
Potent and selective PTP1B inhibition by a platinum(II) complex: possible implications for a new antitumor strategy†
Caixia
Yuan
 a,
Weirong
Wang
a,
Jianwei
Wang
a,
Xinhua
Li
a,
Yan-Bo
Wu
a,
Shaodong
Li
a,
Liping
Lu
*a,
Miaoli
Zhu
*a,
Shu
Xing
*b and
Xueqi
Fu
b
a,
Weirong
Wang
a,
Jianwei
Wang
a,
Xinhua
Li
a,
Yan-Bo
Wu
a,
Shaodong
Li
a,
Liping
Lu
*a,
Miaoli
Zhu
*a,
Shu
Xing
*b and
Xueqi
Fu
b
aInstitute of Molecular Science, Key Laboratory of Chemical Biology and Molecular Engineering of the Education Ministry, Shanxi University, Taiyuan, Shanxi 030006, P. R. China. E-mail: luliping@sxu.edu.cn; miaoli@sxu.edu.cn
bEdmond H. Fischer Signal Transduction Laboratory, College of Life Sciences, Jilin University, Changchun 130023, P. R. China. E-mail: xingshu@jlu.edu.cn
First published on 25th November 2019
Abstract
Showing anti-proliferation activity against MCF7 cells better than cisplatin, a platinum(II) complex, [PtL(DMSO)Cl], was found to potently and selectively inhibit protein tyrosine phosphatase 1B (PTP1B), a putative target for anticancer agents, suggesting a new possible anticancer strategy based on platinum drugs.
In the past few decades, Pt-based anticancer drugs such as cisplatin, carboplatin and oxaliplatin have been vital for the chemotherapy of various malignant tumors.1 However, their clinical success is limited due to their severe side effects and intrinsic or acquired resistance to the treatment,2 leading to the development of improved Pt-based anticancer drugs.3
Though damaging DNA is a widely accepted anticancer mechanism for most Pt(II) drugs, the exploration of new Pt(II) drugs has uncovered the possibility of novel anticancer mechanisms. Specifically, enzyme inhibition was a recently disclosed significant and alternative mechanism for Pt-based anticancer therapeutics.4 For example, Pt complexes were found to be the potent inhibitors for a variety of proteinases associated with tumour development and progression, such as matrix metalloproteinases, glutathione S-transferase, histone deacetylase, mammalian topoisomerases, human thioredoxin reductase 1 and cysteine proteases, etc.5–8 Hence, Pt-based anti-proliferation of cancer cells can be realized by their interactions with various cellular targets, which offers us an immense space to develop Pt-based anticancer drugs.
Protein tyrosine phosphatases (PTPs), a superfamily of enzymes, can participate in the regulation of the intracellular signal transduction pathway by hydrolytically removing the phosphate groups from proteins.9 Dysregulated activities of PTPs are related to the pathogenesis of a number of human diseases such as cancer, diabetes and autoimmune diseases.10 Recently, several PTPs (such as PTP1B, T-cell PTP (TCPTP), and Src homology phosphatase 1 (SHP-1)) were demonstrated to be overexpressed in breast, colorectal, lung, pancreatic, and gastric cancer tissues, suggesting that excessive PTPs may promote tumour development and progression.11 Thus, PTPs are attractive candidates for the development of targeted therapies for multiple cancer types.12 Our recent research has shown that some copper and vanadium complexes could penetrate the cell membrane and selectively interact with their target PTPs, resulting in the enhanced phosphorylation of the related substrates and influencing cellular metabolism.13
Inspired by these previous studies, we wondered whether the anticancer activity of Pt complexes could be realized by the inhibition of PTPs? The answer is positive. Herein, we describe a simple Pt(II) complex (1) possessing an obviously higher anti-proliferation activity than cisplatin against MCF7 cells. Remarkably, the anti-proliferation activity of 1 is probably attributed to its potent inhibition against PTP1B activity, thereby affecting the transduction pathway.
The synthetic route of 1 ([PtL(DMSO)Cl], DMSO = dimethyl sulfoxide and HL = 5-chlorosalicylideneaniline) is shown in Scheme S1 (ESI†). Its structure was characterized by elemental analysis, IR spectra, ESI-MS and X-ray crystallography methods (Fig. S1, S2 and Tables S1, S2, ESI†). As depicted in Fig. 1, 1 adopts the typical square-planar geometry of Pt(II) complexes, where N and O of the chelating Schiff base ligand, S of DMSO and Cl− coordinate towards the platinum(II) center.
