
Collaborative assembly of doxorubicin and galactosyl diblock glycopolymers for targeted drug delivery of hepatocellular carcinoma†
Jianghua
Li‡
a,
Yang
Zhang‡
a,
Chao
Cai‡
 *ab,
Xiaozhi
Rong
ab,
Meng
Shao
a,
Jiarui
Li
a,
Chendong
Yang
a and
Guangli
Yu
*ab
*ab,
Xiaozhi
Rong
ab,
Meng
Shao
a,
Jiarui
Li
a,
Chendong
Yang
a and
Guangli
Yu
*ab
aKey Laboratory of Marine Drugs, Ministry of Education & Shandong Provincial Key Laboratory of Glycoscience and Glycotechnology, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, China. E-mail: caic@ouc.edu.cn; glyu@ouc.edu.cn
bLaboratory for Marine Drugs and Bioproducts, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao 266237, China
First published on 18th November 2019
Abstract
Hepatocellular carcinoma (HCC) patients suffer from severe pain due to the serious systemic side effects and low efficiency of chemotherapeutic drugs, and it is important to develop novel drug delivery systems to circumvent these issues. In this study, a series of galactose-based glycopolymers, poly(N-(prop-2-enoyl)-β-D-galactopyranosylamine)-b-poly(N-isopropyl acrylamide) (pGal(OH)-b-pNIPAA), were prepared through a sequential reversible addition-fragmentation chain transfer (RAFT) polymerization and tetrabutylammonium hydroxide (TBAOH)-mediated removal of acetyl groups. Hydrophilic doxorubicin hydrochloride was introduced to undergo collaborative assembly with poly(N-(prop-2-enoyl)-β-D-peracetylated galactosamine)-b-poly(N-isopropyl acrylamide) (pGal(Ac)-b-pNIPAA) via TBAOH treatment. pGal-b-pNIPAA/doxorubicin (DOX) delivery nanoparticles (GND NPs) formed by collaborative assembly were fully characterized by NMR, TEM and FT-IR, indicating the well-controlled formation of particles with uniform size and high efficiency in terms of drug loading and encapsulation compared with conventional adsorption methods. Meanwhile, the GND NPs were observed to be rapidly disintegrated under acidic conditions and resulted in an increased release of DOX. Cellular experiments showed that pGal-b-pNIPAA/DOX is apparently an asialoglycoprotein receptor (ASGPR)-mediated target of HCC, resulting in enhanced cellular uptake to HepG2 cells and anti-tumor efficacy in vitro. Furthermore, GND NPs III exerted more sustainable and effective anti-tumor effects compared to free DOX on a transgenic zebrafish TO(KrasG12V) model in vivo. These results indicated that the biocompatible nanomaterials developed by collaborative assembly with galactosyl diblock glycopolymers and DOX may serve as a promising candidates for targeting therapy of HCC.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide, and is usually treated with transarterial chemoembolization.1,2 However, chemotherapeutic drugs have various drawbacks.3 Non-targeted broad-spectrum anti-cancer drugs are often rapidly cleared during blood circulation due to the complex circulatory system, poor digestive system, and strong immune system.4–6 Moreover, the serious systemic side effects caused by chemotherapeutic drugs have also become a potential issue during medical therapy.7 Anti-cancer drugs such as doxorubicin hydrochloride were also cleared in a short half-life elimination with poor distribution in vivo.8 To better deliver chemotherapeutic drugs to HCC tissues, numerous pioneering studies have been conducted to develop versatile strategies to resolve the issues in targeted drug delivery.9The development of nanotechnology offers the potential to overcome challenges associated with drug delivery vectors.10,11 Biocompatible nanomaterials serving as vehicles have dramatically enhanced the efficiency of breaking through the barriers during drug delivery.12–14 Tumors typically possess a leaky vasculature compared to healthy vessels in normal organs,15,16 thus the nano-sized agents tend to circulate for a longer period and passively accumulate in solid tumors for enhanced permeability and retention (EPR) when administered intravenously.17,18 Chen et al. investigated size-regulated endocytosis and exocytosis of carboxylated polystyrene (PS) nanoparticles (NPs) and found that particle size is a key factor in regulating intracellular trafficking of NPs.16,19 In addition, nanomaterials can be designed with modified multifunctional moieties20,21 such as targeting ligands,22 bioactive/bio-responsive groups,23,24 and environmentally responsive groups.25,26 Chou et al. combined folic acid with gold NPs to endow the versatile theranostic delivery platform with targeting ability.27 Li et al. prepared sialic acid-imprinted fluorescent conjugated NPs for targeted cancer cell imaging.28 It is a promising strategy for achieving high drug loading efficiency and targeted drug delivery and overcoming other delivery barriers as discussed above regarding the employment of nanomaterials.
Effective cancer research promotes the development of targeted drug delivery.29 Malignant solid tumors tend to have a unique histological microenvironment (hypoxic, acidic) for accelerated proliferation of cancer cells and limited distance for molecular oxygen transport,30,31 which is becoming a novel target for cancer therapy and can be well utilized to develop an environmentally responsive drug release system.32,33 Tsukruk et al. prepared multi-responsive (temperature and pH) microcapsules for intelligent drug delivery via the layer-by-layer assembly of poly(N,N-dimethylaminoethyl methacrylate) and poly-(sodium-4-styrenesulfonate); these smart microcapsules provide a robust and reliable platform to enable controlled loading and release of targeting molecules under multiple stimuli.34,35 Meanwhile, the distinct proteins overexpressed on tumors typically have good affinity with specific carbohydrates compared with normal tissues and organs.36 Carbohydrate-based targeting is of considerable interest for pharmaceutical preparation and drug delivery,37,38 because carbohydrates play a critical role in the occurrence, proliferation, and migration of cancer39,40 and the abnormal glycosylation can be designed for targeted delivery of drugs.41 Asialoglycoprotein receptor (ASGPR), a transmembrane protein overexpressed by hepatocytes as an endocytotic cell surface receptor, has a good binding affinity with asialoglycoproteins which are glycoproteins with terminal galactose residues.42,43 Thus terminal galactose molecules commonly have good binding ability with ASGPR in terms of their structural similarity.44 Wang et al. developed a galactosyl functional fluorescent probe that has good hepatocyte-targeting ability.45 Meanwhile, the development of free radical polymerization methods46,47 makes it convenient to prepare well-defined glycopolymers,48,49 and the “glycoside cluster effect” of the glycopolymers significantly enhances the protein binding affinity.50,51 Jana et al. explored the effect of galactose multivalency on cellular interaction, cellular uptake mechanism, and subcellular targeting and found that the galactose multivalency of nanoparticles directs their interactions with cells, cellular entry/exit mechanism, and cytosolic residency.52 Glycopolymer-based nanomaterial aggregates/assemblies with various morphologies have been obtained through different convenient methods.53–55 Chen et al. proposed a strategy that the chemical process may induce block copolymer micellization.56 Sheng et al. obtained a series of morphology-variable diblock glycopolymers through a sequential reversible addition-fragmentation chain transfer (RAFT) polymerization and deprotection process, which inspired us to develop drug delivery vectors.57 Galactose-based glycopolymers have a better affinity with ASGPR and the special tumor microenvironment and possess considerable potential to be applied in HCC targeting drug delivery.58,59 In the present study, we integrated the advantages of designability on polymers, characteristics of drugs and tumor microenvironments to develop HCC targeting drug delivery systems.
Based on these facts, a series of diblock glycopolymers, poly(N-(prop-2-enoyl)-β-D-galactopyranosylamine)-b-poly(N-isopropyl acrylamide) (GN Vehicle), with galactose/N-isopropyl acrylamide grafts were prepared through a sequential reversible addition-fragmentation chain transfer (RAFT) polymerization and successive TBAOH-mediated deprotection. Multifunctional and smart drug-delivery vectors were successfully constructed with galactose as a targeting molecule and NIPAA as a response reagent of acid and temperature,60 and doxorubicin (DOX) was employed as a drug payload for the treatment of HCC. DOX transforms from hydrophilic to hydrophobic in the presence of TBAOH; thus DOX and galactose polymers would collaboratively assemble under basic treatment. The results of transmission electron microscopy (TEM) analysis showed the homogeneous size and morphology of the drug delivery system. In addition, the GN Vehicle possesses good biocompatibility in vitro and in vivo with uniform dispersion, and the DOX loaded delivery GN Vehicle (GND NPs III) has high drug loading and encapsulation capacity. Most importantly, GND NPs III show the acid-triggered DOX release and exhibit excellent tumor inhibition effect. Furthermore, in vitro cellular experiments showed that GND NPs III apparently target HCC via ASGPR, resulting in enhanced anti-cancer efficacy. Taken together, the collaborative assembly of the drug delivery system has strong targeting effect, high drug loading capacity, sustained and acid triggered drug release characteristics, and enhanced anti-HCC efficacy in vitro and in vivo, which provides the foundation for constructing the ASGPR-mediated targeting HCC drug delivery system (Scheme 1).
