DOI:
10.1039/C9BM01563A
(Paper)
Biomater. Sci., 2020,
8, 201-211
Polyethyleneimine coated Fe3O4 magnetic nanoparticles induce autophagy, NF-κB and TGF-β signaling pathway activation in HeLa cervical carcinoma cells via reactive oxygen species generation†
Received
27th September 2019
, Accepted 4th October 2019
First published on 30th October 2019
Abstract
Fe3O4 magnetic nanoparticles (MNPs), as one of the most intensively researched NPs, have a range of applications in cancer treatments. In current research, we have focused on the influences of MNPs on cancer cells. We chose polyethyleneimine (PEI) coated MNPs (PEI-MNPs) as a model and they are colloidally stable in biological media. It can be proved that PEI-MNPs result in autophagy induction via mTOR-Akt-p70S6 K and ATG7 signaling pathways. For the first time, we have reported that PEI-MNPs activate both NF-κB and TGF-β signaling, two key pro-inflammatory pathways, in cancer cells. More significantly, we have found that autophagy induction and NF-κB and TGF-β activation can be efficiently suppressed through the inhibition of PEI-MNP dependent reactive oxygen species (ROS) over-production. ROS are deemed as a ‘double edge sword’ for cancer cells, owing to the cancer-suppressing and cancer-promoting actions. Our findings would be useful for designing MNPs induced ROS anti-cancer strategies or diminishing long-term toxic effects.
1. Background
Nanosized materials have striking features compared with objects in common sizes.1–4 Magnetite nanoparticles, also called Fe3O4 nanoparticles (NPs) are arguably one of the most researched NPs especially in cancer theranostics.5–7 They are applicable as targeting carriers for anti-cancer drugs or nucleic acids,8–10 or contrast agents for magnetic resonance imaging (MRI)-based diagnosis.11,12 Besides, they can be used to form different types of supramolecular nanoassemblies,13,14 to perform magnetic hyperthermia15 and photothermal16 based therapy for cancer ablation. Also, MNPs are capable of sensitising some anti-cancer compounds or overcoming drug resistance.17,18 Ultrasmall MNPs are reported to induce tumour growth inhibition possibly by stimulation of pro-inflammatory macrophages.19 What's more, the combination of MNPs with immunomodulators is capable of enhancing tumour-localised immunotherapy.20 All these treatments require direct interactions of MNPs with cancerous cells, which yet remain poorly understood. Furthermore, reactive oxygen species (ROS) are termed chemically active oxygen-containing molecules, which exert oxidative stress.21 It is implicated that ROS play crucial roles in cell signaling and homeostasis.22 The escalated ROS production is a common phenomenon in MNPs treated cell models, which has been widely reported.23,24 However, the global functions of ROS are rarely clarified. In the current study, we intended to underpin the understanding of the MNPs elicited cellular responses; more importantly, we tried to establish the relationship of ROS with these observed cellular responses.
2. Materials and methods
2.1 Materials
Rapamycin (RAPA), chloroquine diphosphate salt (CQ), bafilomycin A1 (BAF), N-acetyl-L-cysteine (NAC), monodansylcadaverine (MDC), acridine orange (AO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and diphenyleneiodonium chloride (DPI) were obtained from Sigma-Aldrich (USA). Apocynin and 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF) were purchased from Aladdin Biochemical Technology (China). 6-Amino-4-(4-phenoxyphenylethylamino)quinazoline (QNZ) and schisandrin B (Sch B) were purchased from MedChemexpress Co. Ltd (USA). Reactive oxygen species assay kit C1300 was purchased from Applygen Technologies Inc. (China). MitoTracker Red CMXRos was purchased from Yeasen Biotechnology Co., Ltd (China).
2.2 Methods
2.2.1 Preparation and characterisation of MNPs.
10 nm polyethyleneimine coated Fe3O4 magnetic nanoparticles (PEI-MNPs) were prepared under hydrothermal conditions as described previously.25,26 TEM images were captured using a JEM-2100 transmission electron microscope (JEOL, Japan) at 200 kV. SEM images were obtained using a JEM-6700F scanning electron microscope (ZEISS-SUPRA55, Germany). The micro-elemental analysis of 10 nm PEI-MNPs was performed using an energy-dispersive X-ray device (Oxford-Aztec X-Max80). XRD experiments were performed on a 6100 X-ray diffraction instrument (Shimadzu, Japan). Magnetic measurements were carried out using a Lakeshore 7407 vibrating sample magnetometer (Lakeshore, USA) at 293 K. The DLS and zeta potential measurements were carried out using a ZS90 Zetasizer nanoanalyser (Malvern, UK). The FTIR spectrum was recorded using a Tensor 27 FTIR spectrometer (Bruker, Germany) in transmission mode. Dry nano-particles were mixed with KBr crystals, and then pressed into pellets before measurements.
2.2.2 Cell culture.
HeLa (human cervical cancer), MCF-7 (human breast cancer) and HepG2 (human liver cancer) cells were maintained in DMEM medium. L02 cells were grown in RPMI 1640 medium. Each type of medium was supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin. All cells were incubated in a humidified atmosphere with 5% CO2 at 37 °C. Please note that all cell lines used were preserved by our laboratory and originally obtained from ATCC.
2.2.3 Fluorescence imaging of cells.
For AO, MDC or MitoTracker Red CMXRos staining, HeLa cells were seeded and cultured at 37 °C for 24 h. Different concentrations of agents for cell treatments were added and incubated where necessary. After this, 2 mM AO, 50 mM MDC or 100 nM MitoTracker Red CMXRos was added to the cells and incubated at 37 °C for 15, 40 and 30 min respectively in the dark. The cells were washed three times with Dulbecco's phosphate buffered saline (DPBS). Images were captured by using an Olympus fluorescence microscope.
2.2.4 Observations of autophagic vacuoles (AVs) by transmission electron microscopy (TEM).
HeLa cells with or without PEI-MNP treatment were fixed in 2.5% glutaraldehyde (pH 7.5) at 4 °C for 2 h and post-fixed with 0.1 M osmic acid for 3 h at room temperature. After fixation, cells were dehydrated in a gradient of 50–100% ethanol and embedded in Spurr resin. 70 nm ultra-thin sections were obtained using a microtome (Leica EM UC6). After double staining with lead citrate and uranyl acetate, sections were observed and pictured under a transmission electron microscope (JEM-1230; JEOL) at 40–120 kV voltages.
2.2.5 Western blotting.
The harvested cells were washed with ice-cold PBS twice and then lysed with a RIPA buffer supplemented with 1 mM PMSF (phenylmethylsulphonyl fluoride), protease inhibitors and phosphatase inhibitors where necessary. Protein samples were extracted and then resolved by 10 or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a methanol-activated PVDF membrane. The membrane was then blocked in PBST buffer containing 5% non-fat dry milk for 1 h at room temperature. Blots were then incubated overnight at 4 °C with primary antibodies. HRP-conjugated secondary antibody was incubated for 2 h at room temperature. Finally, the chemiluminescence (ECL) signals were visualised by using a Sage Creation MiniChemi™ imaging system. Primary antibodies against LC3 (cat #L7543, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, Sigma-Aldrich, USA), ATG7 (cat #8558, 1:1000, Cell Signaling Technology, USA), p62 (cat #5114, 1:1000, Cell Signaling Technology, USA), phospho-p70S6 kinase (p-p70s6k) (cat #sc-8418, 1:1000, Santa Cruz Biotechnology, USA), phospho-mTOR (p-mTOR) (cat #5536, 1:1000, Cell Signaling Technology, USA), phospho-Smad2 (p-Smad2) (cat #ab53100, 1:1000, Abcam, USA) and p65 (cat #sc-71675, 1:200, Santa Cruz Biotechnology, USA) were used, and antibodies against Lamin A/C (cat #sc-7293, 1:200, Santa Cruz Biotechnology, USA) and β-actin (cat #KM9001 T, 1:5000, Sungene Biotech, China) were used as loading controls.
:1000, Sigma-Aldrich, USA), ATG7 (cat #8558, 1:1000, Cell Signaling Technology, USA), p62 (cat #5114, 1:1000, Cell Signaling Technology, USA), phospho-p70S6 kinase (p-p70s6k) (cat #sc-8418, 1:1000, Santa Cruz Biotechnology, USA), phospho-mTOR (p-mTOR) (cat #5536, 1:1000, Cell Signaling Technology, USA), phospho-Smad2 (p-Smad2) (cat #ab53100, 1:1000, Abcam, USA) and p65 (cat #sc-71675, 1:200, Santa Cruz Biotechnology, USA) were used, and antibodies against Lamin A/C (cat #sc-7293, 1:200, Santa Cruz Biotechnology, USA) and β-actin (cat #KM9001 T, 1:5000, Sungene Biotech, China) were used as loading controls.
2.2.6 Fluorescence imaging of eGFP-LC3 puncta.
HeLa cells stably expressing eGFP-LC3 were seeded on the coverslips with or without PEI-MNPs. Cells were fixed with cold methanol for 5 min at −20 °C. Then the cells were washed with cold DPBS three times. The coverslips were then sealed with Mowiol in the dark. Images were captured and analysed using an inverted fluorescence microscope (Nikon Eclipse Ti).