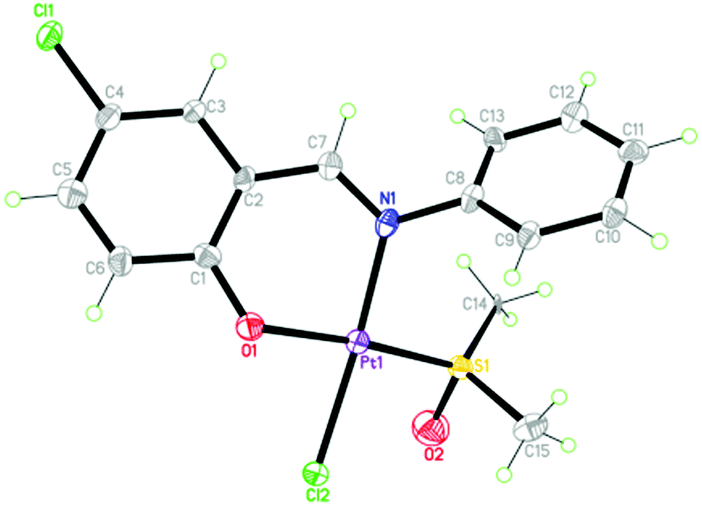 | ||
| Fig. 1 The crystal structure of 1; thermal ellipsoids are given at the 30% probability level. | ||
Using cisplatin as the control, the anticancer potential of 1 was tested on MCF7 cells. As shown in Fig. 2, after MCF7 cells were incubated with 1 for 48 h, the cell viability dramatically decreased from about 73% to 17% with the increase of the concentration of 1 from 0.1 to 10 μM. In contrast, a similar treatment using cisplatin alone caused a decrease from 90% to 50%. Note that the IC50 values of 1 and cisplatin were 0.32 and 9.56 μM, respectively. The value of 0.32 μM was also obviously smaller than those for the reported similar Pt(II) complexes (Table S3, ESI†).14 Overall, 1 exhibited efficient anti-proliferation effect in both dose- and time-dependent manners (Fig. S3, ESI†).
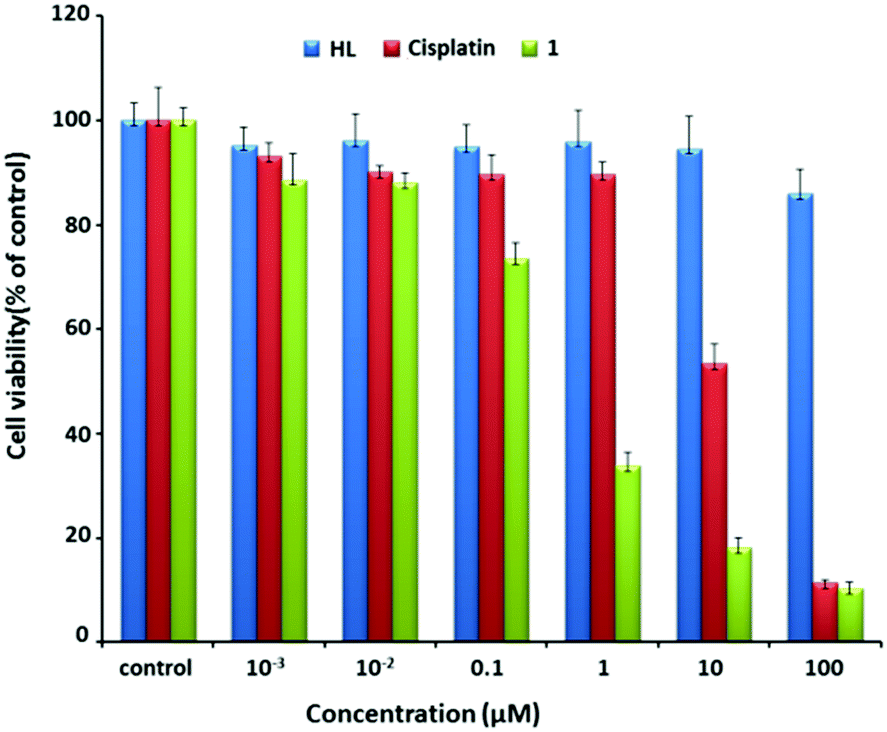 | ||
| Fig. 2 Cell viability after incubation with varying concentrations of HL (blue), cisplatin (red), and 1 (green) for 48 h. | ||
The effect of 1 on the apoptosis of MCF7 cells was also investigated using cisplatin as the control. As shown in Fig. 3, the percentages of cells in early and late apoptosis induced by 1 at the concentrations of 1, 5 and 10 μM were 34, 46 and 53% respectively, suggesting a dose-dependent apoptosis-inducing manner. A concentration of 10 μM of 1 reduces the amount of viable MCF7 cells to 44% by the induction of both early and late apoptosis, while the same concentrations of cisplatin and ligand had insignificant effects within the chosen exposure time (Fig. S4, ESI†), demonstrating that the apoptosis-inducing potency of 1 exceeded that of cisplatin for MCF7 cells.