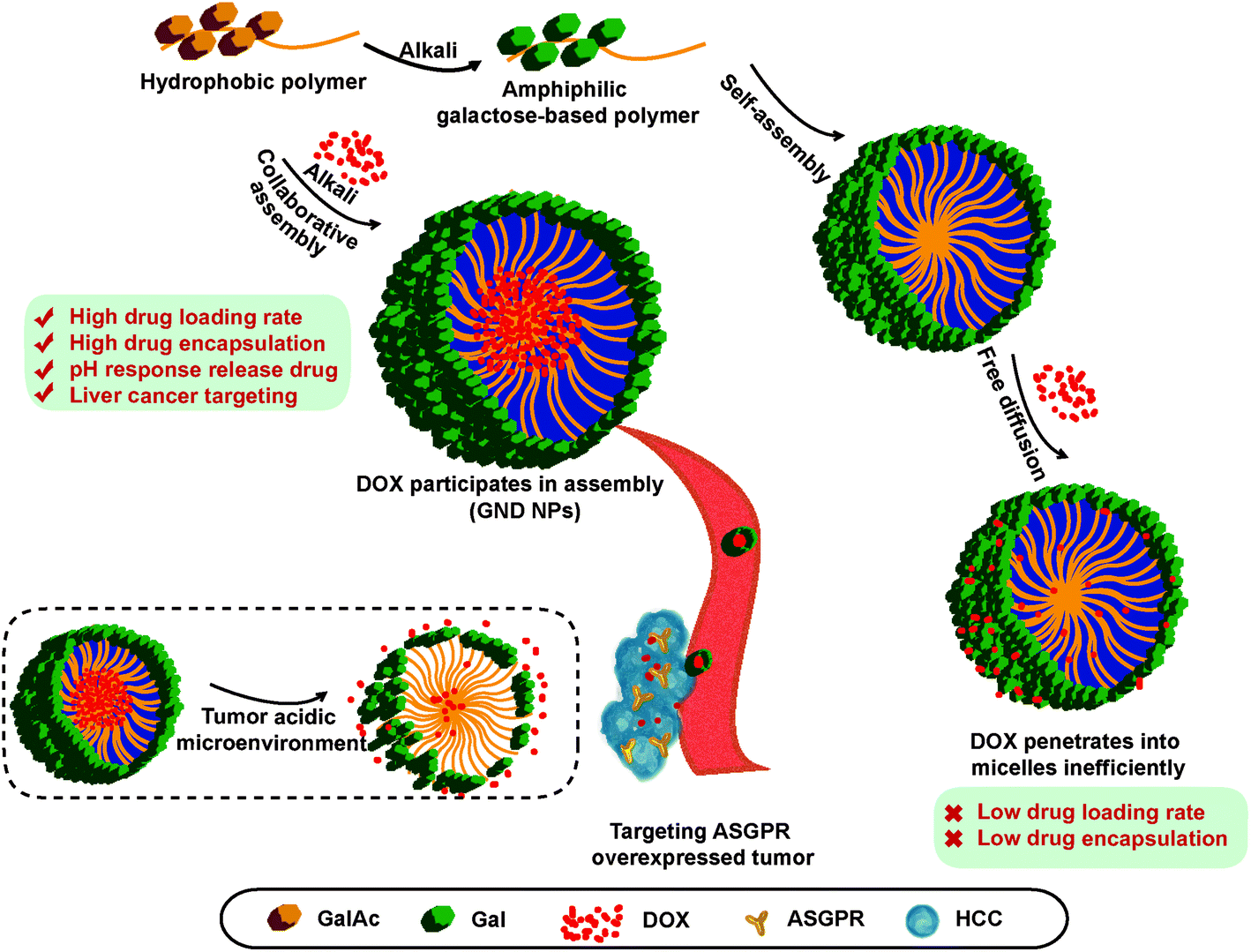 | ||
| Scheme 1 The schematic illustrations of the formation of GND NPs through collaborative assembly, and their cellular uptake and targeted drug-release. | ||
Results and discussion
Synthesis and characterization of diblock glycopolymers
Galactosyl diblock glycopolymers were synthesized via RAFT polymerization as illustrated in Fig. 1a. Free galactose as the starting material was modified with a double bond at the reducing end, which is the prerequisite for free radical polymerization. Likhosherstov et al.61 developed a practical protocol for the selective amination of unprotected sugar derivatives such as galactose by using ammonium carbamate in methanol. β-D-Galactopyranosylamine 2 is stable and convenient to efficiently couple with acryloyl chloride in the presence of Na2CO3 at 0 °C in methanol–water.62 Then, N-(prop-2-enoyl)-β-D-galactopyranosylamine 3, the double bond-substituted galactose, is able to participate in diblock glycopolymer synthesis via RAFT polymerization. However, acrylic acid is a byproduct that is difficult to separate in the process of amidation due to its similar solubility and polarity to the product. Pure N-(prop-2-enoyl)-β-D-galactopyranosylamine 2 was finally obtained after the deprotection of peracetylated N-(prop-2-enoyl)-β-D-galactopyranosylamine 3, which was easily separated from the byproducts (Fig. S1†). Meanwhile, galactosyl diblock glycopolymers pGal(Ac)m-b-pNIPAAn7 can be prepared from peracetylated N-acryloyl galactosamine as the raw material in the presence of 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid, and the obtained pGal(Ac)m-CTA could further initiate N-isopropyl acrylamide as the macro-CTA. Similarly, the macro glycopolymers pGal(OH)m and pGal(OH)m-b-pNIPAAn can also be obtained by the deprotection of acetyl groups (Fig. S2†). The 1H NMR spectrum of a series of pGal-b-pNIPAA is shown in Fig. S3.† Finally, diblock glycopolymer pGal(OH)m-b-pNIPAAn8 could be obtained after the removal of the protecting groups. 1H-NMR and FT-IR spectra of these intermediates, glycomonomers, and glycopolymers are shown in Fig. 1b and c, respectively. The representative signals of pNIPAAm were at around 1.17 ppm for methyl and 3.93 ppm for amino-linked tertiary hydrocarbon in the 1H NMR spectra. The acetyl groups on pGal(Ac)m showed the characteristic peaks at around 2.00 ppm and 5.60–5.00 ppm due to the attachment of the electron withdrawing groups. The successful formation of pGal(OH)m obtained via the deprotection of pGal(Ac)m was confirmed by the representative signals at around 4.96, 4.00, 3.79, and 3.66 ppm with the clear disappearance of the acetyl groups. The polymerization of pGal(OH)m-b-pNIPAAn and pGal(Ac)m-b-pNIPAAn was confirmed by the retention of the representative peaks for specific functional groups. The characteristic peaks at around 3300 cm−1 represent the secondary amino groups in the FT-IR spectra of pNIPAA, pGal(OH)m-b-pNIPAAn and pGal(Ac)m-b-pNIPAAn, the peaks at 3600–3300 cm−1 indicate the hydroxyl groups in pGal(OH)m-b-pNIPAAn and pGal(OH)m, and the signals at around 1750 cm−1 confirmed the presence of carbonyl on the acetyl groups. All characteristic peaks were well assigned, corresponding to their molecular structures and previous relevant reports. TEM results revealed that