2.2.7 Luciferase reporter assay.
HeLa cells were transfected with the NF-κB-Luc reporter or TGF-β-Luc reporter plasmids together with the pRL-TK Renilla luciferase plasmids (Promega). The following steps were carried out based on a previous publication.27
2.2.8 Reactive oxygen species (ROS) analysis.
ROS generation in HeLa cells after 24 h treatment of PEI-MNPs was analysed by staining the cells with 10 μM 2,7-dichlorofluorescin diacetate (H2DCFDA) for 30 min in the dark. For morphological study, the cells were observed and pictured using a fluorescence inverted microscope (Nikon Eclipse).
2.2.9 Reverse transcription polymerase chain reaction (RT-PCR).
Total RNA was isolated from HeLa cells using TRIzol. The synthesis of single-strand complementary DNA (cDNA) was performed using TransScript® All-in-One First-Strand cDNA Synthesis SuperMix (Transgen). Polymerase chain reactions were performed in a final system containing 2 μL of cDNA, 2 μL each of the p62 forward (5′-ATCGGAGGATCCGAGTGT-3′) and reverse (5′-TGGCTGTGAGCTGCTCTT-3′) primers, 25 μL 2 × Taq Master Mix and RNase-free water with denaturation at 94 °C for 15 s, annealing at 57 °C for 30 s, and extension at 68 °C for 15 s for 30 cycles, followed by incubation at 68 °C for 2 min. GAPDH was used as the control with the forward primer 5′-ACAGCAACACAGAAGACCGT-3′ and the reverse primer 5′-GGCAGGTTTCTCAAGACGGA-3′. The PCR products were visualised with GelRed staining under UV light after electrophoretic separation on agarose gel.
2.2.10 Statistical analysis.
The values obtained in the experiments were expressed as the mean ± standard deviation (SD) and were analysed by Student's t-test where necessary. All statistical analyses were performed using SPSS 17.0 software and P < 0.05 was considered statistically significant.
3. Results and discussion
3.1 Characterisation of 10 nm PEI-MNPs
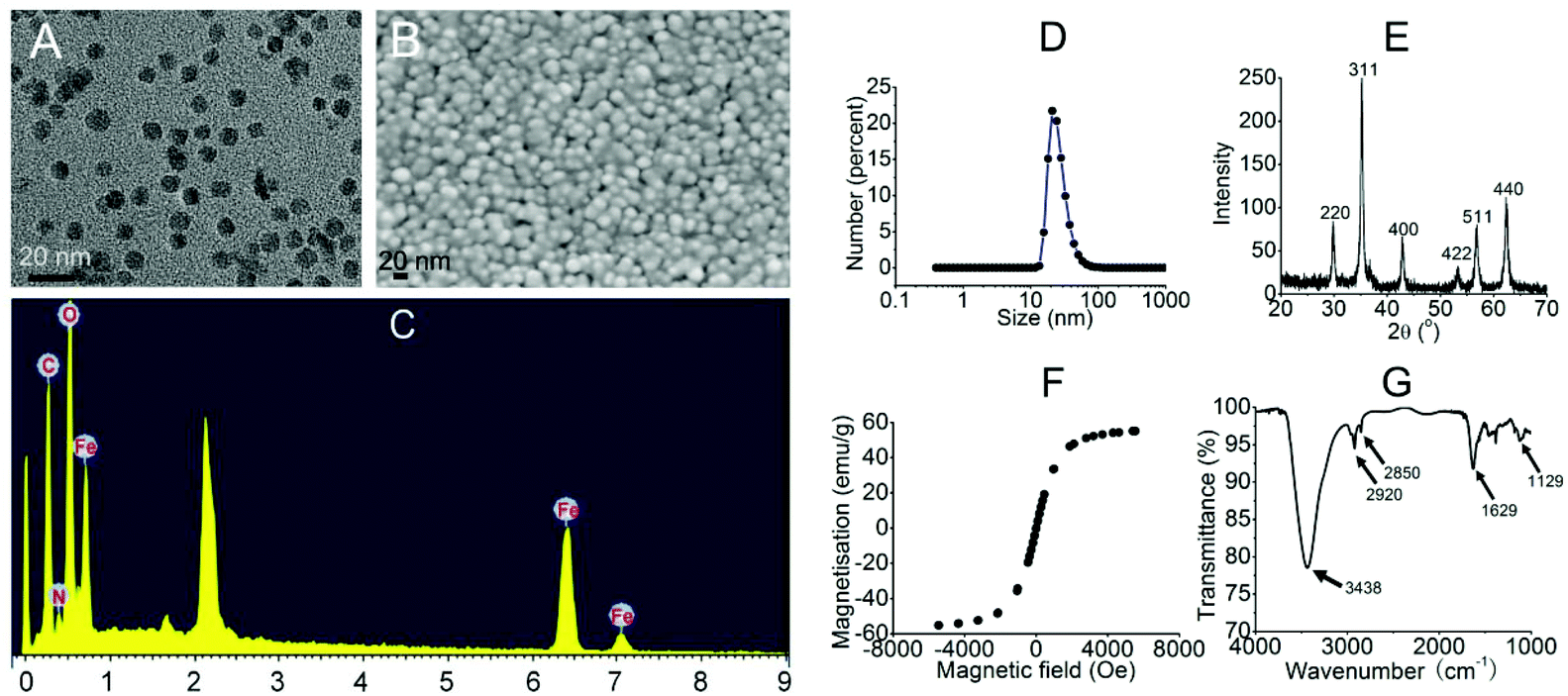
To start with, we intended to use PEI-MNPs throughout the study, as PEI is a well-proved bio-compatible polymer28 and would render MNPs colloidally stable in biological media.29 Thus we firstly synthesised PEI-MNPs and subjected them to biophysical characterisation. As shown in Fig. 1A and B, both TEM and SEM images were captured and the results suggested that the sizes of the PEI-MNPs were generally uniform and approximately 10 nm. Energy dispersive X-Ray spectroscopy (EDX) demonstrated the presence of Fe and O elements (Fig. 1C), which should be ascribed to the presence of PEI-MNPs. The dynamic light scattering (DLS) technique was used to measure the hydrodynamic particle size, which in our case was approximately 26.3 ± 4.42 nm (Fig. 1D). Fig. 1E shows the XRD (X-ray diffraction) pattern of the PEI-MNPs that displayed six characteristic peaks at 2θ of 30.6, 35.2, 42.8, 53.2, 56.8, and 62.5° and they matched very well with the (220), (311), (222), (400), (422), (511) and (440) planes of Fe3O4 crystals. The hysteresis loop of the PEI-MNPs demonstrated fine superparamagnetic properties (Fig. 1F). In the FT-IR spectrum of 10 nm PEI-MNPs (Fig. 1G), the strong absorption bands at 3438 and 1629 cm−1 were ascribed to the –NH2 groups introduced from polyethylenimine. The absorption bands at 2920 and 2850 cm−1 accounted for the –CH2– groups of polyethylenimine. The 1129 cm−1 band was owing to the stretching vibration of the C–N bond. The zeta potential (ζ) of the as-synthesised PEI-MNPs was measured as 25.1 ± 1.23 mV, which accorded well with previous data.30 This zeta potential translated to high colloidal stability in aqueous solution.