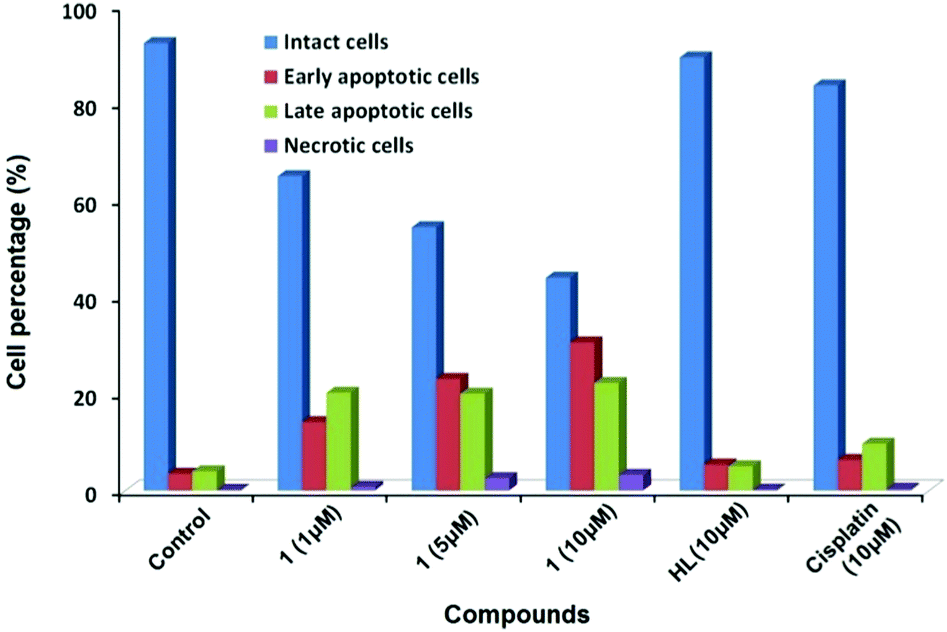 | ||
| Fig. 3 The percentage of intact, early apoptotic, late apoptotic, and necrotic cells in flow cytometric analysis of MCF7 cells after incubation with 1 (1, 5, and 10 μM), HL (10 μM) and cisplatin (10 μM) for 24 h. | ||
DNA is accepted to be a typical target for many Pt(II)-based anticancer drugs, and their interactions are usually studied using UV-Vis spectroscopy.15 As shown in Fig. 4, with increasing amounts of calf thymus DNA (CT-DNA), the absorption spectra of 1 at 285 nm exhibited a maximum hypochromism of 18.8% (−ΔA/A), obviously lower than classical DNA intercalators, such as [Ru(bpy)2(dppz)]2+ showing hypochromism of 40.1%.15 This result possibly implies a kind of weak 1-DNA intercalative interaction. The binding constant (Kb) was 2.17 × 104 M−1, also much lower than that of [Ru(bpy)2(dppz)]2+ (5.0 × 106 M−1) and some reported Pt(II) complexes with a potent anticancer activity (Table S3, ESI†).16
 | ||
| Fig. 4 Absorption spectra of 1 (20 μM) in the absence and in the presence of CT-DNA (0–140 μM) in Tris–HCl–NaCl buffer (pH 7.2); inset: plot of CDNA/(εa − εf) vs. CDNA for absorption titration of CT-DNA with 1. | ||
In addition to UV-vis spectroscopy, the DNA-binding property of 1 was also studied using circular dichroism (CD) spectroscopy and compared with that of cisplatin. As shown in Fig. S5 (ESI†), the characteristic positive and negative bands for CT-DNA appeared at 277 and 246 nm, respectively. In the presence of 1, the intensities of both bands show moderate enhancement without band shifts. By contrast, in the presence of cisplatin, the intensities show an increase at low cisplatin/DNA ratios (rb) but an obvious decrease at the highest ratios (rb = 1) with visible red-shifts (ca. 8 and 2 nm for positive and negative bands, respectively). The results indicate that the binding of 1-DNA is weaker than that of cisplatin–DNA.
Since guanine-N7 is the preferred binding site in DNA for many Pt(II) complexes,17 we monitored the reactivity of both 1 and cisplatin with mononucleotides 5′-GMP through UV-vis and 1H NMR spectroscopy. These spectra show that GMP-cisplatin binding is obviously stronger than the GMP-1 interaction (see Fig. S6 and S7 (ESI†), and corresponding discussion in the ESI† for details).
In brief, the above studies reveal that the binding of 1 to DNA may be weaker than that of cisplatin, which is inconsistent with the higher antiproliferation activity of 1 over cisplatin. We speculate that 1 might interact with other cellular targets apart from DNA.
It is found that PTP1B is overexpressed in MCF7 cells, which may promote tumour development and progression.18 So the inhibitory activity of 1 on recombinant PTP1B was evaluated with three other PTPs as a comparison. As shown in Fig. 5, the IC50 values of 1 inhibiting PTP1B, TCPTP, SHP-1, and hematopoietic protein tyrosine phosphatase (HePTP) are 0.25, 4.51, 4.65, and 31.6 μM, respectively. Specifically, 1 shows much stronger potency against PTP1B than that against TCPTP, SHP-1 (ca. 18-fold) and HePTP (ca. 126-fold). Hence, 1 is a potent and selective PTP1B inhibitor. Considering that the inhibition of cisplatin against PTP1B (IC50 = 1.45 μM) was less than 1 (Fig. S8, ESI†) and the HL ligand itself did not show obvious inhibition activity against four PTPs even at 100 μM, we speculate that the anti-proliferation and apoptosis induction of 1 on MCF7 cells may be associated with its potent inhibition against PTP1B.
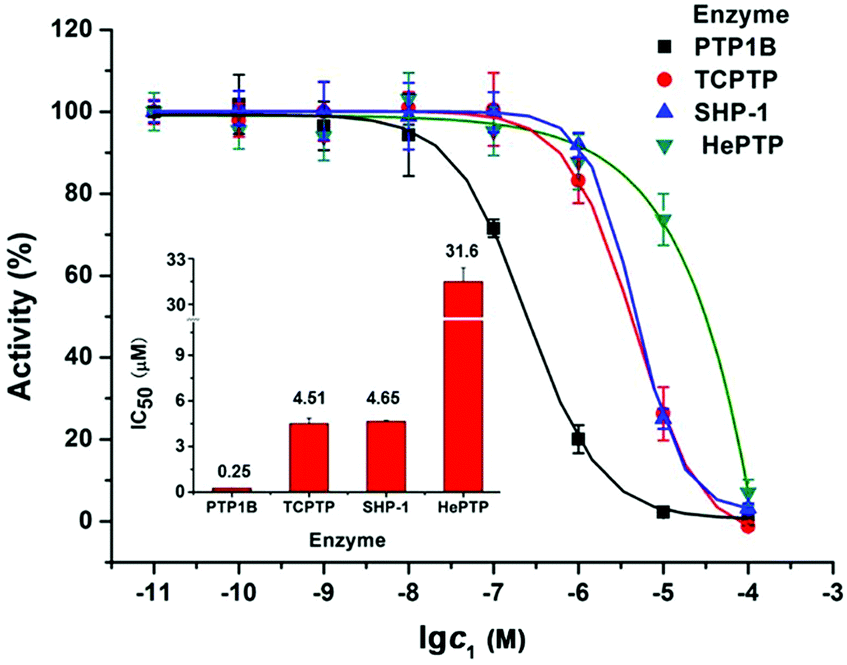 | ||
| Fig. 5 Concentration-dependent inhibition of four tyrosine phosphatases by 1; the inset shows IC50 values. | ||
To identify the possible inhibition mode of 1 on the PTP1B activity, an enzyme kinetics experiment was performed. The lines in the Lineweaver–Burk plots converge on the x-axis left to y-axis which reveals a non-competitive inhibition mechanism (Fig. S9, ESI†), suggesting that 1 may bind to PTP1B at an allosteric site.
Next, the interaction between 1 and PTP1B was investigated by fluorescence spectroscopy. As shown in Fig. S10 and S11 (ESI†), the fluorescence intensity of PTP1B at 335 nm gradually decreased with increasing concentration of 1 or cisplatin, indicating the possible binding between the platinum complexes and PTP1B. Among the three types of amino acid residues of protein emitting fluorescence around 335 nm, Trp is more sensitive to the conformation change of a protein than Tyr and Phe. Therefore, such an intensity decrease may reflect the quenching of Trp fluorescence in PTP1B. The stoichiometries of both 1 and cisplatin binding to PTP1B were calculated to be close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and their binding constants were 1.35 × 108 and 1.86 × 107 M, respectively, revealing stronger binding strength of 1 to PTP1B than that of cisplatin.