the biopolymers self-assemble into micelles when exposed to mild alkaline surroundings and pNIPAAn acts as a hydrophobic core while pGal(OH)m acts as the hydrophilic shell; this phenomenon was consistent with the deprotection-induced micellization recently reported by Chen et al.56 The micelles exhibited great potential as drug vehicles, which were designated as GN Vehicles I–IV, respectively (Fig. 2a). The radius of these micelles has been determined to be <30 nm as characterized by TEM statistics, which did not change with gradient-increased temperature at a low concentration (Fig. S4†).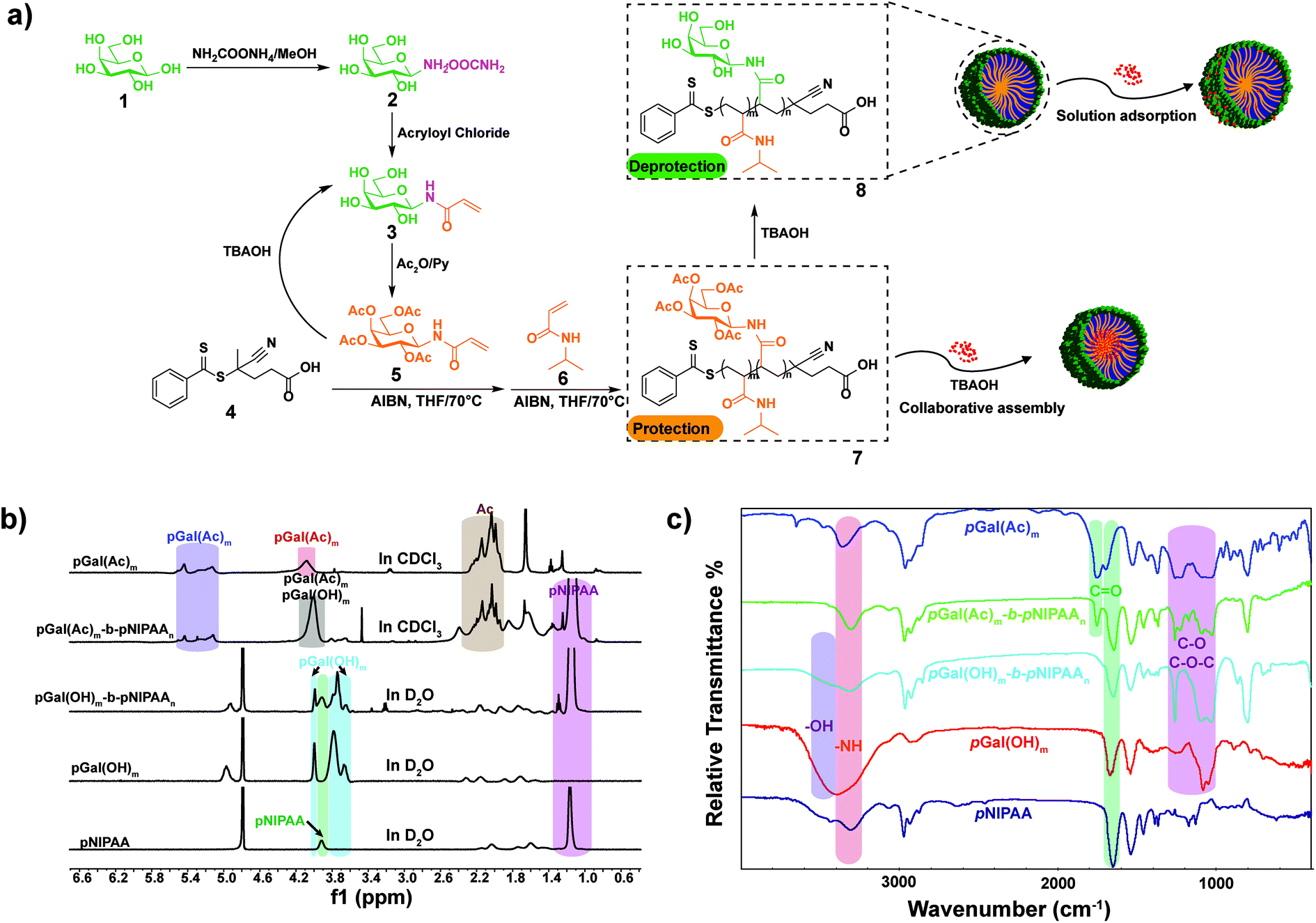 | ||
| Fig. 1 (a) Synthesis of the galactosyl diblock glycopolymers and the alternative approaches to load DOX. (b) 1H NMR spectra and (c) FT-IR spectra of pNIPAA, pGal(OH)m, pGal(OH)m-b-pNIPAAn, pGal(Ac)m-b-pNIPAAn and pGal(Ac)m. | ||
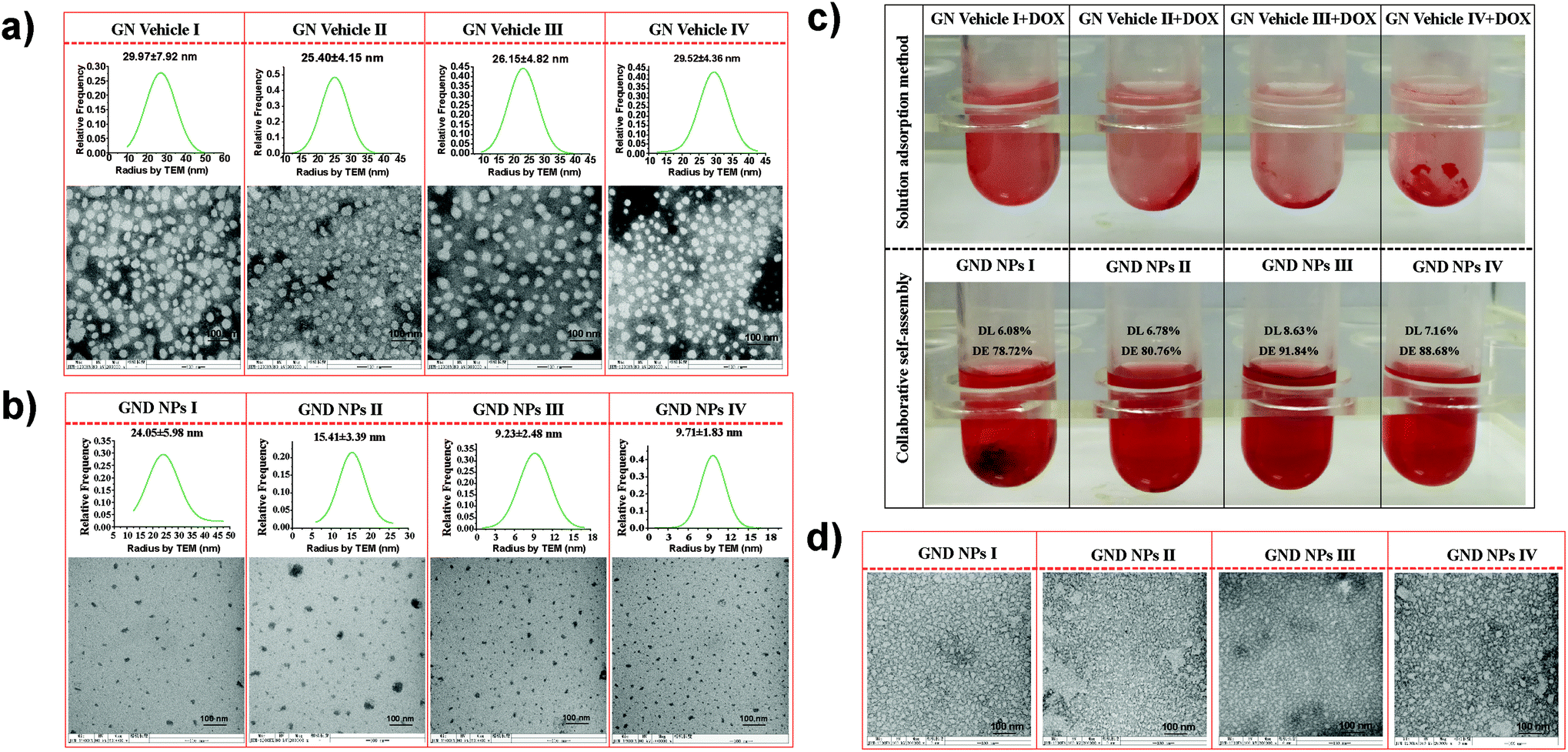 | ||
| Fig. 2 (a) Morphology and the particle size statistics of GN Vehicles I–IV. (b) Morphology and the particle size statistics of GND NPs I–IV in buffer at pH 7.4. (c) Comparison of drug loading effect of the solution adsorption and collaborative assembly methods. (d) Morphology of GND NPs I–IV after being transferred to buffer at pH 5.5 for 4 h. | ||
Preparation and characterization of the drug delivery system
For drug loading, the solution adsorption method was used to adsorb the drug onto the surface or pores of the vehicle after forming the carrier material.63 In this study, the galactosyl diblock glycopolymers self-assembled into micelles due to the phase transition via alkali treatment induction (Fig. 2a), and doxorubicin is an anti-tumor drug that becomes aqueous insoluble after treatment with alkali. The pNIPAA and the hydrophobic doxorubicin aggregated into the hydrophilic core, while the galactosyl polymers were utilized as the hydrophilic shell to stabilize the nanoparticles in aqueous solution. Thus, the galactosyl diblock glycopolymers and doxorubicin can collaboratively assemble into a series of nanoparticles listed as GND NPs I–IV, respectively, and doxorubicin plays an important role as a hydrophobic core during the process of assembly due to the “hydrophobic effect” (Fig. 2b). Furthermore, the unloaded DOX precipitated at the bottom by centrifugation as shown in the experimental results in Fig. 2c, and GND NPs were found to be superior in terms of drug loading efficiency and encapsulation rate compared with the conventional solution adsorption approach. It is the advantage of the collaborative assembly reported in this study, promoting the high drug encapsulation rate of DOX in the hydrophobic core during the self-assembly process. Among these nanoparticles, the GND NPs III were determined to be the optimal delivery system exhibiting 8.63% drug loading efficiency and 91.84% drug encapsulation rate. The results of the quantitative analysis of doxorubicin are shown in Fig. S5 and Tables S1–S3.