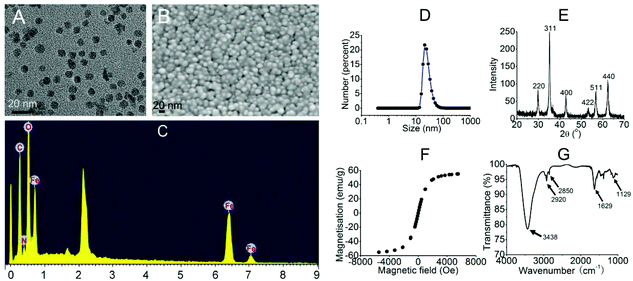 |
| | Fig. 1 The characterisation of the 10 nm PEI-MNPs. A: TEM; B: SEM; C: EDX; D: DLS; E: XRD; F: magnetisation curve; G: FT-IR. | |
3.2 PEI-MNPs cause overproduced ROS in cancer cells
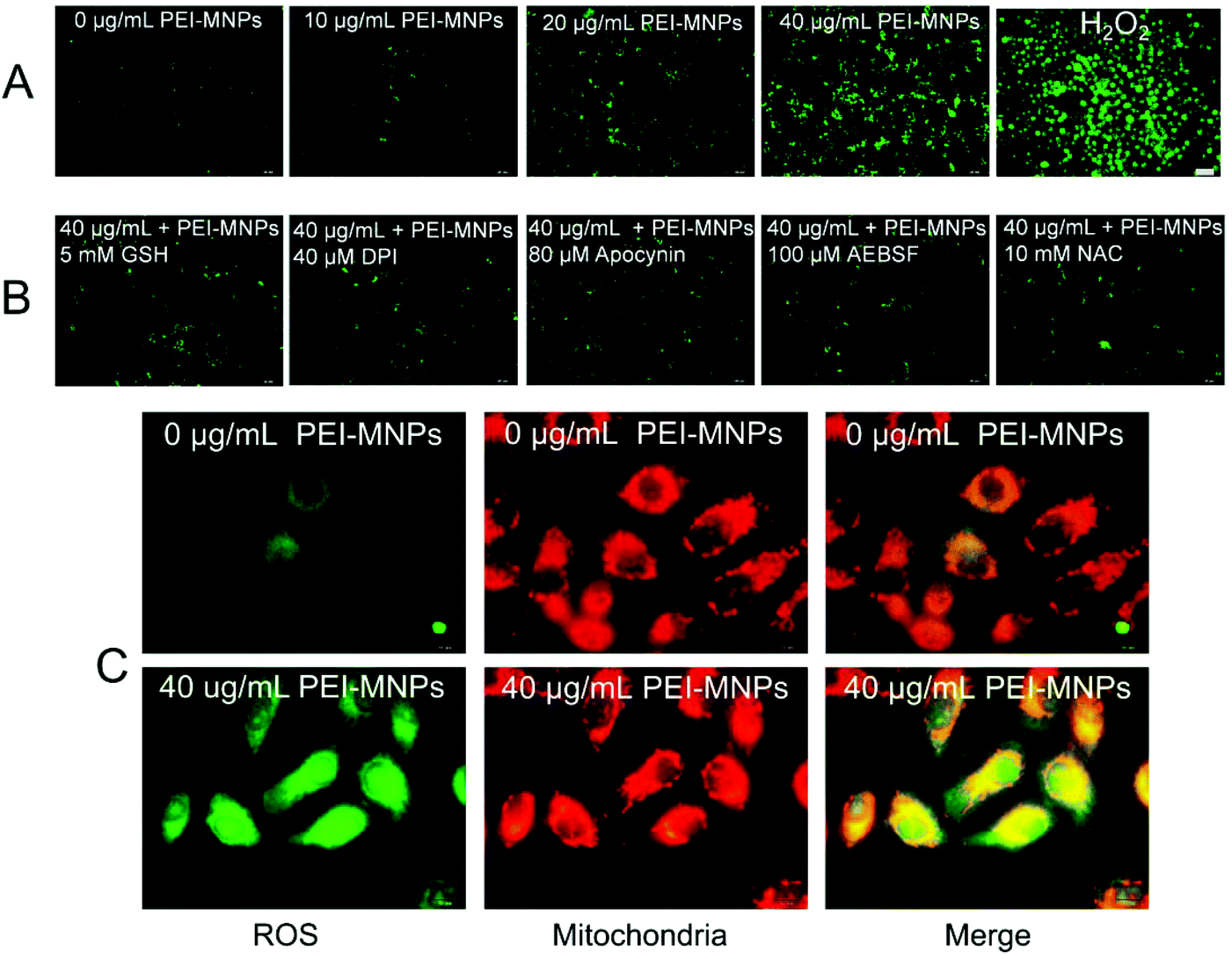
A few studies have confirmed that PEI-MNPs undergo cellular uptake, and we also observed this as shown in Fig. S-1.† It was noticed that the cellular ROS level was evaluated after the exposure of HeLa cells to PEI-MNPs. We used a well-known chemical probe H2DCFDA as a cell permeant indicator and its fluorescence can be specifically turned on by cellular ROS. For the control group, cells displayed weak fluorescence, suggesting a basal ROS level (Fig. 2A). In contrast to this, the treatment of PEI-MNPs to HeLa cells resulted in significant production of ROS in a dose-dependent manner as evidenced by strongly fluoro-stained cells (Fig. 2A). When adding a well-known and well-characterised OH˙ scavenger reduced L-glutathione (GSH)31,32 to the PEI-MNPs treated cells, it was observed that the fluorescence staining of ROS became much less intense (Fig. 2B), indicating that OH˙ greatly accounted for PEI-MNP induced ROS and thus the Fenton reaction contributed to ROS overproduction. It is generally believed that intracellular ROS derive from two sources, namely mitochondria and NADPH oxidases in cells. We proposed that this also applied to PEI-MNPs induced ROS over-production. We firstly employed three well known NADPH oxidase inhibitors DPI, apocynin, AEBSF together with N-acetyl-L-cysteine (NAC), a cell permeable antioxidant and a potent ROS scavenger, to probe ROS clearance. As shown in Fig. 2B, all four chemical agents showed a significant quenching effect toward excessive ROS. This manifested the fact that the generation of PEI-MNPs induced ROS, at least, partially from NADPH oxidases. But it was noted that even maximum inhibition hardly brought ROS to the basal level, implying some other sources existed. Next, we double-stained cells with the ROS specific dye H2DCFDA and MitoTracker Red CMXRos, as previously described.33 As shown in Fig. 2C, for the control group, there was a basal level of ROS, as expected. While for the 40 μg mL−1 (please note all concentrations in the study were used as particle concentration) PEI-MNPs treated group, cancer cells were intensively fluoro-stained; more importantly, significant ROS were co-localised with mitochondria, evidenced by the yellowish puncta (green plus red) in the merged images.
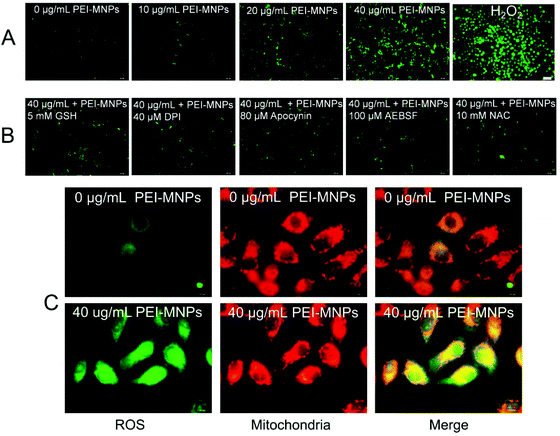 |
| | Fig. 2 PEI-MNPs induced over-production of ROS in cancer cells. A: Fluorescent images of ROS in HeLa cells. H2O2 group was treated with 30% H2O2 in a 1:1000 dilution for 15 min. B: Fluorescence images of ROS in HeLa cells. GSH was added into HeLa cells together with PEI-MNPs for 24 h. DPI, apocynin, and AEBSF were added for 30 min while NAC was added for 1 h respectively prior to the PEI-MNPs exposure. C: Fluorescence images of ROS and mitochondria in HeLa cells. To locate mitochondria, the cells were stained with MitoTracker Red CMXRos. Merged double images were constructed to confirm ROS generated by the mitochondria. Please note for all images, PEI-MNPs were treated for 24 h. | |
3.3 PEI-MNPs stimulate autophagy which can be significantly alleviated by decreasing cellular ROS level
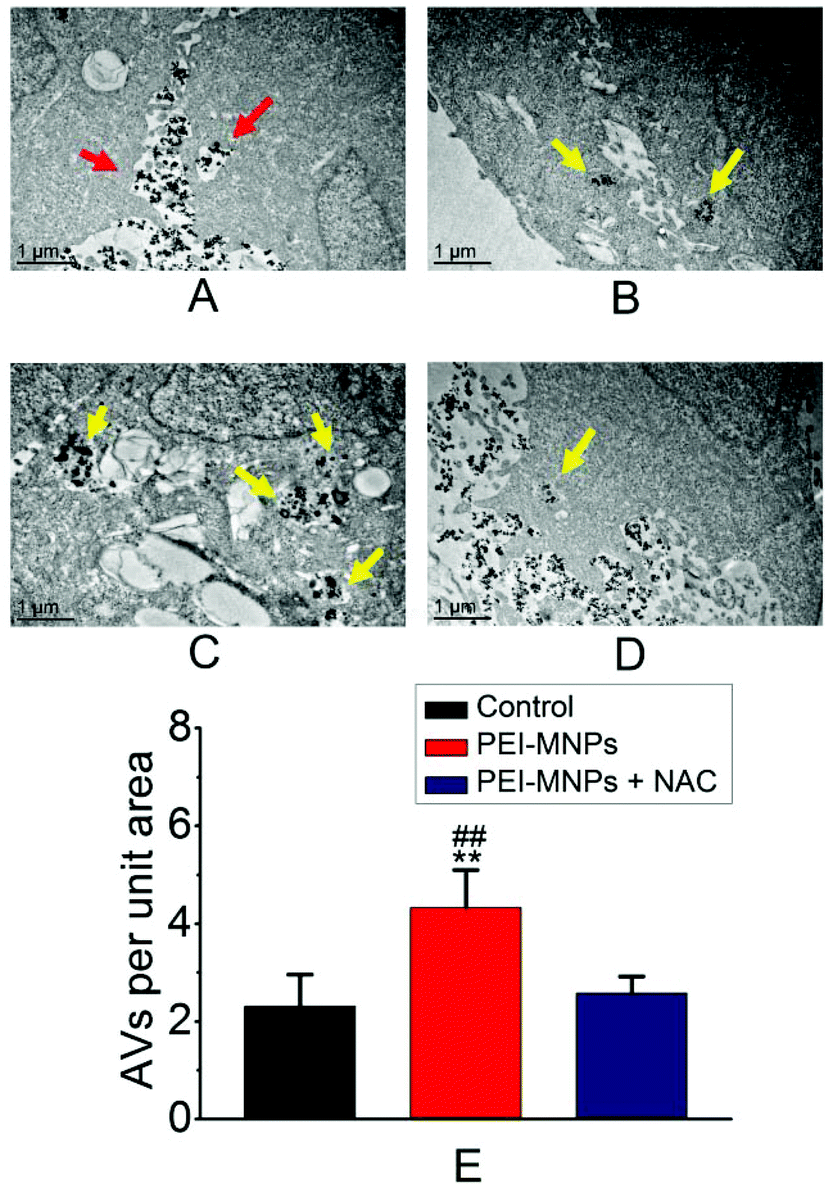
Autophagy can be considered as a ‘self-eating’ cellular process. Certain useless components are wrapped up within a vesicle termed as autophagosome (AP), and subsequently, an AP fuses with a lysosome (LY) to form an autophagolysosome (AL). The contents in AL are supposed to be degraded enzymatically, which generates amino acids, sugars, fatty acids, and nucleosides for macromolecular synthesis and energy provision.34,35 We firstly adopted transmission electron microscopy (TEM) to directly observe autophagic vacuoles (AVs). As shown in Fig. 3A, cells started to take up PEI-MNPs. The AVs stood at the basal level for the control group (Fig. 3B). The numbers of AVs (indicated by yellow arrows) considerably increased in PEI-MNPs treated cancer cells (P < 0.01), as shown in Fig. 3C and E, while the addition of 10 mM NAC 1 h before PEI-MNPs reversed this trend as evidenced by the fact that the AV numbers decreased correspondingly (P < 0.01), reaching a similar level as the control group, as shown in Fig. 3D and E. All these observations led to the conclusion that PEI-MNPs caused the accumulation of AVs and the addition of NAC would reverse this effect. Next, we investigated the change of LC3 (microtubule-associated protein light chain 3). As we know, the cytoplasmic form LC3-I is converted into lipidated form LC3-II. The latter aggregates onto the membranes of autophagic vacuoles (AVs) and thus is a common diagnostic marker of AV formation.
