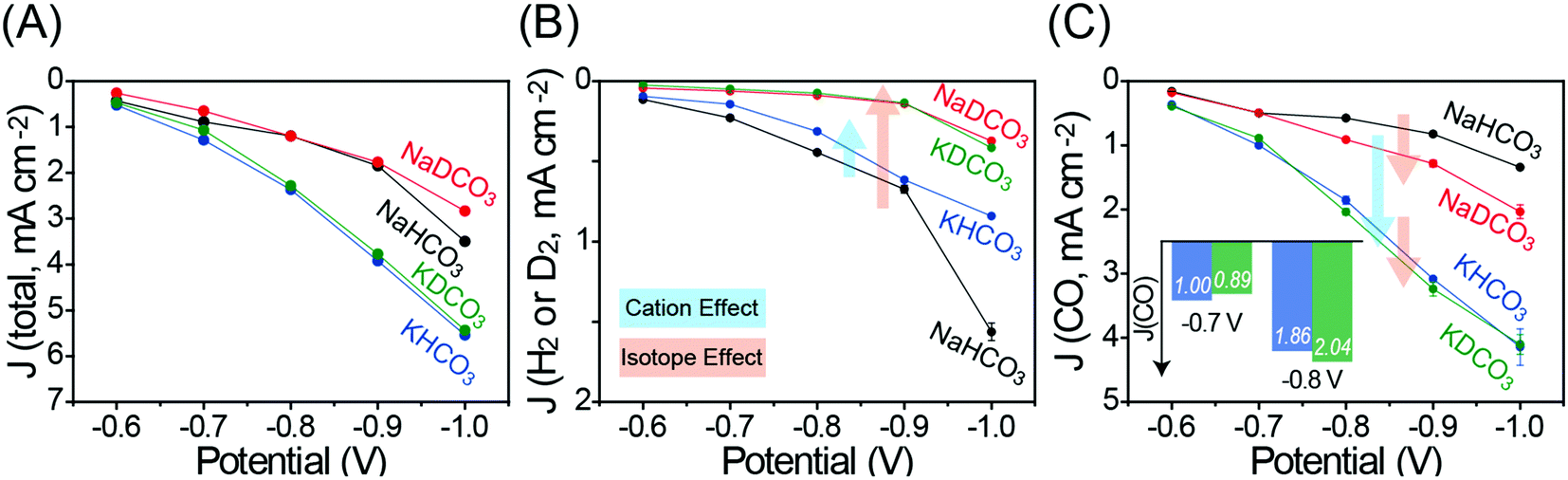 | ||
| Fig. 3 Isotope and cation effects on the CO2RR. (A) Total current densities, (B and C) partial current density of (B) CO and (C) D2 or H2 with different electrolytes. Inset in (C) compares J(CO) values in 0.1 M KHCO3 (blue) and KDCO3 (green) at −0.7 and −0.8 V. | ||
Fig. 3B shows the partial current density of D2 and H2 evolutions (J(D2) or J(H2)) at different potentials. It is noteworthy that, upon replacing H2O with D2O, the DER activity is dramatically decreased compared to the HER activity, which is consistent with the FE trend in Fig. 2. In contrast to the decreased DER activity, it is shown that both the cation and isotopic effects influence the CO2RR activity (Fig. 3C). In particular, the isotopic effect can be clearly observed in J(CO) profiles at −0.8 V and more negative potentials in Na-based electrolytes. This phenomenon can be understood in a way that DER is less likely to take place than HER, again due to the lower [D+] than [H+], promoting more catalytic active sites for the CO2RR and consequently enhancing J(CO). On the other hand, in K-based electrolytes, a relatively small enhancement of J(CO) is observed, indicating that the hydrolysis effect of the K ion is more influential on CO2RR compared to the isotopic effect of D2O.