†Additionally, TEM was conducted to characterize the well-controlled formation of nanoparticles with collaborative assembly of doxorubicin and galactosyl diblock glycopolymers. GN Vehicles I–IV, obtained via deprotection of the galactosyl diblock glycopolymers pGal(Ac)m-b-pNIPAAn7, were observed to form micelles through self-assembly. Subsequently, GND NPs I–IV were constructed via collaborative assembly with pGal(Ac)m-b-pNIPAAn7 and doxorubicin, and the morphology transition from the micelles to the nanoparticles took place under the normal pH conditions. The average diameters of GND NPs I–IV were determined by TEM, which were smaller than those of hollow micelle vehicles (Fig. 2a and b). However, the assembled delivery vehicles clearly disintegrated due to the morphological transformation from nanoparticles to broken micelles when exposed to a buffer at pH 5.5 (Fig. 2d). The zeta potential values indicated that the length of pNIPAA is too short for GN Vehicle I to form stable micelles both at pH 7.4 and 5.5, because micelles with zeta potential values in the range of −10 < 0 < 10 mV are normally considered to be neutral and unstable. In addition, with the extension of pNIPAA, the hydrophilic/hydrophobic effect plays a major role in the stability of the polymer, and the other NP Vehicles were relatively stable at pH 7.4 while they were destroyed due to protonation when soaked at pH 5.5. Meanwhile, the absolute difference in the zeta potential values of GN Vehicle III is the most significant among GN Vehicles at different pH values (Fig. 3a). The pH responsiveness of GN Vehicles may contribute to the pH responsiveness drug release of GND NPs. After comprehensive consideration of drug loading efficiency, drug encapsulation rate and zeta potential analysis, GND NPs III were selected as the optimal drug delivery system for the evaluation of targeted anti-tumor activity.
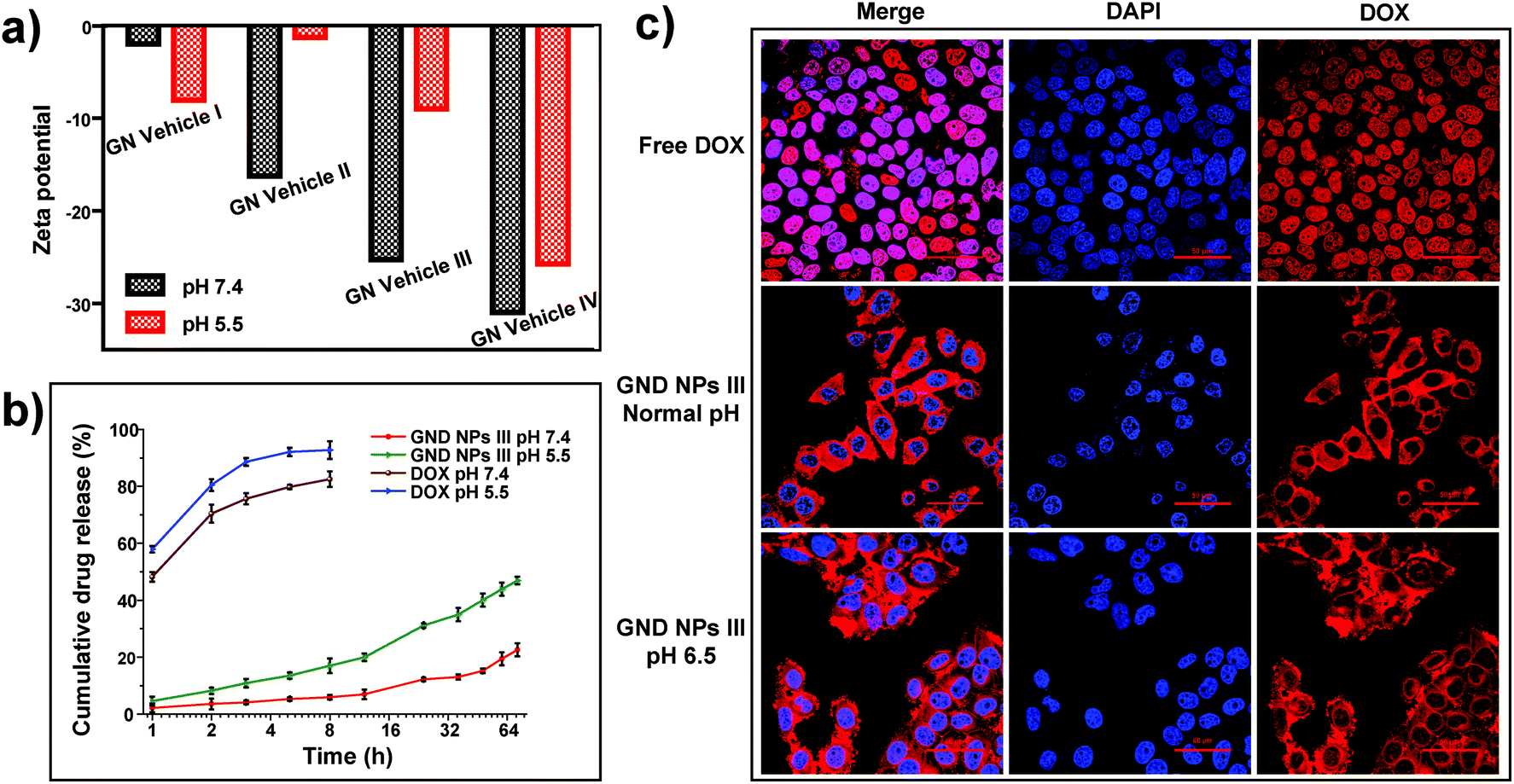 | ||
| Fig. 3 pH responsive drug release. (a) Zeta potential of GN Vehicles I–IV under different pH conditions at a concentration of 1 mg mL−1. (b) Accumulative release (%) of GND NPs III in vitro and free DOX was taken as the control. (c) CLSM images of HepG2 cells incubated with free DOX under normal pH conditions and GND NPs III (5 μg mL−1) under normal or acidic pH conditions for 2 h. For each panel, the images from right to left show DOX fluorescence in cells (red), cell nuclei stained with DAPI (blue) and overlays of the two images. | ||
We evaluated the drug releasing rate under two pH conditions at 5.5 and 7.4, simulating the acidic microenvironment of tumor tissues and the normal physiological media, respectively. As shown in Fig. 3b, free doxorubicin solution was used as the control which measures the amount of drug release at pH 7.4 and 5.5, respectively. The release of free DOX was quick both at pH 7.4 and 5.5, and it is worth noting that the release of free DOX was quicker at pH 5.5. Therefore, the variable solubility of DOX at different pH levels plays an important role in the acid-triggered DOX release and also contributes to the disintegration of NPs. The cumulative drug release ratio of GND NPs III was only 20% at pH 7.4 in PBS buffer for 72 h, while the cumulative release ratio of GND NPs III dramatically increased up to 45.0% at pH 5.5 within 72 h. Obviously, the collaborative assembly of doxorubicin and the diblock glycopolymers achieved sustainable release of drugs. And the phenomenon was consistent with the pH responsiveness of the diblock glycopolymers and the solubility change of doxorubicin under different pH conditions. Accordingly, GND NPs III achieve sustained drug release and can trigger drug release under acidic conditions.