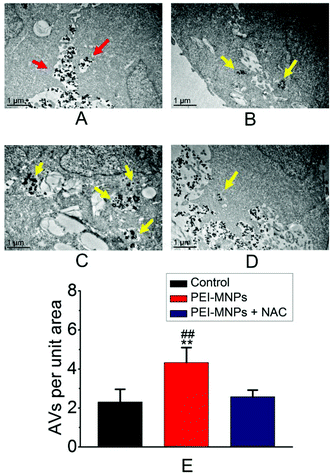 |
| | Fig. 3 PEI-MNPs treatment caused more accumulation of autophagic vacuoles (AVs) in cancer cells revealed by TEM. A: PEI-MNPs were dispersed between two cells and started to go into one cell. B: Control group (buffer treated HeLa cancer cells in the absence of PEI-MNPs). C: 40 μg mL−1 PEI-MNP treated group. D: PEI-MNPs were treated for 24 h and 10 mM NAC was pre-treated for HeLa cells for 1 h before PEI-MNPs addition. E: Statistical analysis of TEM results. Please note PEI-MNPs were treated for 24 h. **P < 0.01 vs. Controls. ##P < 0.01 of ‘40 μg mL−1 PEI-MNPs’ group vs. ‘40 μg mL−1 PEI-MNPs + 10 mM NAC’ group. Student's t-test was used. The red arrows indicate PEI-MNPs, and the yellow arrows indicate AVs. | |
The abundance of LC3-II and the number of LC3-positive puncta are positively related to the amount of AVs within the cells. It was clear that in the presence of PEI-MNPs, the formation of LC3-II puncta markedly increased. This can be confirmed by immunofluorescence using HeLa cells containing ectopically expressed eGFP-LC3 plasmids. As shown in Fig. 4A, B and D, eGFP-LC3 showed a diffused distribution pattern in the controls, whereas, increased numbers of cytoplasmic LC3-positive puncta were observed in 40 μg mL−1 PEI-MNP treated groups (P < 0.005). As shown in Fig. 4C and D, the pre-treatment with NAC considerably decreased the numbers of LC3-positive puncta (P < 0.01). We also measured the abundance of p62, one LC3 binding protein that was supposed to be degraded during autophagy. It is one of the best studied substrates of autophagy therefore it could be used to reflect autophagic flux. We found that the p62 level strikingly declined upon PEI-MNPs treatment. The 40 μg mL−1 PEI-MNPs treatment gave rise to almost complete p62 degradation (Fig. 4E). But it is noticeable that the decline of p62 is possibly owing to two reasons: stimulation of its degradation or down-regulation of its expression. In order to discriminate these two events, we performed a semi-quantitative RT-PCR (reverse transcription polymerase chain reaction) experiment to test the mRNA level of p62 in the presence or absence of PEI-MNPs. As shown in Fig. S-2,† the mRNA level kept almost constant in different groups after 24 h PEI-MNPs treatment, which indicated that the p62 protein decline was mainly owing to its accelerated degradation. This observation would support the conclusion that PEI-MNPs were able to induce autophagy. The LC3-II abundance changed upon PEI-MNPs treatment (Fig. 4E and G), which was in line with fluorescent LC3 puncta images. Moreover, we performed a ‘clamp’ experiment (Fig. S-3†), in which we carried out co-treatment using PEI-MNPs together with an autophagic inhibitor either CQ or BAF. The LC3-II/β-actin levels significantly increased for ‘MNP + BAF’ (P < 0.005) and ‘MNP + CQ’ (P < 0.01) combinations compared to the solely BAF- and CQ-treated groups respectively. Also, we tested the LC3 and p62 proteins in MCF-7 (breast cancer) and HepG2 (liver cancer) cell lines derived from two common solid tumour types. The changes of LC3 and p62 were similar to what happened for HeLa cells (Fig. S-4†). This implied that autophagy induction could be a general phenomenon for PEI-MNPs treated human tumor cells. Contrary to the LC3-II/β-actin and p62/β-actin patterns resulting from PEI-MNPs treatment, pre-incubation of NAC for 1 h caused almost no significant LC3-II/β-actin and p62/β-actin changes (Fig. 4F, G and H), indicating that ROS was vital in PEI-MNPs induced autophagy.
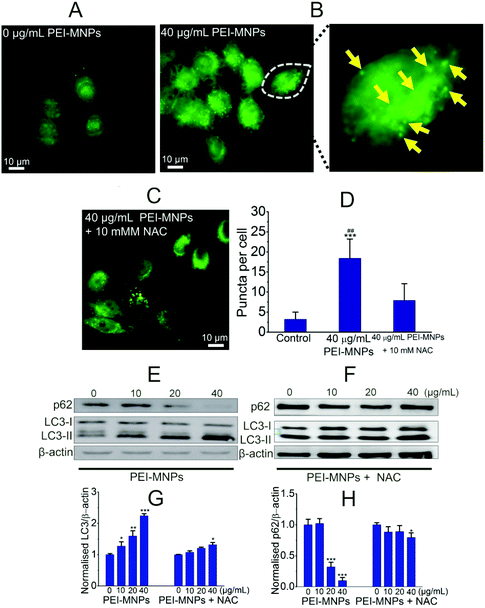 |
| | Fig. 4 PEI-MNPs trigger autophagy, which can be regulated by ROS. A to D: immunofluorescence images (A to C) and statistical analysis (D) showing LC3 puncta in cancer cells. Please note that the yellow arrows indicated LC3-positive puncta. E to H: Western blotting experiments (E and F) and statistical analysis of western blotting results showing the effect of NAC on PEI-MNPs induced autophagy (G and H). For all groups, PEI-MNPs were treated for 24 h and 10 mM NAC was pre-treated for HeLa cells for 1 h before PEI-MNPs addition where necessary. *P < 0.05; **P < 0.01; ***P < 0.005 vs. Controls. ##P < 0.01 of ‘40 μg mL−1 PEI-MNPs’ group vs. ‘40 μg mL−1 PEI-MNPs + 10 mM NAC’ group. Student's t-test was used. | |
3.4 PEI-MNPs acidify cellular compartments, stimulate ATG7 but suppress Akt-mTOR-p70S6K pathways, contributing to their autophagy-inducing effect
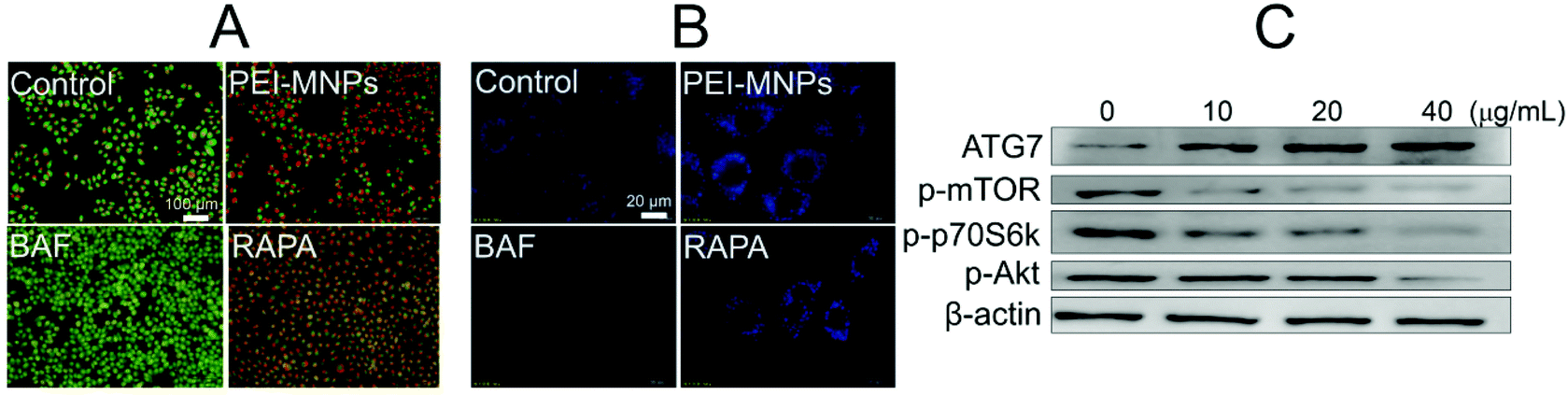
Next, we would like to explore the underlying mechanisms for PEI-MNPs to induce autophagy. To do so, AO (acridine orange) and MDC (monodansylcadaverine) were used in cancer cell fluorostaining (Fig. 5A and B). They are both fluorogenic dyes environmentally sensitive to the acidity of subcellular compartments. Specifically, AO is an acidotropic fluorochrome which changes to orange-red fluorescence at lower pH values, and MDC is a lysosomotropic agent used to identify AVs.36 BAF is a potent inhibitor of vacuolar-type H+-ATPase, thereby significantly boosting the pH value of lysosome (LYs). It strongly de-acidifies vacuoles, which is the main reason for its autophagic inhibition effect.37 RAPA is a well-known autophagy inducer.38 Both BAF and RAPA treated cells were used as controls. For PEI-MNPs treated cancer cells, it can be seen that they caused a marked accumulation of acidic vesicles, similar to RAPA (Fig. 5A). In contrast, BAF almost de-acidified cells as evidenced by the fact that there was hardly orange-red fluorescence observed (Fig. 5A). MDC staining also proved so, since MDC fluorescence intensity was significantly enhanced in PEI-MNPs and RAPA treated cells, whereas dramatically decreased in BAF treated cells (Fig. 5B). The Akt-mTOR-p70S6K cascade is one of the main pathways for autophagy regulation and its inhibition intensifies autophagy.39 As shown in Fig. 5C, PEI-MNPs treated HeLa cells had down-regulated phospho-Akt, phospho-mTOR and phospho-p70S6K. All this indicated the inhibition of the Akt-mTOR-p70S6K pathway. ATG7 (autophagy-related gene 7), an autophagy-essential protein for autophagosome assembly,40 increased in PEI-MNPs treated HeLa cells, indicating that PEI-MNPs probably induced autophagy by facilitating the formation of autophagosomes.
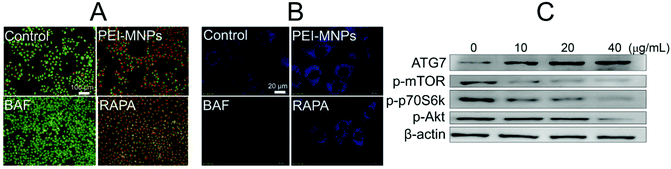 |
| | Fig. 5 PEI-MNPs acidify subcellular compartments but also stimulate ATG7 and Akt-mTOR pathways. A and B: Fluorescence images of cancer cells after AO and MDC staining respectively (PEI-MNPs were used at 40 μg mL−1 for 24 h. BAF and RAPA were used at 200 nM and 25 nM respectively for 12 h). C: Western blotting experiment reveling PEI-MNPs influenced signaling pathways. Please note that PEI-MNPs were treated for 24 h. | |
3.5 PEI-MNPs elicited NF-κB and TGF-β activation, which can be regulated by ROS
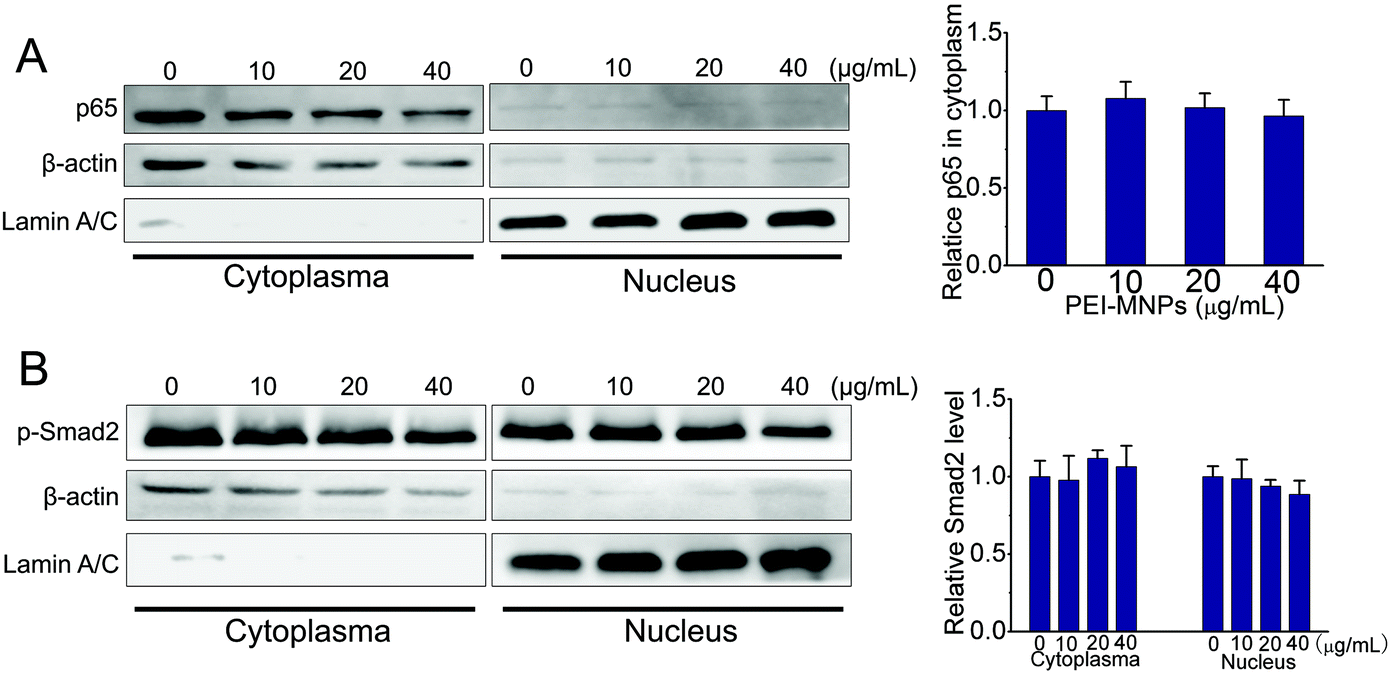
The NF-κB (nuclear factor κB) signaling pathway contributes to cancer initiation and progression in complex ways.41 It is commonly activated in a number of cancers. NF-κB is shown to promote a pro-inflammatory environment, inhibit apoptosis and stimulate cancer cell proliferation. Classically, IκBα is subjected to IκB kinase (IKK)-mediated phosphorylation, which leads to its ubiquitination and degradation. The degradation of the inhibitory molecule IκBα leads to the activation of the NF-κB signaling pathway.42 The transforming growth factor beta (TGF-β) signaling pathway is involved in embryogenesis and tumorigenesis by controlling cell growth, differentiation, migration, apoptosis, cellular homeostasis and other cellular functions.43 Intriguingly, the TGF-β signaling pathway is context-dependent. In the normal cells it functions as an inducer of apoptosis as well as controlling cell differentiation and proliferation. However, at advanced stages of cancers, TGF-β signaling mainly fosters tumour growth, exacerbates tumour metastasis, tumor angiogenesis and local tumor immunosuppression.44 We firstly employed well-established luciferase reporter experiments to examine the effects of PEI-MNPs on NF-κB and TGF-β signaling pathways. As shown in Fig. 6A and B, in the presence of PEI-MNPs, both of the pathways seemed to be activated in a PEI-MNPs dose-dependent manner, as proved by the significantly raised luciferase activity. For both, 24 h treatment with 40 μg mL−1 PEI-MNPs resulted in their activation by more than 50%, indicating a clear up-regulated influence of PEI-MNPs (P < 0.005). However, for the NAC pre-treated cancer cells, they did not display any obvious change in terms of luciferase activity. Additionally, we carried out western blotting investigations. It is well known that p65 and phospho-Smad2 nuclear translocation are symbols for the activation of NF-κB and TGF-β signaling pathways respectively. As shown in Fig. 6C and D, p65 and phospho-Smad2 displayed evident cytosol to nucleus translocation. Besides, PEI-MNPs decreased the level of IKBα (Fig. S-5†). Similarly, the addition of NAC suppressed this trend and there was no clear difference with regard to p65 and phospho-Smad2 abundance in nuclei for all three PEI-MNPs treated groups. Finally, immunofluorescence experiments, as shown in Fig. 6E and F, were carried out and the results accorded with both luciferase and immunoblotting very well. The p65 and p-Smad2 cytoplasm-to-nucleus translocation was clear for 40 μg mL−1 PEI-MNPs treated cancer cells, which was efficiently suppressed by NAC pre-treatment. Moreover, the p65 and p-Smad2 cytoplasm-to-nucleus translocation was also ameliorated by their respective inhibitors 6-amino-4-(4-phenoxyphenylethylamino)quinazoline (QNZ) and schisandrin B (Sch B).45,46
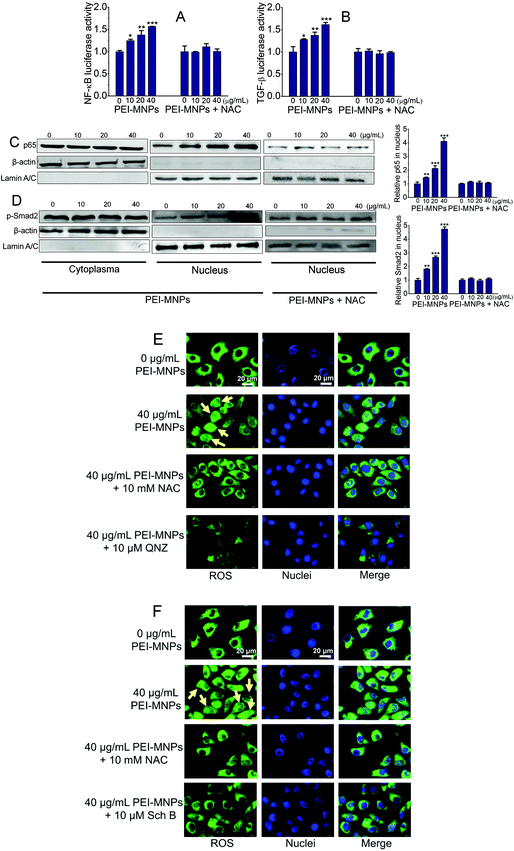 |
| | Fig. 6 PEI-MNPs trigger the activation of NF-κB and TGF-β pathways, which can be alleviated by ROS inhibition. A and B: Luciferase reporter experiments for monitoring the activities of NF-κB and TGF-β pathways respectively. C and D: Western blotting experiments revealing the p65 and p-Smad2 cytosol-nucleus translocation. E and F: representative fluorescent images of cytosol-nucleus translocation of p65 and p-Smad2 respectively. Please note that 10 mM NAC, 10 μM QNZ and 10 μM Sch B were pre-treated for 1 h, 0.5 h and 0.5 h respectively prior to 24 h exposure of PEI-MNPs. *P < 0.05; **P < 0.01; ***P < 0.005 vs. controls; Student's t test was used. Yellow arrows indicate very typical cytosol-nucleus translocation. | |
In summary, all three independent experiments corresponded well as a whole, which highlighted the importance of ROS in the regulation of PEI-MNPs induced NF-κB and TGF-β activation.
In addition to HeLa cancer cells, we also tested the NF-κB and TGF-β activation in normal cells upon PEI-MNP treatment. Based on some previous reports, the liver is one of the organs that MNPs are most heavily distributed in, thus we chose human normal hepatic L02 cells to do so.47 As shown in Fig. 7, it was noticed that p65 stayed almost unchanged in the cell plasma of L02 cells, while it was nearly discernible in nuclei. Thus under current experimental conditions, the cytoplasm-to-nucleus translocation of p65 was undetectable (Fig. 7A). For p-smad2, it had a similar trend, both p-Smad2 in cell plasma and nuclei remained unaffected by PEI-MNPs treatment (Fig. 7B). The above-mentioned results indicated that PEI-MNPs seem not to influence NF-κB and TGF-β pathways in human normal hepatic L02 cells.
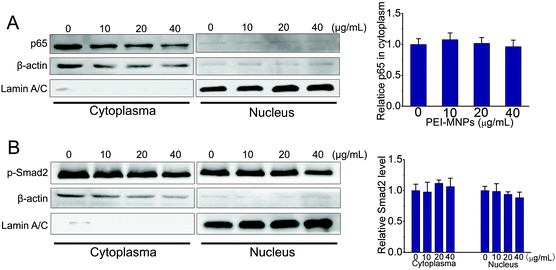 |
| | Fig. 7 PEI-MNPs seem not to influence NF-κB and TGF-β pathways in human normal hepatic L02 cells. A: Western blotting experiments revealing the abundance of p65 in cytoplasma and nucleus of L02 cells. B: Western blotting experiments revealing the abundance of p-Smad2 in cytoplasma and nucleus of L02 cells. Please note that L02 cells were treated with PEI-MNPs for 24 h. | |
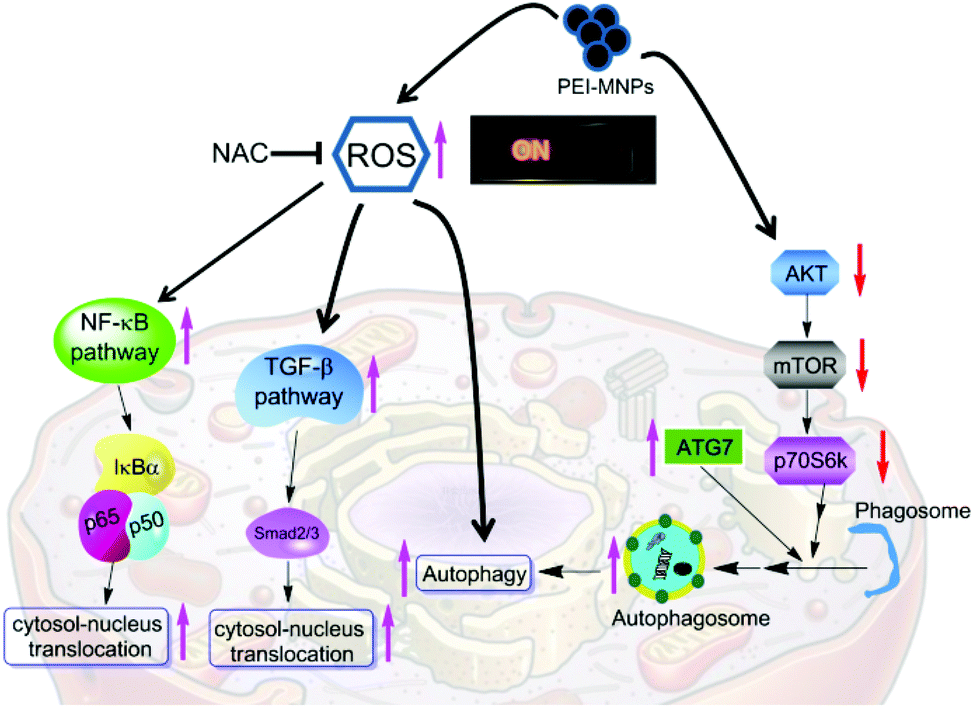
Finally, we summarised the roles of ROS escalation in MNPs-elicited cellular responses as shown in Fig. 8. PEI-MNPs are one type of well proven highly biocompatible nanomaterial and thus bear huge potential in various biomedical applications especially in cancer treatments. In the current study, we have reported that PEI-MNPs result in excessive ROS generation, as well as induce autophagy, which agrees well with previous reports.18 One step further, we have unprecedentedly discovered that ROS act as switches for MNPs-induced autophagy activation. A few types of NPs are capable of autophagy induction.48–50 This can be probably harnessed for treating cancer via autophagic flux blocking.51 It is reported that silver NPs induce autophagy and autophagic inhibition enhances the anticancer activity of silver NPs.52 Though some previous reports show that SiO2, ZnO and CuO NPs stimulate NF-κB,53–55 and silver NPs, pristine graphene and carbon nanotubes stimulate the TGF-β signaling pathway.56–58 It is noteworthy that for the first time, we have reported here that PEI-MNPs could activate both NF-κB and TGF-β pathways. More significantly, we have tried to link ROS over-production with these responses by using the ROS scavenger NAC. The results showed that the inhibition of the ROS level would effectively relieve the above-mentioned three cellular responses, indicating that ROS could be deemed as a ‘master switch’. NF-κB and TGF-β pathways exert negative effects in terms of cancer treatment; they would establish environments to foster tumour growth. This is significant, since some direct contact of PEI-MNPs with cancer cells has already been seen in a number of studies and we postulate that precaution should be taken into consideration if there is a long term exposure of PEI-MNPs to cancer cells. Furthermore, ROS, as a group of some crucial molecules have profound implications in pathophysiology. In fact, there is a great deal of interest in studying ROS regulated molecular signaling such as NF-κB, TGF-β and autophagy, but most of which is under basal physiological conditions.59–64 Our study not only reinforces the importance of ROS but also sheds light on the ‘global controller’ functions of ROS. In terms of MNPs, they also have unique properties such as magnetism, and catalase-like and peroxidase-like activities, which can be used to finely modulate the production of ROS.65–67 Apart from what is reported in our current study, PEI-MNPs potentially incur a range of cellular responses, including membrane leakage, chromosome condensation, impaired mitochondria function, DNA damage and so forth. Although we did not use an NAC based inhibitory method to probe the relationship with all of them, literature mining indicated a close association of them with ROS generation.68 NP-interacted cells generally have higher ROS levels including TiO2, SiO2, and ZnO, Al2O3, CeO2 and photosensitiser-functionalised NPs.69–71 Therefore, we believe that our rationale would be transferable to the research of other NPs. There has been a surge of interest in modulating highly toxic ROS production by MNPs to assist cancer treatments, mainly through amplifying drug or radiation efficacy.72–74 Hence comprehending more functions of ROS would be helpful for designing PEI-MNPs induced ROS anti-cancer strategies.75 Though PEI-MNPs did not demonstrate much acute cytotoxicity on cancer cells (as evidenced by MTT experiment in Fig. S-6†), their long term toxicity for cancer treatment should be paid attention to. The activation of NF-κB and TGF-β pathways needs to be recognised in this aspect.
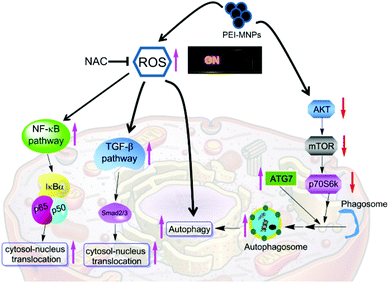 |
| | Fig. 8 The summary of roles of ROS on observed PEI-MNPs elicited cell responses in cancer cells. | |
4. Conclusions
PEI-MNPs are well proven highly biocompatible nanomaterials and bear huge potential in various biomedical applications especially cancer treatment. In current research, we have discovered that PEI-MNPs intensify ROS generation in cancer cells in a dose-dependent manner. Autophagy induction can be observed in PEI-MNPs treated cancer cells, which was proved by TEM examination of AV accumulation, AO and MDC fluorostaining, LC3-positive puncta experiment and p62 abundance measurement. Also PEI-MNPs stimulate NF-κB and TGF-β signaling pathways, evidenced by both the luciferase reporter experiment as well as cytosol to nucleus translocation of p65 and p-Smad2. One step further, we have confirmed that ROS seemed to be a ‘master switch’ for the aforementioned cellular responses, as ROS inhibition effectively quenches all of them. The level of ROS depends on the balance between endogenous ROS and cellular antioxidant systems. It is also noted that cancer cells usually have a higher ROS level than normal cells. Normal cells may be more tolerant to oxidative stress for survival rather than cancer cells since, for the latter, a balanced ROS level would exceed the threshold if the same levels of exogenous ROS exist. Thus it may be wise to take advantage of MNPs induced ROS stress to selectively kill cancer cells. Moreover, ROS are metaphorised as a ‘double edge sword’ for cancer cells, owing to the cancer-suppressing and cancer-promoting actions they bear. Our research furthers this consent by showing the relationship of ROS with NF-κB and TGF-β, two key pro-inflammatory pathways that would contribute to cancer development and progression. This finding would draw attention to notice MNPs induced ROS. In summary, our work not only elucidates some novel cellular mechanisms for MNPs, but also highlights and associates excessive ROS production with MNPs elicited cellular responses, which would be useful in designing MNPs induced ROS anti-cancer strategies or in diminishing long-term adverse effects.
Conflicts of interest
All of the authors would like to declare that there are no conflict of interest.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 81503086 and 21672161), a research funding of ‘1000 Talents Plan’ of Tianjin (to LM) and a funding from the Foundation of Key Laboratory of Industrial Fermentation Microbiology of Ministry of Education and Tianjin Key Lab of Industrial Microbiology (2017YC003), and Tianjin Municipal Science and Technology Committee (118PTSYJC00140, 19JCYBJC27800).
References
- O. V. Salata, J. Nanobiotechnol., 2004, 2, 3 CrossRef.
- L. Ma and S. L. Cockroft, ChemBioChem, 2010, 11, 25–34 CrossRef CAS.
- L. Ma, N. Sun, Y. Meng, C. Tu, X. Cao, Y. Wei, L. Chu and A. Diao, Anal. Chim. Acta, 2018, 1036, 107–114 CrossRef CAS.
- L. Ma, G. Wu, Y. Li, P. Qin, L. Meng, H. Liu, Y. Li and A. Diao, Nanoscale, 2015, 7, 18044–18048 RSC.
- D. Yoo, J.-H. Lee, T.-H. Shin and J. Cheon, Acc. Chem. Res., 2011, 44, 863–874 CrossRef CAS.
- A. J. Cole, V. C. Yang and A. E. David, Trends Biotechnol., 2011, 29, 323–332 CrossRef CAS.
- K. Fan, C. Cao, Y. Pan, D. Lu, D. Yang, J. Feng, L. Song, M. Liang and X. Yan, Nat. Nanotechnol., 2012, 7, 459–464 CrossRef CAS.
- R. Liu, Y. Guo, G. Odusote, F. Qu and R. D. Priestley, ACS Appl. Mater. Interfaces, 2013, 5, 9167–9171 CrossRef CAS.
- S. Huth, J. Lausier, S. W. Gersting, C. Rudolph, C. Plank, U. Welsch and J. Rosenecker, J. Gene Med., 2004, 6, 923–936 CrossRef CAS.
- F. M. Kievit, F. Y. Wang, C. Fang, H. Mok, K. Wang, J. R. Silber, R. G. Ellenbogen and M. Zhang, J. Controlled Release, 2011, 152, 76–83 CrossRef CAS.
- H. E. Daldrup-Link, D. Golovko, B. Ruffell, D. G. DeNardo, R. Castaneda, C. Ansari, J. Rao, G. A. Tikhomirov, M. F. Wendland and C. Corot, Clin. Cancer Res., 2011, 17, 5695–5704 CrossRef CAS.
- V. I. Shubayev and S. Jin, Adv. Drug Delivery Rev., 2009, 61, 467–477 CrossRef CAS PubMed.
- Q. Yu, Y.-M. Zhang, Y.-H. Liu, X. Xu and Y. Liu, Sci. Adv., 2018, 4, eaat2297 CrossRef CAS.
- Q. Yu, Y.-M. Zhang, Y.-H. Liu and Y. Liu, Adv. Ther., 2019, 2, 1800137 CrossRef CAS.
- R. Di Corato, G. Béalle, J. Kolosnjaj-Tabi, A. Espinosa, O. Clement, A. K. Silva, C. Menager and C. Wilhelm, ACS Nano, 2015, 9, 2904–2916 CrossRef CAS.
- S. Shen, S. Wang, R. Zheng, X. Zhu, X. Jiang, D. Fu and W. Yang, Biomaterials, 2015, 39, 67–74 CrossRef CAS.
- B. Chen, Y. Liang, W. Wu, J. Cheng, G. Xia, F. Gao, J. Ding, C. Gao, Z. Shao and G. Li, Int. J. Nanomed., 2009, 4, 251–259 CrossRef CAS.
- B. Chen, S. Qian, X. Wang, G. Feng, Y. Dai, Y. Yan, J. Ding, G. Chong, C. Jian and J. Li, Int. J. Nanomed., 2008, 3, 277–286 CAS.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman and M. Moseley, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS.
- C.-S. Chiang, Y.-J. Lin, R. Lee, Y.-H. Lai, H.-W. Cheng, C.-H. Hsieh, W.-C. Shyu and S.-Y. Chen, Nat. Nanotechnol., 2018, 13, 746–754 CrossRef CAS PubMed.
- P. T. Schumacker, Cancer Cell, 2006, 10, 175–176 CrossRef CAS.
- M. I. Khan, A. Mohammad, G. Patil, S. Naqvi, L. Chauhan and I. Ahmad, Biomaterials, 2012, 33, 1477–1488 CrossRef CAS.
- C. M. Sims, S. K. Hanna, D. A. Heller, C. P. Horoszko, M. E. Johnson, A. B. Montoro, V. Reipa, K. R. Riley and B. C. Nelson, Nanoscale, 2017, 9, 15226–15251 RSC.
- L. Ma, N. Sun, J. Zhang, C. Tu, X. Cao, D. Duan, A. Diao and S. Man, Nanoscale, 2017, 9, 17699–17703 RSC.
- C.-L. Chiang, C.-S. Sung, T.-F. Wu, C.-Y. Chen and C.-Y. Hsu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 822, 54–60 CrossRef CAS.
- L. Ma, H. Liu, P. Qin, C. Hu, S. Man, Y. Li, Z. Liu, Z. Liu and A. Diao, Biochem. Biophys. Res. Commun., 2017, 483, 779–785 CrossRef CAS PubMed.
- H. Cai, X. An, J. Cui, J. Li, S. Wen, K. Li, M. Shen, L. Zheng, G. Zhang and X. Shi, ACS Appl. Mater. Interfaces, 2013, 5, 1722–1731 CrossRef CAS.
- Y. He, J. Qin, S. Wu, H. Yang, H. Wen and Y. Wang, Biomater. Sci., 2019, 7, 2759–2768 RSC.
- L. Zhang, T. Wang, L. Li, C. Wang, Z. Su and J. Li, Chem. Commun., 2012, 48, 8706–8708 RSC.
- A. S. Arbab, L. A. Bashaw, B. R. Miller, E. K. Jordan, B. K. Lewis, H. Kalish and J. A. Frank, Radiology, 2003, 229, 838–846 CrossRef.
- C. B. Pocernich, M. La Fontaine and D. A. Butterfield, Neurochem. Int., 2000, 36, 185–191 CrossRef CAS.
- H. P. Indo, M. Davidson, H.-C. Yen, S. Suenaga, K. Tomita, T. Nishii, M. Higuchi, Y. Koga, T. Ozawa and H. J. Majima, Mitochondrion, 2007, 7, 106–118 CrossRef CAS.
- N. Mizushima, T. Yoshimori and B. Levine, Cell, 2010, 140, 313–326 CrossRef CAS.
- H. Liu, C. Hu, N. Sun, Y. Li, S. Man, Z. Liu, A. Diao and L. Ma, RSC Adv., 2017, 7, 24291–24297 RSC.
- C. L. Vazquez and M. I. Colombo, Methods Enzymol., 2009, 452, 85–95 CAS.
- A. Yamamoto, Y. Tagawa, T. Yoshimori, Y. Moriyama, R. Masaki and Y. Tashiro, Cell Struct. Funct., 1998, 23, 33–42 CrossRef CAS.
- C. H. Jung, S. H. Ro, J. Cao, N. M. Otto and D. H. Kim, FEBS Lett., 2010, 584, 1287–1295 CrossRef CAS PubMed.
- S. Saiki, Y. Sasazawa, Y. Imamichi, S. Kawajiri, T. Fujimaki, I. Tanida, H. Kobayashi, F. Sato, S. Sato and K.-I. Ishikawa, Autophagy, 2011, 7, 176–187 CrossRef CAS.
- Z. Xie and D. J. Klionsky, Nat. Cell Biol., 2007, 9, 1102–1109 CrossRef CAS.
- B. Hoesel and J. A. Schmid, Mol. Cancer Ther., 2013, 12, 1–15 CrossRef.
- P. Viatour, M.-P. Merville, V. Bours and A. Chariot, Trends Biochem. Sci., 2005, 30, 43–52 CrossRef CAS.
- P. T. Dijke and C. S. Hill, Trends Biochem. Sci., 2004, 29, 265–273 CrossRef.
- R. J. Akhurst and R. Derynck, Trends Cell Biol., 2001, 11, S44–S51 CAS.
- M. Tobe, Y. Isobe, H. Tomizawa, T. Nagasaki, H. Takahashi and H. Hayashi, Bioorg. Med. Chem., 2003, 11, 3869–3878 CrossRef CAS.
- E.-J. Park, J. N. Chun, S.-H. Kim, C. Y. Kim, H. J. Lee, H. K. Kim, J. K. Park, S. W. Lee, I. So and J.-H. Jeon, Biochem. Pharmacol., 2012, 83, 378–384 CrossRef CAS.
- J. Wang, Y. Chen, B. Chen, J. Ding, G. Xia, C. Gao, J. Cheng, N. Jin, Y. Zhou and X. Li, Int. J. Nanomed., 2010, 5, 861 CAS.
- O. Zabirnyk, M. Yezhelyev and O. Seleverstov, Autophagy, 2007, 3, 278–281 CrossRef CAS PubMed.
- H. Eidi, O. Joubert, C. Némos, S. Grandemange, B. Mograbi, B. Foliguet, J. Tournebize, P. Maincent, A. Le Faou and I. Aboukhamis, Int. J. Pharm., 2012, 422, 495–503 CrossRef CAS.
- K.-N. Yu, T.-J. Yoon, A. Minai-Tehrani, J.-E. Kim, S. J. Park, M. S. Jeong, S.-W. Ha, J.-K. Lee, J. S. Kim and M.-H. Cho, Toxicol. in Vitro, 2013, 27, 1187–1195 CrossRef CAS.
- X. Ren, Y. Chen, H. Peng, X. Fang, X. Zhang, Q. Chen, X. Wang, W. Yang and X. Sha, ACS Appl. Mater. Interfaces, 2018, 10, 27701–27711 CrossRef CAS.
- J. Lin, Z. Huang, H. Wu, W. Zhou, P. Jin, P. Wei, Y. Zhang, F. Zheng, J. Zhang and J. Xu, Autophagy, 2014, 10, 2006–2020 CrossRef.
- X. Liu and J. Sun, Biomaterials, 2010, 31, 8198–8209 CrossRef CAS.
- E.-J. Park and K. Park, Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro, Toxicol. Lett., 2009, 184, 18–25 CrossRef CAS.
- C. Monteiller, T. Lang, W. Macnee, S. Faux, A. Jones, B. Miller and K. Donaldson, Occup. Environ. Med., 2007, 64, 609–615 CrossRef CAS.
- E.-J. Park, E. Bae, J. Yi, Y. Kim, K. Choi, S. H. Lee, J. Yoon, B. C. Lee and K. Park, Environ. Toxicol. Pharmacol., 2010, 30, 162–168 CrossRef CAS.
- Y. Li, Y. Liu, Y. Fu, T. Wei, L. Le Guyader, G. Gao, R.-S. Liu, Y.-Z. Chang and C. Chen, Biomaterials, 2012, 33, 402–411 CrossRef CAS.
- L. Lin, L. Liu, B. Zhao, R. Xie, W. Lin, H. Li, Y. Li, M. Shi, Y. G. Chen and T. A. Springer, Nat. Nanotechnol., 2015, 10, 465–471 CrossRef CAS.
- M. J. Morgan and Z.-G. Liu, Cell Res., 2010, 21, 103–115 CrossRef.
- G. Gloire, S. Legrand-Poels and J. Piette, Biochem. Pharmacol., 2006, 72, 1493–1505 CrossRef CAS.
- H. Sauer, M. Wartenberg and J. Hescheler, Cell. Physiol. Biochem., 2001, 11, 173–186 CrossRef CAS.
- H. Wang and I. E. Kochevar, Free Radicals Biol. Med., 2005, 38, 890–897 CrossRef CAS.
- R. Scherzshouval and Z. Elazar, Trends Biochem. Sci., 2011, 36, 30–38 CrossRef CAS PubMed.
- R. Scherzshouval and Z. Elazar, Trends Cell Biol., 2007, 17, 422–427 CrossRef CAS.
- L. Wang, Z. Wang, X. Li, Y. Zhang, M. Yin, J. Li, H. Song, J. Shi, D. Ling, L. Wang, N. Chen and C. Fan, Nano Res., 2018, 11, 2746–2755 CrossRef CAS.
- Z. Chen, J. J. Yin, Y. T. Zhou, Y. Zhang, L. Song, M. Song, S. Hu and N. Gu, ACS Nano, 2012, 6, 4001–4012 CrossRef CAS.
- M. Domenech, I. Marrero-Berrios, M. Torres-Lugo and C. Rinaldi, ACS Nano, 2013, 7, 5091–5101 CrossRef CAS.
- G. Liu, J. Gao, H. Ai and X. Chen, Small, 2013, 9, 1533–1545 CrossRef CAS.
- L. K. Adams, D. Y. Lyon and P. J. J. Alvarez, Water Res., 2006, 40, 3527–3532 CrossRef CAS.
- Y. Li, S. Yu, Q. Wu, M. Tang, Y. Pu and D. Wang, J. Hazard. Mater., 2012, 219–220, 221–230 CrossRef CAS.
- M. S. Wason, J. Colon, S. Das, S. Seal, J. Turkson, J. Zhao and C. H. Baker, Nanomedicine, 2013, 9, 558–569 CrossRef CAS.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang and Z. Gu, Nano Lett., 2017, 17, 928–937 CrossRef CAS.
- G. Huang, H. Chen, Y. Dong, X. Luo, H. Yu, Z. Moore, E. A. Bey, D. A. Boothman and J. Gao, Theranostics, 2013, 3, 116–126 CrossRef CAS.
- M. Huo, L. Wang, Y. Chen and J. Shi, Nat. Commun., 2017, 8, 357 CrossRef.
- J.-Y. Tang, A. A. Farooqi, F. Ou-Yang, M.-F. Hou, H.-W. Huang, H.-R. Wang, K.-T. Li, S. Fayyaz, C.-W. Shu and H.-W. Chang, Semin. Cancer Biol., 2018, 58, 109–117 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01563a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*