:1, and their binding constants were 1.35 × 108 and 1.86 × 107 M, respectively, revealing stronger binding strength of 1 to PTP1B than that of cisplatin.
Besides the studies on the recombinant PTP1B inhibition activity, the cellular efficacy of 1 selectively inhibiting PTP1B was also assessed by examining its effects on the phosphorylation levels of some PTP substrates using Western blotting analysis in MCF7 cells. Previous studies have shown that PTP1B and TCPTP could remove the phosphate from p-Src529 and p-Src418, respectively.19 Treatment of cells with 1 led to a dose-dependent increase in the phosphorylation level of p-Src(Y529), while no obvious change in p-Src(Y418) was observed (Fig. 6a), which implied the significant inhibition of PTP1B function. This can be the result of two possible reasons: inhibition of the PTP1B activity or/and PTP1B expression. To clarify this, we measured the total amount of PTP1B in the presence of 1 using a PTP1B-antibody by the Western blot assay. The results indicate that 1 scarcely changes the PTP1B expression in cancer cells (Fig. 6b). Thus the increased phosphorylation level of p-Src(Y529) may be attributed to the potent and selective inhibition of 1 against cellular PTP1B activity. Apart from the PTP1B specific substrate p-Src(Y529), the phosphorylation levels of the other PTP1B substrates (such as p-IRS-1(Y896), p-IR/IGF1R(pYpYpY1158/1162/1163) and p-EGFR(Y1092))20 were also distinctly improved with the increased concentration of 1. As a positive control, cisplatin has no effect on the phosphorylation levels of p-Src(Y529) and p-Src(Y418) even at 100 μM (Fig. S12, ESI†), indicating that cisplatin doesn’t distinctly inhibit the activities of cellular PTP1B and TCPTP.
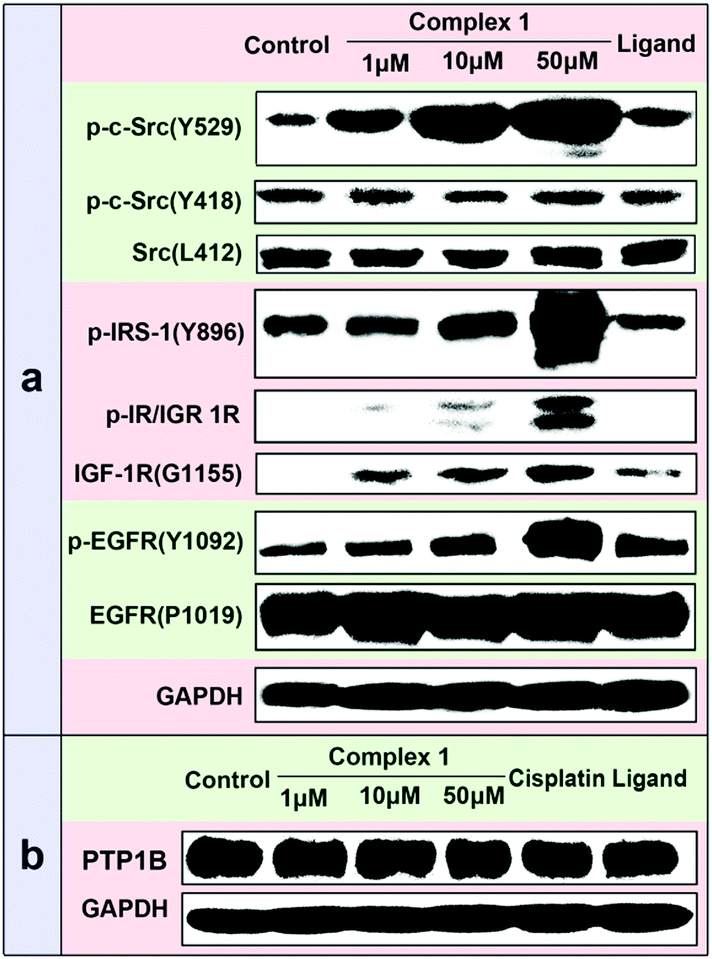 | ||
| Fig. 6 (a) Effects of different concentrations of 1 on the phosphorylation levels of several PTP substrates among MCF7. From left to right, lane 1: control; lane 2–4: 1 (1, 10 and 50 μM); lane 5: ligand (50 μM). (b) Effects of 1, cisplatin and ligand on the level of cellular PTP1B expression among MCF7. From left to right, lane 1: control; lane 2–4: 1 (1, 10 and 50 μM); lane 5: cisplatin (50 μM); lane 6: ligand (50 μM). | ||
To further explain the relationship between the anticancer potential of 1 and its inhibition against PTP1B, the cell lines HepG2 with high PTP1B expression and A549 with low PTP1B expression were chosen for comparison with MCF7 (Fig. S13, ESI†). The results show that 1 efficiently inhibits the proliferation of HepG2 cells (IC50 = 0.37 μM), similar to its effect on MCF7, but hardly affects the proliferation of A549 cells (Fig. S14, ESI†). Such results indicate again that the anti-proliferative ability of 1 may be related to the potent inhibition of the PTP1B activity.
We also measured the uptakes of 1 and cisplatin by determining the Pt contents in three cells using ICP-MS (Table S4, ESI†). The Pt contents in MCF7, HepG2, and A549 are 26.23, 18.30, and 19.18 ng per 106 cells, respectively. The cellular uptakes of 1 in the three cells are comparable, indicating that there is no direct connection between the anti-proliferation effect of 1 and its cellular uptake. Nevertheless, the uptakes of 1 are 17-, 7-, and 11-fold stronger than those of cisplatin for MCF7, HepG2, and A549 cells, respectively. The higher cellular uptake may in-part account for the much higher anti-proliferation activity of 1 than cisplatin.
In summary, we have found that Pt(II) complex 1 possesses much higher anti-proliferation activity than cisplatin. The mechanism of anti-proliferation of 1 was demonstrated to be potent inhibition of PTP1B, which significantly influences the cellular phosphorylation level and thus may further influence the intracellular signal transduction pathway. Such a mechanism is distinctly different from the famous DNA-damaging mechanism for cisplatin, thereby providing a new clue for designing novel platinum-based anticancer drugs with PTP1B as the potential targets.
The study is supported financially by NSFC (Grant No. 21471092, 21571118, 21671124, and 21001070), Shanxi “1331 Project” Engineering Research Center (PT201807), Scientific Instrument Center of Shanxi University, and the beamline 3W1A of BSRF.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des., Dev. Ther., 2017, 11, 599 CrossRef CAS PubMed; (b) K. Suntharalingam, J. J. Wilson, W. Lin and S. J. Lippard, Metallomics, 2014, 6, 437 RSC.
- S. Dilruba and G. V. Kalayda, Cancer Chemother. Pharmacol., 2016, 77, 1103 CrossRef CAS.
- (a) A. I. Matesanz, C. Hernandez, A. Rodriguez and P. Souza, Dalton Trans., 2011, 40, 5738 RSC; (b) A. Casini and J. Reedijk, Chem. Sci., 2012, 3, 3135 RSC; (c) N. Muhannad, N. Sadia, C. C. Zhu, C. Luo, Z. J. Guo and X. Y. Wang, Chem. Commun., 2017, 53, 9971 RSC; (d) N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106 RSC; (e) O. Pinato, C. Musetti and C. Sissi, Metallomics, 2014, 6, 380 RSC; (f) D. Hu, C. Yang, C. N. LoK, F. Xing, P. Y. Lee, Y. M. E. Fung, H. Jiang and C. M. Che, Angew. Chem., Int. Ed., 2019, 58, 10914 CrossRef CAS PubMed; (g) Z. Zhu, Z. Wang, C. Zhang, Y. Wang, H. Zhang, Z. Gan, Z. Guo and X. Wang, Chem. Sci., 2019, 10, 3089 RSC.
- D. Griffith, J. P. Parker and C. J. Marmion, Anti-Cancer Agents Med. Chem., 2010, 10, 354 CrossRef CAS.
- (a) R. Sasanelli, A. Boccarelli, D. Giordano, M. Laforgia, F. Arnesano, G. Natile, C. Cardellicchio, M. A. M. Capozzi and M. Coluccia, J. Med. Chem., 2007, 50, 3434 CrossRef CAS; (b) F. Arnesano, A. Boccarelli, D. Cornacchia, F. Nushi, R. Sasanelli, M. Coluccia and G. Natile, J. Med. Chem., 2009, 52, 7847 CrossRef CAS.
- (a) L. J. Parker, L. C. Italiano, C. J. Morton, N. C. Hancock, D. B. Ascher, J. B. Aitken, H. H. Harris, P. Campomanes, U. Rothlisberger, A. D. Luca, M. L. Bello, W. H. Ang, P. J. Dyson and M. W. Parker, Chem. – Eur. J., 2011, 17, 7806 CrossRef CAS; (b) W. H. Ang, I. Khalaila, C. S. Allardyce, L. Juillerat-Jeanneretand and P. J. Dyson, J. Am. Chem. Soc., 2005, 127, 1382 CrossRef CAS.
- (a) D. Griffith, M. P. Morgan and C. J. Marmion, Chem. Commun., 2009, 6735 RSC; (b) D. M. Griffith, B. Duff, K. Y. Suponitsky, K. Kavanagh, M. P. Morgan, D. Egan and C. J. Marmion, J. Inorg. Biochem., 2011, 105, 793 CrossRef CAS.
- (a) K. Becker, C. Herold-Mende, J. J. Park, G. Lowe and R. H. Schirmer, J. Med. Chem., 2001, 44, 2784 CrossRef CAS; (b) Y. Lo, T. Ko, W. Su, T. Su and A. H. J. Wang, J. Inorg. Biochem., 2009, 103, 1082 CrossRef CAS PubMed; (c) Y. Lo, W. Su, T. Ko, N. Wang and A. H. J. Wang, J. Biomol. Struct. Dyn., 2011, 29, 267 CrossRef CAS PubMed.
- N. K. Tonks, Nat. Rev. Mol. Cell Biol., 2006, 7, 833 CrossRef CAS.
- (a) L. P. Lu and M. L. Zhu, Antioxid. Redox Signaling, 2014, 20, 2210 CrossRef CAS; (b) Y. R. Xu and G. J. Fisher, J. Cell Commun. Signal., 2012, 6, 125 CrossRef CAS; (c) D. P. Labbe, S. Hardy and M. L. Tremblay, Prog. Mol. Biol. Transl. Sci., 2012, 106, 253 CAS; (d) W. J. A. J. Hendriks, A. Elson, S. Harroch, R. Pulido, A. Stoker and J. den Hertog, FEBS J., 2013, 280, 708 CrossRef CAS PubMed.
- (a) J. R. Wiener, B. J. Kerns, E. L. Harvey, M. R. Conaway, J. D. Iglehart, A. Berchuck and R. C. Bast Jr., J. Natl. Cancer Inst., 1994, 86, 372 CrossRef CAS; (b) R. He, Z. Yu, R. Zhang and Z. Zhang, Acta Pharmacol. Sin., 2014, 35, 1227 CrossRef CAS; (c) S. Lin, Y. Lai, C. Wang, M. Tsai, S. Yu, G. Chang and J. J. W. Chen, Mol. Cell. Biol., 2012, 72, 4183 Search PubMed; (d) M. K. Joo, J. Park, H. S. Yoo, B. J. Lee, H. J. Chun, S. W. Lee and Y. Bak, Tumor Biol., 2016, 37, 4603 CrossRef CAS.
- (a) L. R. Bollu, A. Mazumdar, M. I. Savage and P. H. Brown, Clin. Cancer Res., 2017, 23, 2136 CrossRef CAS; (b) M. T. Rukert, P. V. de Andrade, V. S. Santos and V. S. Silveira, Cell. Mol. Life Sci., 2019, 76, 2571 CrossRef; (c) J. S. Lazo, K. E. McQueeney, J. C. Burnett, P. Wipf and E. R. Sharlow, Int. J. Biochem. Cell Biol., 2018, 96, 171 CrossRef CAS.
- (a) C. X. Yuan, M. L. Zhu, Q. M. Wang, L. P. Lu, S. Xing, X. Q. Fu, Z. Jiang, S. Zhang, Z. W. Li, Z. Y. Li, R. T. Zhu, L. Ma and L. Q. Xu, Chem. Commun., 2012, 48, 1153 RSC; (b) Y. Q. Jia, L. P. Lu, M. L. Zhu, C. X. Yuan, S. Xing and X. Q. Fu, Eur. J. Med. Chem., 2017, 128, 287 CrossRef CAS.
- F. Rahman, A. Ali, R. Guo, Y. Zhang, H. Wang, Z. Lia and D. Zhang, Dalton Trans., 2015, 44, 2166 RSC.
- (a) Q. Gan, C. L. Zhang, B. F. Wang, Y. H. Xiong, Y. L. Fu, Z. W. Mao and X. Y. Le, RSC Adv., 2016, 6, 35952 RSC; (b) S. S. Bhat, A. S. Kumbhar, P. Lönnecke and E. Hey-Hawkins, Inorg. Chem., 2010, 49, 4843 CrossRef CAS; (c) J. G. Liu, Q. L. Zhang, X. F. Shi and L. N. Ji, Inorg. Chem., 2001, 40, 5045 CrossRef CAS; (d) Z. F. Chen, Y. F. Shi, Y. C. Liu, X. Hong, B. Geng, Y. Peng and H. Liang, Inorg. Chem., 2012, 51, 1998 CrossRef CAS.
- (a) M. J. Waring, J. Mol. Biol., 1965, 13, 269 CrossRef CAS; (b) A. Bielawska, B. Poplawska, A. Surayński, R. Czarnomysy and K. Bielawski, Eur. J. Pharmacol., 2010, 643, 34 CrossRef CAS; (c) M. Jamshidi, R. Yousefi, S. M. Nabavizadeh, M. Rashidi, M. G. Haghighi, A. Niazi and A. Moosavi-Movahedi, Int. J. Biol. Macromol., 2014, 66, 86 CrossRef CAS.
- (a) Z. Chen, S. Zhang, Z. Zhu and Y. Zhang, New J. Chem., 2017, 41, 6340 RSC; (b) A. M. J. Fichtinger-Schepman, J. L. van der Veer, J. H. J. den Hartog, P. H. M. Lohman and J. Reedijk, Biochemistry, 1985, 24, 707 CrossRef CAS.
- (a) S. G. Julien, N. Dubé, M. Read, J. Penney, M. Paquet, Y. Han, B. P. Kennedy, W. J. Muller and M. L. Tremblay, Nat. Genet., 2007, 39, 338 CrossRef CAS; (b) C. Blanquart, S. Karouri and T. Lssad, Biochem. Biophys. Res. Commun., 2009, 387, 748 CrossRef CAS; (c) M. Yu, Z. Liu, Y. Liu, X. Zhou, F. Sun, Y. Liu, L. Li, S. Hua, Y. Zhao, H. Gao, Z. Zhu, M. Na, Q. Zhang, R. Yang, J. Zhang, Y. Yao and X. Chen, FEBS J., 2019, 286, 1136 CrossRef CAS.
- (a) J. D. Bjorge, A. Pang and D. J. Fujita, J. Biol. Chem., 2000, 275, 41439 CrossRef CAS; (b) S. Zhang, L. Chen, Y. Luo, A. Gunawan, D. S. Lawrence and Z. Y. Zhang, J. Am. Chem. Soc., 2009, 131, 13072 CrossRef CAS; (c) F. Liang, S. Y. Lee, J. Liang, D. S. Lawrence and Z. Y. Zhang, J. Biol. Chem., 2005, 280, 24857 CrossRef CAS.
- (a) A. M. Valverde, J. Revuelta-Cervantes, L. Menes, A. Gonzalez-Rodriguez, V. Pardo, P. de la Villa, D. J. Burks and A. I. Arroba, Eur. J. Ophthalmol., 2013, 23, 450 Search PubMed; (b) E. N. Gurzov, Me. Tran, M. A. Fernandez-Rojo, T. L. Merry, X. Zhang, Y. Xu, A. Fukushima, M. J. Waters, M. J. Watt, S. Andrikopoulos, B. G. Neel and T. Tiganis, Cell Metab., 2014, 20, 85 CrossRef CAS; (c) C. Bousquet, N. Delesque, F. Lopez, N. Saint-Laurent, J. Estève, K. Bedecs, L. Buscail, N. Vaysse and C. Susini, J. Biol. Chem., 1998, 273, 7099 CrossRef CAS; (d) H. Zhang and H. J. Forman, Free Radical Biol. Med., 2015, 89, 701 CrossRef CAS; (e) T. Tiganis, A. M. Bennett, K. S. Ravichandran and N. K. Tonks, Mol. Cell. Biol., 1998, 18, 1622 CrossRef CAS; (f) T. Yuan, H. Ma, Z. Du, X. Xiong, H. Gao, Z. Fang, L. He, H. Li and H. Gu, Biochem. Biophys. Res. Commun., 2017, 488, 439 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reactants and physical measurements, synthesis and characterization (X-ray, ESI-MS, IR and UV-vis) of 1, crystal structure refinements and crystallographic data, DNA-binding assays (CD, UV-vis and 1HNMR), PTP inhibition assays, the binding constant and the stoichiometry calculations, and cell biological assays. CCDC 1580462. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc06972k |
| This journal is © The Royal Society of Chemistry 2020 |