Simultaneously, we observed that GND NPs III were mainly distributed in the cytoplasm of HepG2 cells and less in the nucleus, whereas free doxorubicin was mainly distributed in the nucleus. This phenomenon may be attributed to the size of GND NPs III that is small to enter the cells but not small enough to enter the nucleus. Considering the drug acid responsive release of GND NPs III and the acid microenvironment of tumors, the cells cultured under neutral pH conditions could hardly simulate the tumor microenvironment and the distribution of GND NPs III in HepG2 cells. Hence, HepG2 cells were first cultured under neutral pH conditions until they were completely attached to the substrate, then the cultural medium was replaced with the buffer at pH 6.5 and the normal pH, and the cells were incubated for another 2 h separately. We observed that the distribution of GND NPs III gradually increased in the nucleus with decreasing buffer pH (Fig. 3c). The present data of CLSM coincided with the abovementioned results of drug release. It could be concluded that the GND NPs III could enhance the increased intracellular accumulation of DOX in the nucleus under an acidic tumor microenvironment.
In vitro anti-tumor efficacy of GND NPs III
To evaluate the HCC targeting ability and anti-tumor efficacy of the GND NPs III, HepG2 cells (hepatoma cells) were used as galactose receptor overexpressed (ASGPR+) cells, whereas A549 cells (lung cancer cells), HeLa cells (cervix cancer cells), and Chang liver cells (normal hepatocytes) were used as the negative control and normal cells, respectively. Hollow micelle vehicle GN Vehicle III showed no cytotoxicity against the HepG2, A549, HeLa, and Chang liver cells even at high concentrations of up to 1 mg mL−1 for 48 h incubation as depicted in Fig. 4a (72 h incubation as depicted in Fig. S6†), which demonstrated that the galactosyl diblock glycopolymers have good biocompatibility to serve as drug vehicles. In addition, the A549, HeLa, and HepG2 cells were subsequently incubated with GND NPs III at varying concentrations ranging from 1 to 32 μg mL−1 for 72 h. Compared with the A549 and HeLa cells, GND NPs III exhibited higher inhibitory effects on the proliferation of HepG2 cells, due to the overexpression of ASGPR in the cells (Fig. 4b). To evaluate the targeted therapy for hepatic carcinoma, a confocal laser scanning microscope (CLSM) was employed to examine the intracellular distribution of DOX after incubation of versatile cells with GND NPs III (in the presence or absence of galactose). As shown in Fig. 4c, significant fluorescence was detected for HepG2 after incubation with GND NPs III, whereas almost no DOX fluorescence was observed in the A549 and HeLa cells. Free galactose as a competitive inhibitor was applied for the assessment of ASGPR targeting, and a weaker fluorescence was clearly observed in HepG2, whereas no significant change was detected in the A549 and HeLa cells. Accordingly, we speculated that the HepG2 cells have tight binding affinity with GND NPs III through overexpressed ASGPR on their cell surface, and free galactose in cell culture medium may bind to ASGPR and inhibit the cellular uptake of GND NPs III in a competitive manner. The results reported in this study showed that the galactose-based diblock glycopolymers could be potentially developed as a drug targeting therapy for HCC.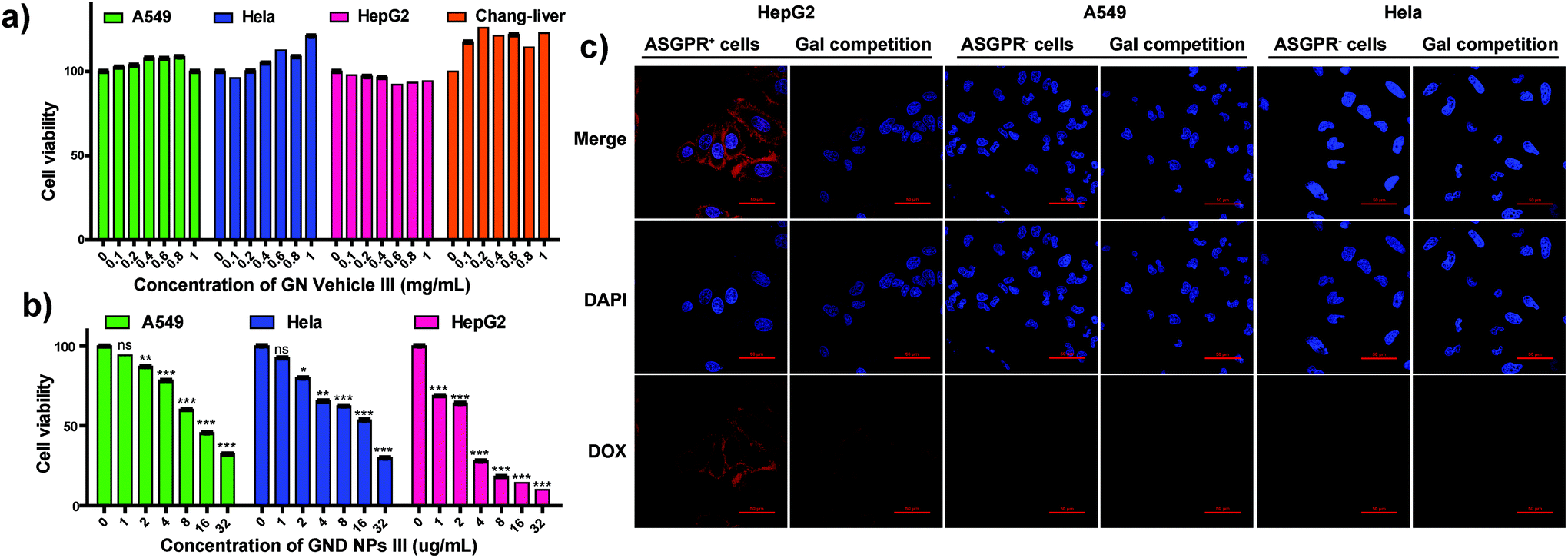 | ||
| Fig. 4 (a) Cell viability assay for GN Vehicle III with A549, Hela, HepG2 and Chang-liver cells after 48 h incubation (n = 4). (b) Cell viability assay for the DOX-loaded GND NPs III with the A549, HeLa and HepG2 cells for 72 h incubation; values are means ± SD. *p < 0.05; **p < 0.01; and ***p < 0.001. Unpaired t-test, two-tailed (n = 4). (c) Representative CLSM microimages of the HepG2, A549 and HeLa cells incubated with GND NPs III for 1 h: in the absence or presence of 20 mM galactose. The cellular nuclei were stained with DAPI (blue). The scale bar is 50 μm. | ||
In vivo anti-tumor efficacy of GND NPs III
Approximately 76% of the human genes have orthologues in zebrafish at the genomic level.64 The use of zebrafish as the model animal showed comprehensive advantages including low costs of zebrafish husbandry, availability in large quantity, optical transparency of zebrafish larvae, and specific creation of new transgenic zebrafish lines.65,66 The wild-type embryos were placed in a 12-well plate (50 embryos in 3 mL of ERS (embryo rearing solution) per well) at 10 hpf (hours post fertilization) and were cultured with GN vehicle III-ERS medium from 10–72 hpf (Fig. 5a). Compared with the control group, no obvious abnormal phenotypes were observed in the exposed groups of 50, 100, 200, and 400 μg mL−1 (Fig. 5b). In addition, we observed a higher embryo hatching ratio in GN vehicle-treated groups compared with the control group, and no obvious abnormal phenotypes were observed in the exposed groups of 50, 100, 200, and 400 μg mL−1 (Fig. 5b and c). In addition, we observed a higher embryo hatching ratio in GN Vehicle-treated groups compared with the control group (Fig. S7†). These results indicated that GN Vehicle III has good biocompatibility for drug delivery.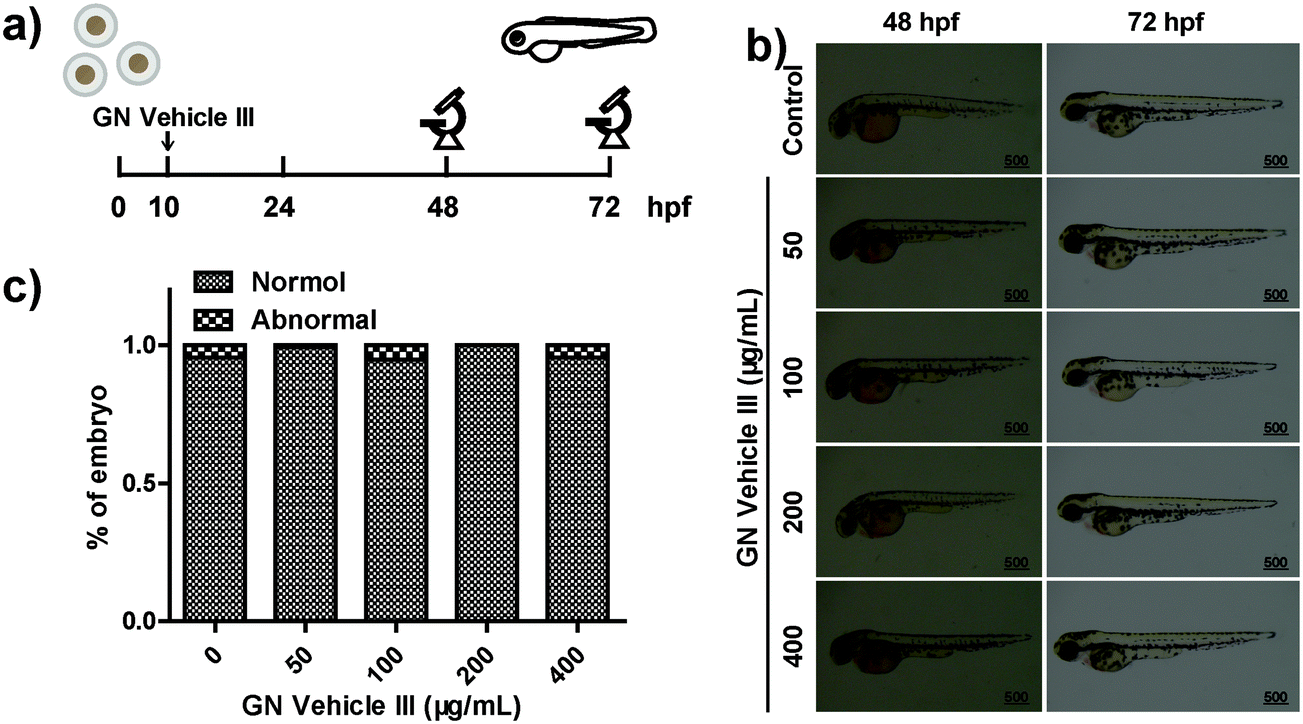 | ||
| Fig. 5 (a) Schematic illustration of the experimental procedure to study the biocompatibility of GN Vehicle III in vivo. (b) Phenotype of zebrafish embryos following exposure to GN vehicle III. The results are from at least three independent experiments and photographs taken at 48 hpf and 72 hpf. (c) The survival situation of zebrafish embryos following exposure to GN Vehicle III at 72 hpf (n = 102). | ||