It is interesting that J(CO) at low overpotentials in D2O-based electrolytes is lower than or comparable to that in H2O-based electrolytes under the same cation salt conditions. For example, at −0.7 V, the J(CO) value in 0.1 M KDCO3 is lower than that in 0.1 M KHCO3. However, with increasing overpotential more negatively, the J(CO) values in 0.1 M KDCO3 get higher than those in 0.1 M KHCO3 (inset in Fig. 3C). That is, at low overpotentials, higher [H+] is likely to facilitate higher CO yield in 0.1 M KHCO3 since the protonation step is also essential for the CO2RR. As the overpotential increases and J(CO) consequently increases, the CO yield is limited by the transport of CO2. The J(CO) in 0.1 M KDCO3 surpasses that in 0.1 M KHCO3 in this case because the DER, a main competing reaction, is less favorable than the HER.
In summary, we demonstrate the isotope influence of D2O on the CO2RR using a Pd catalyst. In situ X-ray characterization confirms PdD formation, similar to the case of PdH formation in H2O during the CO2RR. It is revealed that a simple replacement of H2O with D2O can not only decrease the DER rate due to the low D+ concentration but also facilitate the CO2RR. Results from the current study provide a potentially useful methodology to tune the product distribution in other types of CO2RR reactions.
This research was supported by the US Department of Energy, Office of Basic Energy Sciences, Catalysis Science Program (Grant No. DE-FG02-13ER16381). The authors acknowledge technical support with 7-BM (QAS) at the National Synchrotron Light Source-II (NSLS-II) and Center for Functional Nanomaterials (CFN) in Brookhaven National Laboratory (Contract No. DE-SC0012704) and 17-BM at Advanced Photon Source (APS) in Argonne National Laboratory (Contract No. DE-AC02-06CH11357). J. H. L. acknowledges the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant No. NRF-2017R1A6A3A03004202).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Hori, Modern Aspects of Electrochemistry, Springer, 2008, ch. 3, vol. 42, pp. 89–189 Search PubMed.
- M. Meinshausen, N. Meinshausen, W. Hare, S. C. Raper, K. Frieler, R. Knutti, D. J. Frame and M. R. Allen, Nature, 2009, 458, 1158–1162 CrossRef CAS PubMed.
- B. M. Tackett, E. Gomez and J. G. Chen, Nat. Catal., 2019, 2, 381–386 CrossRef CAS.
- C. W. Lee, K. D. Yang, D. H. Nam, J. H. Jang, N. H. Cho, S. W. Im and K. T. Nam, Adv. Mater., 2018, 30, 1704717 CrossRef.
- W. Zhu, B. M. Tackett, J. G. Chen and F. Jiao, Top. Curr. Chem., 2018, 376, 41 CrossRef.
- J. H. Lee, S. Kattel, Z. Jiang, Z. Xie, S. Yao, B. M. Tackett, W. Xu, N. S. Marinkovic and J. G. Chen, Nat. Commun., 2019, 10, 3724 CrossRef.
- W. Sheng, S. Kattel, S. Yao, B. Yan, Z. Liang, C. J. Hawxhurst, Q. Wu and J. G. Chen, Energy Environ. Sci., 2017, 10, 1180–1185 RSC.
- D. Gao, H. Zhou, F. Cai, D. Wang, Y. Hu, B. Jiang, W.-B. Cai, X. Chen, R. Si, F. Yang, S. Miao, J. Wang, G. Wang and X. Bao, Nano Res., 2017, 10, 2181–2191 CrossRef CAS.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CrossRef CAS.
- J. H. Lee, S. Kattel, Z. Xie, B. M. Tackett, J. Wang, C.-J. Liu and J. G. Chen, Adv. Funct. Mater., 2018, 28, 1804762 CrossRef.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS.
- M. Cho, J. T. Song, S. Back, Y. Jung and J. Oh, ACS Catal., 2018, 8, 1178–1185 CrossRef CAS.
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- A. Kreżel and W. Bal, J. Inorg. Biochem., 2004, 98, 161–166 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS.
- W. Zhu, R. Michalsky, O. N. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS.
- Y. Katayama, F. Nattino, L. Giordano, J. Hwang, R. R. Rao, O. Andreussi, N. Marzari and Y. Shao-Horn, J. Phys. Chem. C, 2019, 123, 5951–5963 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray diffraction pattern, EXAFS results, electrochemical data, and TEM analyses. See DOI: 10.1039/c9cc07611e |
| This journal is © The Royal Society of Chemistry 2020 |