To investigate the in vivo therapeutic efficacy, transgenic HCC (HCC) zebrafish model, TO(KrasG12V) constructed by Gong,67 was used to evaluate the anti-tumor efficiency. The TO(KrasG12V) zebrafish model developed HCC driven by oncogenic Kras signaling after doxycycline exposure, and Lipan zebrafish served as the normal control for liver area analysis.65 To critically evaluate the anti-tumor effect, the appropriate concentration of doxycycline was first determined. According to the previous report,67 TO(KrasG12V) zebrafish embryos were reared with standard ERS with doxycycline at final concentrations of 0, 7.5, 15, 30, and 60 μg mL−1 from 3 dpf to 7 dpf, respectively (Fig. S8a†). Green fluorescence was observed in all doxycycline-treated embryos, indicating the induced oncogene expressions in liver (Fig. S8b†). At the concentration of 15 μg mL−1, doxycycline can induce a fluorescent area similar to a normal liver area, indicated by red fluorescence in LiPan embryos. The doxycycline-induced tumor areas at the concentrations of 30 and 60 μg mL−1 were significantly larger than the normal liver area, suggesting the outgrowth in an induced liver tumor (Fig. S8c†). However, fluorescence intensity showed no significant difference between 30 μg mL−1 and 60 μg mL−1 doxycycline-treated groups (Fig. S8d†). Additionally, doxycycline has no obvious effect on the zebrafish embryo development except for the liver outgrowth at any tested concentrations. Hence, appropriate inducing conditions for liver tumors in transgenic zebrafish TO(KrasG12V) embryos were treatment with doxycycline at a final concentration of 30 μg mL−1 for 4 days from 3 dpf to 7 dpf.
The TO(KrasG12V) zebrafish embryos were divided randomly into groups treated with doxycycline at the final concentration of 30 μg mL−1 from 3 dpf to 7 dpf, and free DOX or GND NPs III were given at the concentrations of 0, 1, 2, 4, 8, and 16 μg mL−1 between 3 dpf to 5 dpf (Fig. 6a). Induced liver tumor areas and fluorescence intensities were evaluated (Fig. 6b and c). The results showed that the anti-tumor effect was gradually enhanced with the increasing concentrations of doxorubicin in both free DOX and GND NPs III groups. However, GND NPs III treatment significantly reduced induced tumor area and fluorescence intensity at a lower working concentration compared with that of free DOX. As shown in Fig. 6c, GND NPs III exhibited a significant anti-tumor effect at 2 μg mL−1 while free doxorubicin showed a similar effect at 4 μg mL−1. Furthermore, the tumor inhibition rate of GND NPs III is approximately 75% while that of free DOX is only 40% at the concentration of 8 μg mL−1. The above results suggested a better anti-tumor effect of GND NPs III than free DOX, which should be attributed to the liver-specific targeting and sustainable drug release function of GND NPs III.
 | ||
| Fig. 6 Anti-tumor effect evaluation in vivo. (a) Schematic illustration of the experimental procedure to study the anti-tumor effects of GND NPs III in vivo. (b) Phenotype of zebrafish embryos following the treatment with free doxorubicin and GND NPs III. (c) Liver fluorescence area and relative fluorescence intensity of the zebrafish with hepatocellular carcinoma treated with GND NPs III while free doxorubicin was taken as the control (n ≥ 25). The results are from at least three independent experiments. Total embryo number for each group is given at the top. Values are means ± SD. *p < 0.05; **p < 0.01; and ***p < 0.001. Unpaired t-test, two-tailed. | ||
Conclusions
In summary, a series of galactosyl diblock glycopolymers with N-isopropylacrylamide grafts were designed and prepared via RAFT polymerization. GN Vehicle III aggregated by self-assembly indicated the low cytotoxicity up to 1 mg mL−1 by MTT assay towards multiple different cells in vitro and zebrafish model in vivo. The glycopolymers and doxorubicin collaboratively assembled into nanoparticles with uniform size due to the phase transition triggered by alkali conditions. Among these drug delivery systems, GND NPs III exhibited higher drug loading efficiency and encapsulation rate with acid-triggered DOX release. Furthermore, the galactosyl diblock glycopolymers developed as drug delivery vehicles are apparently mediated by ASGPR targeting HCC, resulting in significantly enhanced anti-cancer efficacy in vitro and in vivo. The present study may provide a promising strategy to establish the multifunctional drug delivery system for targeted HCC treatment.Experimental section
Materials
D-(+)-Galactose (99%), acryloyl chloride (96%), ammonium carbamate (97%), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (>97%), and tetrabutylammonium hydroxide (25% in H2O, TBAOH) were purchased from Aladdin and used without further purification. 2,2′-Azobis(2-methylpropionitrile) (99%, AIBN) and N-isopropylacrylamide (98%, NIPAA) were purchased from Aladdin and further purified via recrystallization, respectively, with methanol and hexane. Doxorubicin, 4′,6-diamidino-2-phenylindole (DAPI) and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich. SephadexTM LH-20 was purchased from GE Healthcare. Other chemical reagents were purchased from Energy Chemical and used directly. Water was purified using a Milli-Q® water purification system. HepG2 and A549 cells were purchased from Chinese Academy of Sciences (Shanghai, China) and Hela cells and Chang-liver cells were kindly provided by Marine Biomedical Research Institute of Qingdao. Dulbecco's Modified Eagle's Medium (DMEM), minimum Eagle's medium (MEM) and Roswell Park Memorial Institute 1640 medium (RPMI-1640) were purchased from Gino Biomedical Technology Co., Ltd. All the cell lines were maintained at 37 °C in a humidified incubator with 5% CO2. Wild-type zebrafish, LiPan zebrafish and TO(krasG12V) transgenic zebrafish (kindly provided by Dr Gong67) were applied in this study. Embryos were incubated in ERS (embryo rearing solution) medium (1% NaCl, 1% KCl, 1% CaCl2, 1% MgSO4, and 0.01% methylene blue, pH 7.2) and 1-phenyl-2-thiourea (PTU) was added in ERS medium to prevent the growth of melanin. Adult zebrafish were kept at 28.5 °C with a 14-hour light/10-hour dark cycle. All experimental protocols were approved by and conducted in accordance with the Ethical Committee of Experimental Animal Care, Ocean University of China.Synthesis and self-assembly of galactosyl diblock glycopolymers via RAFT polymerization
Drug loading and releasing
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min until all liquid separated, and then the concentration of unencapsulated DOX in the centrifugal fluid was determined using the microplate reader. Finally, the efficiency of drug loading and drug encapsulation was calculated according to the following formulas:
000 rpm for 20 min until all liquid separated, and then the concentration of unencapsulated DOX in the centrifugal fluid was determined using the microplate reader. Finally, the efficiency of drug loading and drug encapsulation was calculated according to the following formulas: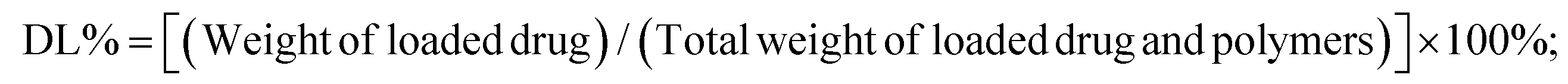
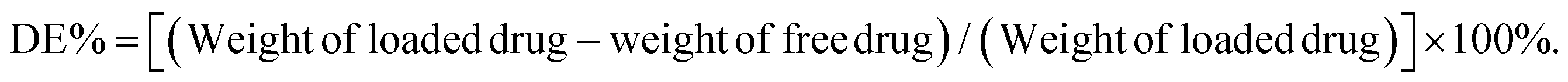
All the release experiments were conducted under dark in triplicate. The cumulative amount of released DOX was calculated accordingly.
In vitro anti-tumor evaluation
| Cell viability (%) = (OD570 test/OD570 control) × 100%. |
In vivo anti-tumor evaluation
The zebrafish HCC model, TO(KrasG12V) constructed by Gong,67 was used to evaluate the anti-tumor efficiency. The zebrafish model develops cancer driven by oncogenic Kras signalling after doxycycline exposure. All experimental protocols were approved by and conducted in accordance with the Ethical Committee of Experimental Animal Care, Ocean University of China.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China, the NSFC-Shandong Joint Fund for Marine Science Research Centers (21602212, 31670811, 81991522 and U1606403), the National Science and Technology Major Project for Significant New Drugs Development (2018ZX09735004), the Shandong Provincial Major Science and Technology Innovation Project (2018SDKJ0404), the National Key Research and Development Program of China (2018YFC0310900), the Fundamental Research Funds for the Central Universities (201762002) and the Taishan Scholar Project Special Funds.Notes and references
- A. Forner, M. Reig and J. Bruix, Lancet, 2018, 391, 1301–1314 CrossRef
.
- J. D. Yang, P. Hainaut, G. J. Gores, A. Amadou, A. Plymoth and L. R. Roberts, Nat. Rev. Gastroenterol. Hepatol., 2019, 16, 589–604 CrossRef PubMed
- L. Peng, Z. Li, X. Li, H. Xue, W. Zhang and G. Chen, Macromol. Rapid Commun., 2017, 38, 1600548–1600554 CrossRef PubMed
- E. Markovsky, H. Baabur-Cohen, A. Eldar-Boock, L. Omer, G. Tiram, S. Ferber, P. Ofek, D. Polyak, A. Scomparin and R. Satchi-Fainaro, J. Controlled Release, 2012, 161, 446–460 CrossRef CAS PubMed
- G. Chen, D. Svirskis, W. Lu, M. Ying, Y. Huang and J. Wen, J. Controlled Release, 2018, 277, 142–153 CrossRef CAS PubMed
- C. G. Zhang, W. J. Zhu, Y. Liu, Z. Q. Yuan, S. D. Yang, W. L. Chen, J. Z. Li, X. F. Zhou, C. Liu and X. N. Zhang, Sci. Rep., 2016, 6, 23859–23870 CrossRef CAS PubMed
- H. Wang, S. Mukherjee, J. Yi, P. Banerjee, Q. Chen and S. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 18639–18649 CrossRef CAS PubMed
- A. Gabizon, H. Shmeeda and Y. Barenholz, Clin. Pharmacokinet., 2003, 42, 419–436 CrossRef CAS PubMed
- J. M. Llovet, R. Montal, D. Sia and R. S. Finn, Nat. Rev. Clin. Oncol., 2018, 15, 599–616 CrossRef PubMed
- S. G. Klochkov, M. E. Neganova, V. N. Nikolenko, K. Chen, S. G. Somasundaram, C. E. Kirkland and G. Aliev, Semin. Cancer Biol., 2019 DOI:10.1016/j.semcancer.2019.08.028
- F. Leng, F. Liu, Y. Yang, Y. Wu and W. Tian, Nanomaterials, 2018, 8, 202–216 CrossRef PubMed
- S. Zhang, R. Geryak, J. Geldmeier, S. Kim and V. V. Tsukruk, Chem. Rev., 2017, 117, 12942–13038 CrossRef CAS PubMed
- R. Xia, X. Zheng, X. Hu, S. Liu and Z. Xie, ACS Appl. Mater. Interfaces, 2019, 11, 5782–5790 CrossRef CAS PubMed
- W. L. Liu, T. Liu, M. Z. Zou, W. Y. Yu, C. X. Li, Z. Y. He, M. K. Zhang, M. D. Liu, Z. H. Li, J. Feng and X. Z. Zhang, Adv. Mater., 2018, 30, 1802006–1802015 CrossRef PubMed
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS
- M. Bouchoucha, M.-F. Côté, R. C. Gaudreault, M.-A. Fortin and F. Kleitz, Chem. Mater., 2016, 28, 4243–4258 CrossRef CAS
- F. Kamil and J. H. Rowe, J. Gastrointest. Oncol., 2018, 9, 180–195 CrossRef PubMed
- A. V. Nascimento, A. Singh, H. Bousbaa, D. Ferreira, B. Sarmento and M. M. Amiji, Acta Biomater., 2017, 47, 71–80 CrossRef CAS PubMed
- T. Wang, L. Wang, X. Li, X. Hu, Y. Han, Y. Luo, Z. Wang, Q. Li, A. Aldalbahi, L. Wang, S. Song, C. Fan, Y. Zhao, M. Wang and N. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 18619–18625 CrossRef CAS PubMed
- J. Li, C. Cai, J. Li, J. Li, J. Li, T. Sun, L. Wang, H. Wu and G. Yu, Molecules, 2018, 23, 2661–2686 CrossRef PubMed
- V. Biju, Chem. Soc. Rev., 2014, 43, 744–764 RSC
- X. Zhang, L. Li, C. Li, H. Zheng, H. Song, F. Xiong, T. Qiu and J. Yang, Carbohydr. Polym., 2017, 155, 407–415 CrossRef CAS PubMed
- M. Khanal, T. Vausselin, A. Barras, O. Bande, K. Turcheniuk, M. Benazza, V. Zaitsev, C. M. Teodorescu, R. Boukherroub, A. Siriwardena, J. Dubuisson and S. Szunerits, ACS Appl. Mater. Interfaces, 2013, 5, 12488–12498 CrossRef CAS PubMed
- H. Yang, C. Tang and C. Yin, Acta Biomater., 2018, 73, 400–411 CrossRef CAS PubMed
- F. Yin, Z. Wang, Y. Jiang, T. Zhang, Z. Wang, Y. Hua, Z. Song, J. Liu, W. Xu, J. Xu, Z. Cai and J. Ding, Nanomedicine, 2020, 23, 102085–102096 CrossRef PubMed
- M. Hoop, F. Mushtaq, C. Hurter, X. Z. Chen, B. J. Nelson and S. Pane, Nanoscale, 2016, 8, 12723–12728 RSC
- Y. J. Tseng, S. W. Chou, J. J. Shyue, S. Y. Lin, J. K. Hsiao and P. T. Chou, ACS Nano, 2016, 10, 5809–5822 CrossRef CAS PubMed
- R. Liu, Q. Cui, C. Wang, X. Wang, Y. Yang and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 3006–3015 CrossRef CAS PubMed
- L. Amicone and A. Marchetti, Transl. Gastroenterol. Hepatol., 2018, 3, 24 CrossRef PubMed
- Y. Wang, S. Song, T. Lu, Y. Cheng, Y. Song, S. Wang, F. Tan, J. Li and N. Li, Biomaterials, 2019, 220, 119405–119418 CrossRef CAS PubMed
- Q. Yu, T. Huang, C. Liu, M. Zhao, M. Xie, G. Li, S. Liu, W. Huang and Q. Zhao, Chem. Sci., 2019, 10, 9091–9098 RSC
- L. Chen, S. Xu, W. Li, T. Ren, L. Yuan, S. Zhang and X.-B. Zhang, Chem. Sci., 2019, 10, 9351–9357 RSC
- Z. Zhang, Q. Lv, X. Gao, L. Chen, Y. Cao, S. Yu, C. He and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 8404–8411 CrossRef CAS PubMed
- S. B. Yong, J. Y. Chung, Y. Song, J. Kim, S. Ra and Y. H. Kim, Biomaterials, 2019, 219, 119401–119413 CrossRef CAS PubMed
- W. Xu, P. A. Ledin, F. A. Plamper, C. V. Synatschke, A. H. E. Müller and V. V. Tsukruk, Macromolecules, 2014, 47, 7858–7868 CrossRef CAS
- X. Gao, S. Liu, Y. Shi, Z. Huang, Y. Mi, Q. Mi, J. Yang and Q. Gao, Eur. J. Med. Chem., 2017, 125, 372–384 CrossRef CAS PubMed
- X. Wu, B. Lin, M. Yu, L. Yang, J. Han and S. Han, Chem. Sci., 2015, 6, 2002–2009 RSC
- R. Adamo, A. Nilo, B. Castagner, O. Boutureira, F. Berti and G. J. Bernardes, Chem. Sci., 2013, 4, 2995–3008 RSC
- H. Wang, R. Wang, K. Cai, H. He, Y. Liu, J. Yen, Z. Wang, M. Xu, Y. Sun, X. Zhou, Q. Yin, L. Tang, I. T. Dobrucki, L. W. Dobrucki, E. J. Chaney, S. A. Boppart, T. M. Fan, S. Lezmi, X. Chen, L. Yin and J. Cheng, Nat. Chem. Biol., 2017, 13, 415–424 CrossRef CAS PubMed
- S. H. Christiansen, X. Zhang, K. Juul-Madsen, M. L. Hvam, B. S. Vad, M. A. Behrens, I. L. Thygesen, B. Jalilian, J. S. Pedersen, K. A. Howard, D. E. Otzen and T. Vorup-Jensen, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 425–437 CrossRef CAS PubMed
- L. Han, C. Tang and C. Yin, ACS Appl. Mater. Interfaces, 2016, 8, 23498–23508 CrossRef CAS PubMed
- E. I. Rigopoulou, D. Roggenbuck, D. S. Smyk, C. Liaskos, M. G. Mytilinaiou, E. Feist, K. Conrad and D. P. Bogdanos, Asialoglycoprotein receptor (ASGPR) as target autoantigen in liver autoimmunity: lost and found, Autoimmun. Rev., 2012, 12, 260–269 CrossRef CAS PubMed
- B. Shi, M. Abrams and L. Sepp-Lorenzino, Expression of asialoglycoprotein receptor 1 in human hepatocellular carcinoma, J. Histochem. Cytochem., 2013, 61, 901–909 CrossRef CAS PubMed
- J. Tanaka, A. S. Gleinich, Q. Zhang, R. Whitfield, K. Kempe, D. M. Haddleton, T. P. Davis, S. Perrier, D. A. Mitchell and P. Wilson, Biomacromolecules, 2017, 18, 1624–1633 CrossRef CAS PubMed
- Y. Zhang, Z. Li, H. Ge, X. Zhu, Z. Zhao, Z.-Q. Qi, M. Wang and J. Wang, Sens. Actuators, B, 2019, 285, 584–589 CrossRef CAS
- X. Guo, B. Choi, A. Feng and S. H. Thang, Macromol. Rapid Commun., 2018, 39, 1800479–1800498 CrossRef PubMed
- V. Vazquez-Dorbatt, J. Lee, E. W. Lin and H. D. Maynard, ChemBioChem, 2012, 13, 2478–2487 CrossRef CAS PubMed
- F. Fan, C. Cai, L. Gao, J. Li, P. Zhang, G. Li, C. Li and G. Yu, Polym. Chem., 2017, 8, 6709–6719 RSC
- Y. Luo, Y. Gu, R. Feng, J. Brash, A. M. Eissa, D. M. Haddleton, G. Chen and H. Chen, Chem. Sci., 2019, 10, 5251–5257 RSC
- F. Fan, C. Cai, W. Wang, L. Gao, J. Li, J. Li, F. Gu, T. Sun, J. Li, C. Li and G. Yu, ACS Macro Lett., 2018, 7, 330–335 CrossRef CAS
- C. von der Ehe, A. Rinkenauer, C. Weber, D. Szamosvari, M. Gottschaldt and U. S. Schubert, Macromol. Biosci., 2016, 16, 508–521 CrossRef CAS PubMed
- C. Dalal and N. R. Jana, J. Phys. Chem. C, 2018, 122, 25651–25660 CrossRef CAS
- K. J. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed
- C. Rest, R. Kandanelli and G. Fernandez, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC
- K. Ferji, P. Venturini, F. Cleymand, C. Chassenieux and J.-L. Six, Polym. Chem., 2018, 9, 2868–2872 RSC
- L. Su, C. Wang, F. Polzer, Y. Lu, G. Chen and M. Jiang, ACS Macro Lett., 2014, 3, 534–539 CrossRef CAS
- Z. Wang, T. Luo, A. Cao, J. Sun, L. Jia and R. Sheng, Nanomaterials, 2018, 8, 136 CrossRef PubMed
- X. Wu, Y. J. Tan, H. T. Toh, L. H. Nguyen, S. H. Kho, S. Y. Chew, H. S. Yoon and X. W. Liu, Chem. Sci., 2017, 8, 3980–3988 RSC
- K. W. Huang, Y. T. Lai, G. J. Chern, S. F. Huang, C. L. Tsai, Y. C. Sung, C. C. Chiang, P. B. Hwang, T. L. Ho, R. L. Huang, T. Y. Shiue, Y. Chen and S. K. Wang, Biomacromolecules, 2018, 19, 2330–2339 CrossRef CAS PubMed
- M. Papadakis, P. Muller-Buschbaum and A. Laschewsky, Langmuir, 2019, 35, 9660–9676 CrossRef PubMed
- L. M. Likhosherstov, O. S. Novikova and V. N. Shibaev, Dokl. Chem., 2002, 383, 89–92 CrossRef CAS
- J. Tang, E. Ozhegov, Y. Liu, D. Wang, X. Yao and X.-L. Sun, ACS Macro Lett., 2017, 6, 107–111 CrossRef CAS
- K. Yang, T. Gao, Z. Bao, J. Su and X. Chen, J. Mater. Chem. B, 2013, 1, 6442–6448 RSC
- Y.-J. H. Jeng-Wei Lu, Yi.-Ju Yang, H.-An Liao, S.-Ci Ciou, L.-In Lin and Da.-L. Ou, World J. Gastroenterol., 2015, 21, 12042–12058 CrossRef PubMed
- Y. Chen, W. Liu, Y. Shang, P. Cao, J. Cui, Z. Li, X. Yin and Y. Li, Int. J. Nanomed., 2019, 14, 57–74 CrossRef CAS PubMed
- S. Sangabathuni, R. V. Murthy, P. M. Chaudhary, B. Subramani, S. Toraskar and R. Kikkeri, Sci. Rep., 2017, 7, 4239 CrossRef PubMed
- T. W. Chew, X. J. Liu, L. Liu, J. M. Spitsbergen, Z. Gong and B. C. Low, Oncogene, 2014, 33, 2717–2727 CrossRef CAS PubMed
- S. Kumar, B. Maiti and P. De, Langmuir, 2015, 31, 9422–9431 CrossRef CAS PubMed
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, preparation and characterization. See DOI: 10.1039/c9bm01604j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |