
CD44 targeted redox-triggered self-assembly with magnetic enhanced EPR effects for effective amplification of gambogic acid to treat triple-negative breast cancer†
Mangmang
Sang
a,
Lingfei
Han
 a,
Renjie
Luo
a,
Wei
Qu
bc,
Feng
Zheng
a,
Kaigang
Zhang
b,
Fulei
Liu
bc,
Jingwei
Xue
b,
Wenyuan
Liu
*ad and
Feng
Feng
*be
a,
Renjie
Luo
a,
Wei
Qu
bc,
Feng
Zheng
a,
Kaigang
Zhang
b,
Fulei
Liu
bc,
Jingwei
Xue
b,
Wenyuan
Liu
*ad and
Feng
Feng
*be
aKey Laboratory of Drug Quality Control and Pharmacovigilance (China Pharmaceutical University), Ministry of Education, Nanjing 210009, China. E-mail: liuwenyuan8506@163.com; Fax: +86 025 83271038;; Tel: +86 025 83271038
bThe Joint Laboratory of Chinese Pharmaceutical University and Taian City Central Hospital, Taian City Central Hospital, Taian, 271000, China. E-mail: fengfeng@cpu.edu.cn; Tel: +86 13952045795
cDepartment of Natural Medicinal Chemistry, China Pharmaceutical University, Nanjing 211198, China
dHangzhou Institute of Pharmaceutical Innovation, China Pharmaceutical University, 291 Fucheng Lu, Hangzhou 310018, China
eJiangsu Food & Pharmaceutical Science College, 4 Meicheng Donglu, Huaian 223003, China
First published on 23rd October 2019
Abstract
Gambogic acid (GA) is a natural anti-tumor drug whose application is restricted by its poor aqueous solubility and inefficient bioavailability. Developing nanomaterials with excellent biocompatibility can amplify the therapeutic effects of GA. In this study, a tumor-targeted redox controllable self-assembled nano-system with magnetic enhanced EPR effects (mPEG-HA/CSO-SS-Hex/SPION/GA) was developed to improve the anticancer efficacy of GA. The nano-system is constituted by three layers: the outer layer is mono-aminated poly(ethylene glycol) grafted hyaluronic acid (mPEG-HA), which can target the CD44 receptor in breast cancer cells; the middle layer consists of disulfide linked hexadecanol (Hex) and chitosan oligosaccharide (CSO) to control the drug release by reduction response; the core layer is superparamagnetic iron oxide nanoparticles (SPION), which can enhance the EPR effect by magnetic guidance and contribute to GA entrapment. Different experiments were performed to characterize the complex self-assembly, and the cytotoxicity, pharmacokinetics, and in vivo antitumor activity of the self-assembly were investigated to evaluate its anti-tumor effects. The results revealed that mPEG-HA/CSO-SS-Hex/SPION/GA is an excellent nanosystem with appropriate size and sensitive responsiveness; it can accumulate in tumor sites and achieve excellent therapeutic effects on triple-negative breast cancer (TNBC). In summary, a CD44-targeted redox-triggered self-assembly nanosystem with magnetic enhanced EPR effects was developed for effective amplification of GA; it has potential to act as an effective carrier in drug delivery for chemotherapy of TNBC.
1. Introduction
To date, numerous nanomedicines have been developed and applied in clinical or non-clinical areas. Nanomedicines show essential characteristics, such as delivery of specific drugs, higher accumulation in target tissues, improved pharmacokinetic/pharmacodynamics properties, and greater safety and biocompatibility.1 Therefore, nanomedicines are available for treatment of specific diseases which have not been efficiently treated by conventional medicines. The current nanomedicines can be summarized in various categories, including polymeric nanoparticles, solid lipid nanoparticles, liposomes, micelles, and dendrimers.2Self-assembled polymeric micelles (PMs) demonstrate excellent targeting activity to tumors. Furthermore, PMs can improve the efficacy and accurate delivery of drugs via releasing the drugs after specific stimuli, such as acid response or redox reactions.3–5 PMs are an amphiphilic material; they can encapsulate small hydrophobic drugs in their hydrophobic cores and deliver the drugs to their destinations.6–8 Thus, PMs can improve the efficiency of anticancer drugs by selectively accumulating at the tumor site via the EPR effect, prolonging blood circulation time, and enhancing the water solubility of drugs.9,10 HA can be conjugated with hydrophobic polymers to obtain CD44 targeting ability.11,12 Additionally, HA can encase positively charged nanocarriers to adjust the charge distribution and affect the cell uptake of the nanoparticles.13
Breast cancer is one of the most frequent cancers worldwide; it is a serious threat to the health of women. Especially, triple-negative breast cancer (TNBC), which is non-expression of three breast cancer markers, namely estrogen receptor (ER), estrogen progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2).14–16 Currently, there is no effective targeting therapy for triple-negative breast cancer patients, who can only choose systemic chemotherapy; however, systemic chemotherapy has unavoidable adverse effects.15,17 Although the development of adjuvant chemotherapy decreases the mortality of TNBC patients by a rate of 30%,18 there has been limited research on incorporating additional systemic therapies.19 Therefore, it is a pressing matter to develop new drugs or novel formulation strategies to overcome TNBC.
Gambogic acid (GA), a recently explored polyprenylated xanthene small molecule, is the main active ingredient of gamboge resin, which is exuded from the Garcinia hanburyi tree in Southeast Asia. Gamboge resin has multiple therapeutic actions and has been used in China for hundreds of years.20,21 Numerous studies have confirmed that GA has significant anti-tumor effects; it is effective against multitudinous types of malignant tumors, including lung cancer, hepatocarcinoma, gastric carcinoma, prostate, and breast carcinoma.22–24 The China Food and Drug Administration has also approved the second phase of a clinical trial of GA.25 However, its poor water solubility (<5 ppm) and short biological half-life (less than 20 minutes in rats and 1 hour in dogs)26 have proved to be major hurdles for the clinical application of GA.
Tumor double-targeted redox-responsive hybrid multifunctional self-assembled polymeric micelles (mPEG-HA/CSO-SS-Hex/SPION/GA) have been developed for targeted delivery of GA to the TNBC microenvironment, which increased the antitumor activity of the chemotherapeutic drug GA. As shown in Scheme 1, the core of the complex self-assembly is a reducible copolymer based on coupling CSO to disulfide-containing carboxylic acid (CSO-SS-Hex). Then, the tumor magnetic passive targeting SPION nanoparticles and GA were encapsulated into the core of the self-assembly. As a tumor-active targeting ligand of CD44, HA was coated on the surface of the self-assembly via electrostatic absorption. The obtained magnetic complex self-assembly could accumulate in tumor issue and be selectively taken up by tumor cells with the effective aid of both magnetism-enhanced EPR and CD44-receptor-mediated endocytosis.
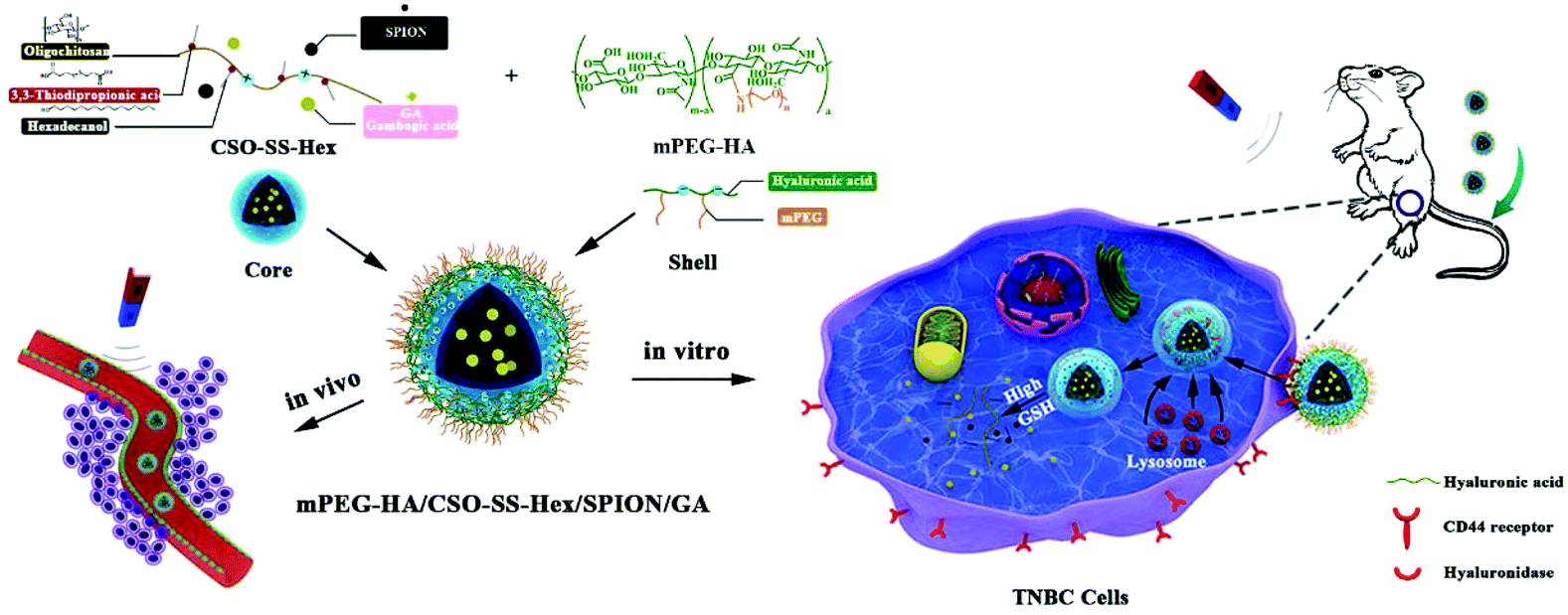 | ||
| Scheme 1 Schemes of mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly preparation and the magnetism-enhanced EPR in vivo and in vitro trafficking pathways of the polymeric self-assembly. The intracellular trafficking pathway involves CD44 receptor-meditated cellular internalization and redox-responsive triggered self-assembly disassembly and drug release. | ||
The in vitro cytotoxicity results showed that mPEG-HA/CSO-SS-Hex/SPION/GA exhibited significantly higher cytotoxicity to cancer cells (4T1, MCF-7 and MDA-MB-231 cell lines, which were obtained from the Shanghai Institutes for Biological Sciences) compared with free GA. Similarly, the complex polymeric self-assembly showed remarkably higher tumor cell uptake than other self-assemblies (HA/CSO-SS-Hex/SPION/GA and CSO-SS-Hex/SPION/GA) over the incubation period due to the dual effects of magnet targeting and CD44 receptor-mediated internalization, which endowed the formulation with tumor double-targeted function. Fluorescence imaging studies demonstrated that the DiR-loaded mPEG-HA/CSO-SS-Hex/SPION/DiR self-assembly accumulated more efficiently in the tumor site, which is also because of the tumor double-targeted function. The pharmacokinetic evaluation results showed that the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly significantly prolonged the half-life and circulatory time of GA in vivo. Moreover, the complex self-assemblies showed better efficiency than free GA to inhibit tumor growth. The overall results indicate that the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly is an effective targeting system for GA transport and treatment of triple-negative breast cancer.
2. Experimental section
2.1. The preparation and characterization of the mPEG-HA/CSO-SS-Hex/SPION/GA complex self-assembly
The synthesis route and the 1H NMR spectra of mono-aminated poly(ethylene glycol) grafted hyaluronic acid (mPEG-HA) are shown in Fig. S1A of the ESI.†The method of the preparation of the GA and SPION-loaded CSO-SS-Hex self-assembly was as follows. The method of the preparation of the CSO-SS-Hex self-assembly was previously reported.27 The SPION-loaded CSO-SS-Hex self-assembly (CSO-SS-Hex/SPION) was prepared using a solvent evaporation method.27 GA was entrapped in the CSO-SS-Hex/SPION self-assembly using a dialysis technique; the specific method is given in the ESI.† Then, mPEG-HA aqueous solution was added to a solution of CSO-SS-Hex/SPION/GA dropwise under vigorous stirring for 1 h at room temperature. The mixture was ultra-sonicated for 15 min with an ice-bath using a probe-type ultrasonicator at 150 W. Then, it was dialyzed against DDW using a dialysis bag (MWCO = 1 kDa) for 24 h and filtered through a 0.45 μm membrane to obtain the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly.
Transmission electron microscopy (TEM) was used to detect the particle sizes of the different complex self-assemblies. In order to investigate the redox-response of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly, the self-assemblies were studied by TEM after being incubated in pH 7.4 ± 10 mM GSH PBS solution at 37 °C for 2 h. The zeta potentials of the different self-assemblies were analyzed using dynamic light scattering (DLS). The drug loading (DL) and entrapment efficiency (EE) of GA in the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly were detected using an HPLC method with UV detection at 360 nm. The content of SPION loaded in the self-assembly was determined using an Iron Assay Kit.28
2.2. In vitro drug release and stability assay
The in vitro drug release was studied by dialysis. The mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly (1.0 mg mL−1, containing 1% tween 80) and free GA were placed in a dialysis bag (MWCO = 3500 Da) with 150 mL of pH 7.4 ± 10 mM GSH PBS buffer (0.01 M, containing 1% tween 80) at 37 °C under gentle shaking (200 rpm). 20 μL of the bag medium was withdrawn and replaced by an equal volume of fresh medium at 0, 1, 2, 4, 8, 12, 24 and 48 h.29 The collected samples were analyzed by HPLC after demulsification to determine the amount of released GA. To investigate the dispersion stability of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly in phosphate buffer saline (0.01 M PBS, pH 7.4) and FBS, the changes in particle size were monitored by DLS for 24 h.2.3. Hysteresis loop measurements
To investigate the superparamagnetism of SPION, the hysteresis loops were generated by a magnetic field (±1.5 T) and detected by a vibrating sample magnetometer. An N38 NiCuNi-Fe magnet (d = 15 mm, h = 6 mm; 0.2 T) was placed in an aqueous solution of mPEG-HA/CSO-SS-Hex/SPION/GA.2.4. In vitro cytotoxicity
Cytotoxicity against MCF-7 cells, MDA-MB-231 cells, 4T1 cells and RAW 264.7 cells was evaluated using a standard MTT/CCK-8 cell viability experiment. 96-well plates with 5 × 103 cells per well were incubated in DMEM containing 10% FBS for 24 h. After incubation of GA, CSO-SS-Hex, CSO-SS-Hex/SPION/GA, HA/CSO-SS-Hex/SPION/GA and mPEG-HA/CSO-SS-Hex/SPION/GA self-assemblies at serial concentration dilutions for 48 h, MTT and CCK-8 solution were added for another 3 h. DMSO (100 μL) was used to dissolve the formazan precipitate, and the results were analyzed and recorded using a microplate reader; then, the IC50 values for each compound were calculated by GraphPad software.2.5. Apoptotic experiment
4T1 cells were seeded in 6-well plates, and then incubated with the mPEG-HA/CSO-SS-Hex/SPION/GA (100 μg mL−1) complex self-assembly for 12 h and 24 h; flow cytometry was used to evaluate the apoptotic cells.2.6. Cell uptake and intracellular release
To investigate the cellular uptake mechanism of the magnetic complex self-assembly, 4T1 cells (overexpressing CD44 receptor) incubated with FITC-labeled CSO-SS-Hex self-assembly were observed via the isothiocyanate group of FITC by fluorescence microscopy.26 Nile red (NR) was used as a model drug and fluorescent probe; it is a stain that is used for the detection of intracellular lipid droplets.30 Because the drug in the free molecule form can interact with target molecules and exert pharmacological action, we further investigated the intracellular drug release behaviors of self-assembly against 4T1 cells using NR, which was loaded into the self-assembly in accordance with the protocol for the preparation of the complex self-assembly.4T cells were treated with the different self-assemblies (FITC-mPEG-HA/CSO-SS-Hex/SPION/NR, FITC-HA/CSO-SS-Hex/SPION/NR and FITC-CSO-SS-Hex/SPION/NR) for 3 h, and a laser scanning confocal microscope was used to evaluate the specific uptakes of the self-assemblies. To investigate the HA-receptor-mediated endocytosis of 4T1 cells, HA (10 mg mL−1)/anti-CD44 antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :250 dilute) were incubated for 1 h before the FITC-mPEG-HA/CSO-SS-Hex/SPION/NR self-assembly was added. The FITC and NR-labeled CSO-SS-Hex/SPION/NR self-assemblies and HA/CSO-SS-Hex/SPION/NR self-assembly were processed as control groups to investigate the influence of HA on cell uptake.
:250 dilute) were incubated for 1 h before the FITC-mPEG-HA/CSO-SS-Hex/SPION/NR self-assembly was added. The FITC and NR-labeled CSO-SS-Hex/SPION/NR self-assemblies and HA/CSO-SS-Hex/SPION/NR self-assembly were processed as control groups to investigate the influence of HA on cell uptake.
The flow cytometry analysis was also studied by FACS analysis to investigate the uptake of the different self-assemblies by 4T1 cells. 4T1 cells were treated with FITC-labeled mPEG-HA/CSO-SS-Hex/SPION/GA (FITC-mPEG-HA/CSO-SS-Hex/SPION/GA), HA/CSO-SS-Hex/SPION/GA (FITC-HA/CSO-SS-Hex/SPION/GA) and CSO-SS-Hex/SPION/GA (FITC-CSO-SS-Hex/SPION/GA) self-assemblies. All flow cytometry was performed and analyzed using a MACS Quant flow cytometer.
To investigate the drug release mediated by disulfide bonds, 4T1 cells were pretreated with 1 mM NEM for 20 min and then divided into two groups: in one group, the culture medium was removed and supplemented with 1 mM GSH,31 while in the other, DMEM was used instead; then, the two groups were incubated with NR-labeled mPEG-HA/CSO-SS-Hex/SPION for 2 h. The cells were also visualized using laser scanning confocal microscopy.
In order to investigate the different substitution degrees of mPEG-HA, 4T1 and raw 264.7 cells were incubated for 24 h; 100 μg mL−1 of three different NR-loaded mPEG-HA/CSO-SS-Hex/SPION/GA (mPEG-HA/CSO-SS-Hex/SPION/NR) self-assemblies with different substitution degrees (18.6%, 15.1%, and 8.8%) were added respectively, as indicated in Table S1.† After incubation at 37 °C for 3 h, the culture medium was removed and the cells were washed twice with cold PBS to remove the self-assemblies which were not ingested by the cells; next, the cellular uptake of the magnetic polymeric self-assemblies in the cells was visualized using laser scanning confocal microscopy.
2.7. In vitro magnetic target assay
A 100 mm Petri dish was used to culture 4T1 cells with 8 mL 10% FBS DMEM medium. After incubation with the mPEG-HA/CSO-SS-Hex/SPION/NR self-assembly (100 μg mL−1) for 24 h, a magnet (field strength approximately 0.2 T) was pasted on the bottom wall of the Petri dish, which was incubated for an additional 3 h. A reflection fluorescence microscope was used to analyze the NR fluorescence and Prussian blue staining of the cells.2.8. Pharmacokinetic evaluation
Male Sprague-Dawley rats (180 to 200 g) were used for the pharmacokinetic study. The rats were randomly divided into two groups (GA and mPEG-HA/CSO-SS-Hex/SPION/GA). After intravenous administration of different samples, blood samples were collected at specified times (0.033, 0.083, 0.167, 0.333, 0.5, 1, 2, 4, 8, 12 and 24 h). 0.5 M HCl was added to the plasma immediately and the samples were extracted with acetonitrile, and then analyzed by LC-MS. The pharmacokinetic parameters were calculated using DAS software (Version 2.0).2.9. In vivo imaging
BALB/c female mice (18 to 20 g) were subcutaneously inoculated with 4T1 cells in the right flank breast.32 When the tumors had grown to volumes of 200 to 300 mm3, the mice were divided into two groups (6 mice per group): mPEG-HA/CSO-SS-Hex/SPION/GA with a magnetic field (MF) and mPEG-HA/CSO-SS-Hex/SPION/GA with no MF, respectively. DiR-loaded mPEG-HA/CSO-SS-Hex/SPION (mPEG-HA/CSO-SS-Hex/SPION/DiR) self-assemblies were administrated33,34 to the mice via the caudal vein. Then, a magnet (approximately 0.2 T) was attached to the tumor using tape. After 0.083, 0.5, 1, 2, 4, 8, and 24 h post-injection, a near infrared fluorescence imaging system was used to acquire fluorescence images of the whole body and 7 organs (tumor, heart, liver, spleen, lung, kidney, intestine) with wavelengths of Ex = 748 nm and Em = 780 nm.2.10. Anti-tumor activity
15 days after the 4T1 cell injection, the tumor-bearing mice whose tumor volumes reached around 100 mm3 were divided into five groups (5 mice per group): saline, GA, CSO-SS-Hex/SPION/GA, mPEG-HA/CSO-SS-Hex/SPION/GA–MF and mPEG-HA/CSO-SS-Hex/SPION/GA + MF (field strength approximately 0.2 T). After 13 days of administration with 6 mg kg−1 of GA, the tumors were harvested. A caliper was used to measure the tumor sizes every two days, and the volumes of the tumors were evaluated by the formula: tumor volume = length × width× width/2.342.11. In vivo biosafety evaluation
Institute of Cancer Research (ICR) mice (18 to 20 g) were randomly divided into three groups: saline, GA and mPEG-HA/CSO-SS-Hex/SPION/GA (concentration of GA: 6 mg kg−1). After 13 days of administration, five organs (heart, liver, spleen, lung, and kidney) and blood were collected. H&E staining of the five organs was performed to evaluate the toxicity of the self-assemblies. The alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in serum were evaluated to determine the liver function. Creatinine (CRE) and urea nitrogen (BUN) levels in serum were evaluated to determine the renal function.2.12. Statistical analysis
Unless otherwise indicated, the statistical analysis was processed using GraphPad Prism software (Version 7.01) or Origin Pro software (Version 2015). The quantitative data were presented as mean ± standard deviation (SD).3. Results and discussion
3.1. Characterization of GA-loaded magnetic complex self-assembly
The synthesis route of mPEG-HA was divided into two steps (Fig. S1†). The substitution degrees of mPEG and HA were determined by calculating the peak area ratios of the methylene units in mPEG (–OCH2CH2–: δ 3.72) to the methyl groups of HA (–COCH3: δ 2.02) in the 1H NMR spectra (Fig. S1†). Three different substitution degrees were investigated (Table S1†). With the decrease of the feed ratio of mPEG to HA, the polymerization degree was also relatively low; thus, the particle size and zeta potential are the reasons for the decreased steric hindrance of mPEG. The substitution degrees of the 8.8% self-assembly showed the highest particle size and zeta potential; this self-assembly may be easily cleared by the immune system.35 However, the zeta potential of the self-assembly with substitution degrees of 8.8% did not increase significantly compared to the substitution degrees of 15.1%; these results indicate that the steric hindrance of mPEG does not play a major role in the properties of self-assemblies with substitution degrees below 15.1%. Although the highest substitution degrees show lower particle sizes and charges, mPEG occupies the active sites of hyaluronic acid and may weaken the targeting effect of HA.36Fig. 1A shows that the size of SPION was less than 10 nm and was uniform. Magnetization curves of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly and SPION are displayed in Fig. 1B; the saturation magnetization of the magnetic complex self-assembly was 5.12 emu g−1, considerably less than that of the SPION nanoparticles (42.83 emu g−1). The loss of magnetization should be attributed to the coating of the surface of SPION nanoparticles by the copolymer. These results indicate that the self-assembly has potential for use as a magnetically guided system for drug delivery.
 | ||
| Fig. 1 Characterization of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly. (A) TEM image and picture of SPION in chloroform. (B) Hysteresis loops of SPION and mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly solutions. (C) TEM image, size distribution and picture in water. (D) In vitro release profiles of GA from the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly under different simulated conditions at 37 °C (n = 3). (E) TEM images of the self-assembly incubated in pH 7.4 PBS and pH 7.4 PBS + 10 mM GSH for 12 h. (F) Stability of mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly in FBS and PBS for 24 h. | ||
TEM imaging showed that the magnetic self-assembly was homogeneously distributed, with a mean diameter of 60 to 100 nm (Fig. 1C); this was smaller than the DLS result (Table S1†). The zeta potential was about 17.5 mV; however, the zeta potential of CSO-SS-Hex/SPION/GA was about 25.8 mV. According to the effective shielding effect of mPEG-HA,37 this implies that the positively charged core were closely covered with the mPEG-HA. Fig. 1C shows that the particles of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly incubated in pH 7.4 PBS + 10 mM GSH at 37 °C for 2 h were much larger than those incubated in PBS alone. The drug loading (DL) and entrapment efficiency (EE) were 23.7% and 85.1%, approximately, as shown in Table S2.† The loading of SPION (DL: 17.24%) could improve the DL and EE of GA; this can be attributed to the stronger hydrophobic interactions of GA, the SPION nanoparticles and the hydrophobic fragments of the copolymer.
To investigate the reduction response of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly, in vitro GA release experiments were performed. Fig. 1D shows that only 40% of the drug was released from the complex self-assembly without GSH. However, the GA release significantly increased up to about 90% within 48 h in presence of GSH. Meanwhile, the free GA was released within 10 h completely in different conditions. These results indicate that the mPEG-HA/CSO-SS-Hex/SPION/GA complex self-assembly facilitates GA release by the reduction response of the self-assembly. Fig. 1E demonstrates the same conclusion. The results of the stability test of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly showed that the self-assembly remained stable with slight size changes over 24 h in both PBS and FBS (Fig. 1F). Fig. S2† shows that the size and zeta potential of the complex assembly in PBS were stable for 7 days; however, the assembly was not stable after 4 days in FBS, due to the reductive substances in FBS.
3.2. Cellular uptake and intracellular drug release
To investigate the cellular uptake and intracellular drug release mechanisms of mPEG-HA/CSO-SS-Hex/SPION/GA, 4T1 cells (overexpressing CD44 receptor) incubated with FITC and NR38-labeled mPEG-HA/CSO-SS-Hex/SPION/GA, HA/CSO-SS-Hex/SPION/GA and CSO-SS-Hex/SPION/GA self-assemblies (mPEG-HA/FITC-CSO-SS-Hex/SPION/NR, FITC-HA/FITC-CSO-SS-Hex/SPION/NR and FITC-CSO-SS-Hex/SPION/NR) were observed by fluorescence microscope.30,39 The results (Fig. 2A and Fig. S3†) show that the fluorescence intensity (FITC) of 4T1 cells treated with the mPEG-HA/FITC-CSO-SS-Hex/SPION/NR (HA−) self-assembly was significantly stronger than that of cells treated with the mPEG-HA/FITC-CSO-SS-Hex/SPION/NR (HA+) self-assembly; the same result was observed in the anti-CD44 block experiment, as shown in Fig. S5.† Also, the fluorescence of the HA/FITC-CSO-SS-Hex/SPION/NR self-assembly-treated cells was stronger than that of cells treated with the FITC-CSO-SS-Hex/SPION/NR self-assembly under the same imaging parameters during the cell imaging process. This finding reveals that HA can competitively bind to CD44 receptors against the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly and play a critical role in increasing cell uptake via receptor-mediated endocytosis, which results in different fluorescence intensities. These results demonstrate that the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly specifically binds to CD44 via CD44 receptor-mediated endocytosis. As shown in Fig. 2A and Fig. S6,† in 4T1 cells, the red fluorescence signal results from the gradual release of NR from the self-assemblies. The red fluorescence of the mPEG-HA/FITC-CSO-SS-Hex/SPION/NR (HA−) self-assembly was also stronger than that of the mPEG-HA/FITC-CSO-SS-Hex/SPION/NR (HA+) self-assembly with incubation time, which also proves the tumor targeting ability of HA. All these results indicate a sharp redox-dependent response of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly with the capacity to quickly release loaded NR into cancer cells.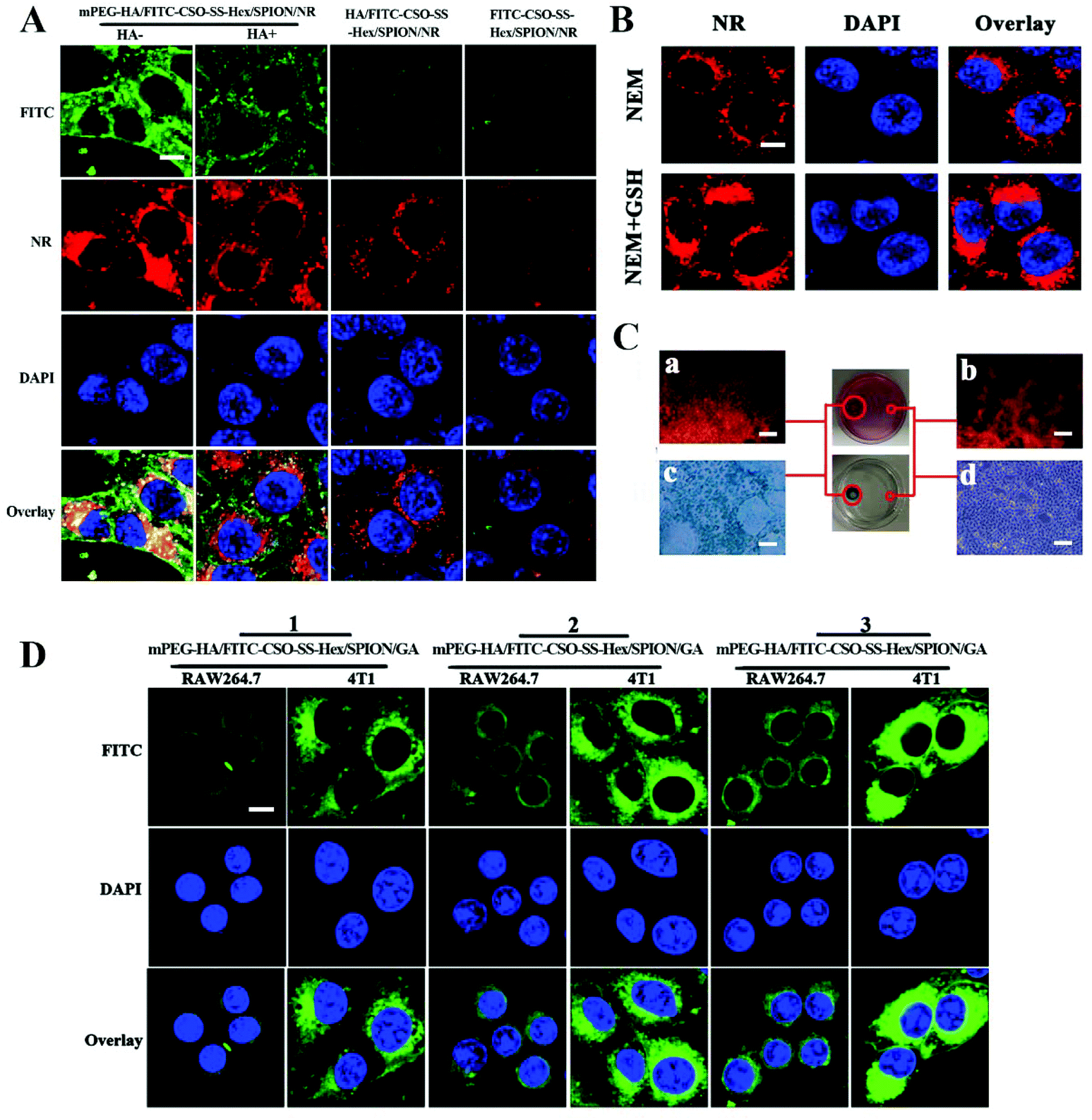 | ||
| Fig. 2 Cell uptake and intracellular drug release of complex self-assemblies in 4T1 and raw 264.7 cells. (A) 4T1 cells were incubated with mPEG-HA/FITC-CSO-SS-Hex/SPION/NR, HA/FITC-CSO-SS-Hex/SPION/NR and FITC-CSO-SS-Hex/SPION/NR after pretreatment with HA (HA+) or no HA (HA−) under different conditions for 3 h, then labeled with DAPI (blue) to identify cell nuclei (scale bar: 5 μm). (B) 4T1 cells were incubated with NR-loaded mPEG-HA/CSO-SS-Hex/SPION for 3 h after pretreatment with NEM (1 mM) and NEM (1 mM) + GSH (10 mM); these were also labeled with DAPI (blue) to identify cell nuclei (scale bar: 5 μm). (C) Micrographs of the mPEG-HA/CSO-SS-Hex/SPION/NR self-assembly after 3 h incubation in an external magnetic field: (a and b) show the NR fluorescence intensities of the cells located in the upper left and right circles; (c and d) show the Prussian blue staining of the cells located in the bottom left and right circles. (a and c) indicate the magnet-targeted area, whereas (b and d) indicate the control area (scale bar: 100 μm). (D) 4T1 and raw 264.7 cells were incubated with FITC-labeled mPEG-HA/CSO-SS-Hex/SPION/GA with different substitution degrees (see Table S1†) for 2 h and labeled with DAPI (blue) to identify cell nuclei (scale bar: 5 μm). | ||
We further investigated the cell uptake of the FITC-labeled FITC-mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly in 4T1 cells at different times of 2 h, 4 h, 6 h, and 8 h by flow cytometry (Fig. S4†). The fluorescence intensities of 4T1 cells incubated with all the self-assembles (FITC-mPEG-HA/CSO-SS-Hex/SPION/GA, FITC-HA/CSO-SS-Hex/SPION/GA and FITC-CSO-SS-Hex/SPION/GA) time-dependently increased. The FITC-HA/CSO-SS-Hex/SPION/GA self-assembly showed higher fluorescence intensity than FITC-CSO-SS-Hex/SPION/GA, which is dependent on the role of HA. The overall results supported the same conclusion that the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly developed in this study appears to be a highly effective, targeted, and traceable multifunctional magnetic complex self-assembly for the effective treatment of cancer at the cellular level.
In order to investigate the drug release mediated by disulfide bonds, we incubated 4T1 with the NR-labeled mPEG-HA/CSO-SS-Hex/SPION self-assembly after pretreatment with NEM, a thiol depletion agent, and NEM + GSH. Fig. 2B and Fig. S6† show that the red fluorescence signal of the NEM + GSH group was stronger than that of the other group, which indicates that the drug release of the self-assembly is mainly mediated by disulfide bonds.
Magnetic targeting experiments were performed in vitro to evaluate the magnetic targeting properties of the NR-loaded mPEG-HA/CSO-SS-Hex/SPION/NR self-assembly. A magnet (0.2 T) was placed on the outer surface of a Petri dish; this area is referred to as the targeting area (left circle), and the non-magnet area as the control (right circle). After washing with PBS, staining with the fluorescent dye NR and Prussian blue was implemented. The results showed that the cells located inside the left circle were dyed deeper than those in the right circle after 3 h of incubation (Fig. 2C); a higher self-assembly concentration was observed in the applied magnetic field, which implies that the magnetic self-assembly efficiently carries cargo to the magnetic guidance-targeted area.
In order to investigate the influence of different substitution degrees of mPEG to HA on the uptake of the self-assemblies, 4T1 and raw 264.7 cells were both incubated with three different FITC37-labeled mPEG-HA/CSO-SS-Hex/SPION/GA assemblies (mPEG-HA/FITC-CSO-SS-Hex/SPION/GA) with different substitution degrees (Table S1†). The results showed that the 4T1 and raw 264.7 cells both demonstrated increased uptake of the self-assemblies with decreasing substitution degrees according to the increased fluorescence intensity (Fig. 2D and Fig. S7†). Because of the increase of the stereospecific blockade of mPEG along with the increase of zeta potential, the self-assembles were more likely to be phagocytized by macrophages. Considering all factors, we chose the middle substitution degree of mPEG to HA as the material to prepare the final self-assembly.
3.3. In vitro efficacy of complex self-assembly
The cell viabilities of GA, CSO-SS-Hex, CSO-SS-Hex/SPION/GA, HA/CSO-SS-Hex/SPION/GA and mPEG-HA/CSO-SS-Hex/SPION/GA self-assemblies against the 4T1, MCF-7 and MDA-MB-231 cell lines were evaluated by CCK-8 kit and MTT assay, as shown in Fig. 3A–C and Fig S8A–C.† The IC50 values of various formulations of GA against the three cell lines are displayed in Tables S3 and S4,† and obvious cytotoxicity was observed for the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly in all three cell lines. Fig. 3D and Fig. S8D† show that the complex self-assembly mPEG-HA/CSO-SS-Hex/SPION/GA demonstrated lower cytotoxicity to RAW 264.7 cells compared to 4T1 cells. These results indicate that the self-assembly is not easily scavenged by macrophages, which reflects the stability of the self-assembly and may enable its long cycle in vivo. Apoptosis of 4T1 cells, showed in Fig. 3E and Fig. S9,† indicated that the complex self-assembly caused cell death more easily compared to free GA. The overall results indicate that the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly appears to be the most effective treatment for tumors at the cellular level.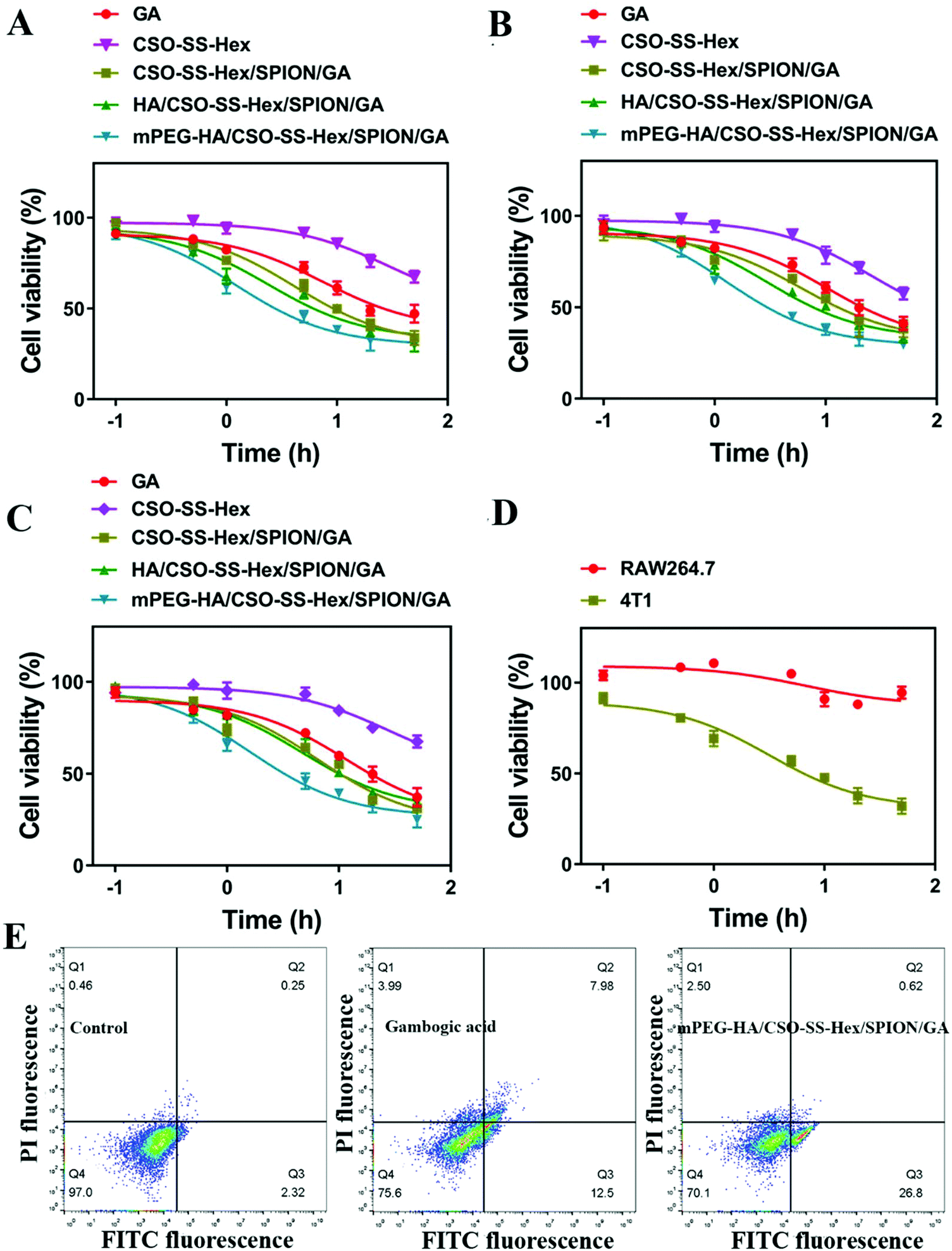 | ||
| Fig. 3 In vitro pharmacodynamics evaluation of the mPEG-HA/CSO-SS-Hex/SPION/GA complex self-assembly. (A–C) Cell viability of 4T1 cells, MCF-7 cells and MDA-MB-231 cells treated with GA and different self-assemblies (GA, CSO-SS-Hex, CSO-SS-Hex/SPION/GA, HA/CSO-SS-Hex/SPION/GA, mPEG-HA/CSO-SS-Hex/SPION/GA) for 48 h (n = 3). (D) Cell viability of 4T1 cells and RAW 264.7 treated with mPEG-HA/CSO-SS-Hex/SPION/GA for 48 h (n = 3). (E) Apoptosis of 4T1 cells incubated with DMEM, GA and mPEG-HA/CSO-SS-Hex/SPION/GA for 12 h. | ||
3.4. In vivo imaging
The tumor-targeting ability of mPEG-HA/CSO-SS-Hex/SPION/GA was evaluated in a 4T1 breast tumor model. DiR, a NIR dye,40 entrapped in the complex self-assembly as mPEG-HA/CSO-SS-Hex/SPION/DiR was prepared to visualize the time-dependent biodistribution in mice. Fig. 4A shows that the fluorescence signal of mPEG-HA/CSO-SS-Hex/SPION/DiR exhibited in the mouse whose tumor was exposed (MF+) to a magnetic field was obviously stronger than that of the mouse that was not exposed (MF−), indicating the good tumor magnetic targeting effect of the self-assembly. After 24 h post injection of the complex self-assembly, we found that the fluorescence was sustained at the tumor location, as shown in Fig. 4B and Fig. S10;† this indicates tumor-specific accumulation of the drug by tumor magnetic targeting of the complex self-assembly.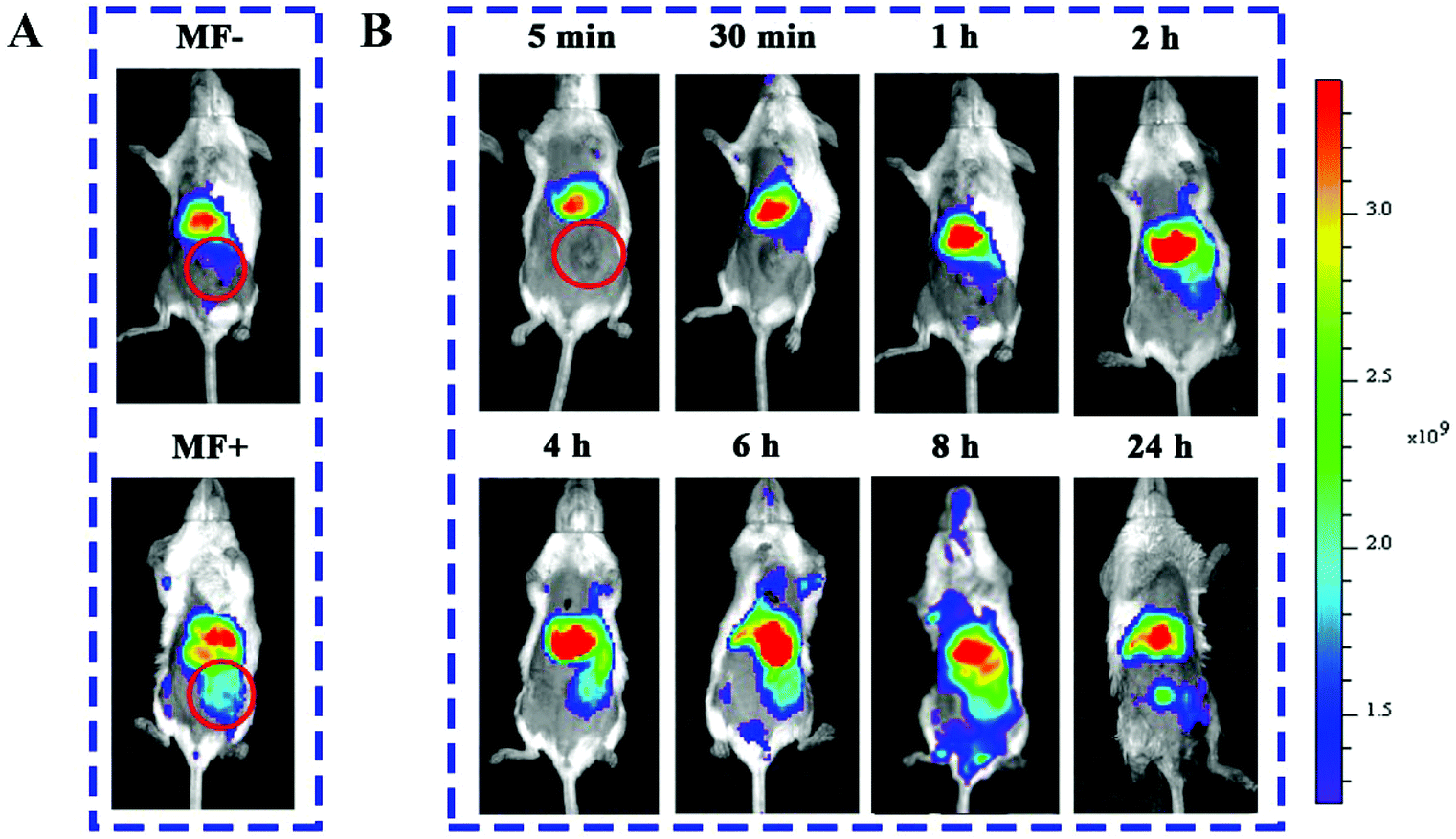 | ||
| Fig. 4 In vivo fluorescence images of tumor-bearing mice acquired after injecting the DiR-conjugated mPEG-HA/CSO-SS-Hex/SPION/DiR self-assembly. (A) Tumors not exposed (MF−) or exposed (MF+) to magnetic fields at 24 h. (B) The tumors exposed to magnetic fields at different times (the red circle indicates the area of a tumor position). | ||
3.5. Anti-tumor activity of mPEG-HA/CSO-SS-Hex/SPION/GA in vivo
A pharmacokinetic study was performed to evaluate the biocompatibility of the mPEG-HA/CSO-SS-Hex/SPION/GA complex self-assembly in vivo. The methodological experiment was evaluated at low, medium and high concentrations, and the results indicated that the method is accurate, precise and reliable for the measurement of GA in plasma. Fig. 5A showed good linearity of the calibration curve in the concentration range of 0.01 to 100 μg mL−1; the correlation coefficient (R2) was 0.9996, and the standard curve equation was calculated by A = 0.7406C + 0.0008. The pharmacokinetic parameters are demonstrated in Table S5.†Cmax was 4.19 ± 2.18 μg mL−1 GA in the complex self-assembly, which was higher than that in free GA. The t1/2 of 2.35 ± 0.11 h of GA from the complex self-assembly in plasma significantly increased by about 4 times compared to that of free GA. The AUC0–t, AUC 0–∞ and MRT values of the complex self-assembly group were all significantly higher than those of the free GA group. These results may be due to the maintenance of the complex self-assembly for a longer time within a pharmacologically effective range. In conclusion, the magnetic complex self-assembly system successfully improved the biocompatibility of GA.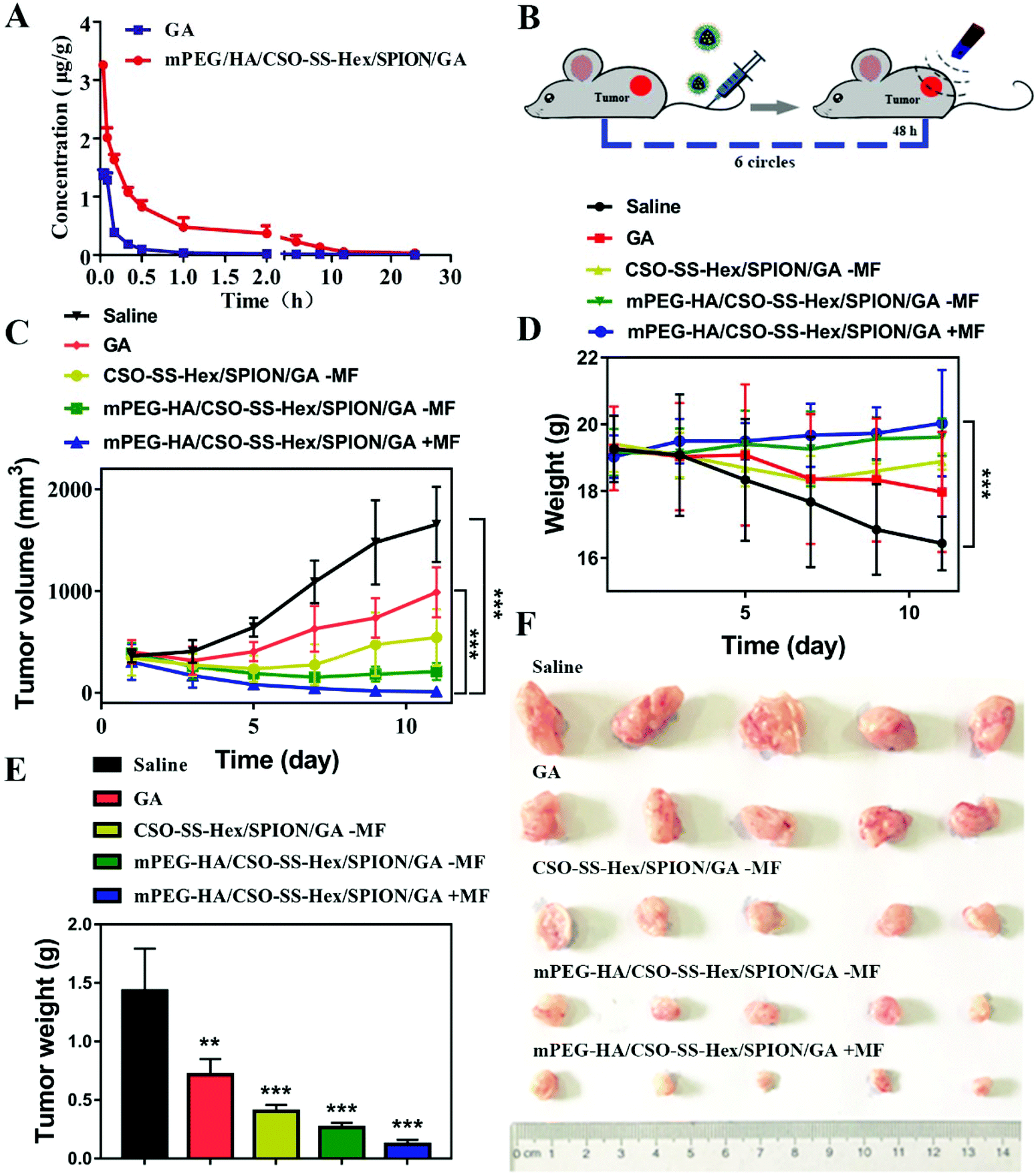 | ||
| Fig. 5 In vivo anti-tumor effects of the mPEG-HA/CSO-SS-Hex/SPION/GA complex self-assembly. (A) Mean blood concentration–time curves of the rats administrated GA and mPEG-HA/CSO-SS-Hex/SPION/GA self-assembles (n = 6). (B) Schematic of the administration. (C) Changes in tumor volume in 4T1 cell-bearing mice after treatment with different formulations for different times (n = 6). (D) Changes in body weight in 4T1-bearing mice (n = 6). (E) Tumor weights of 4T1-bearing mice treated with different self-assemblies for 13 days (n = 6). (F) Images of tumors harvested from all groups after 13 days of treatment (n = 6). | ||
After we studied the anticancer activities of mPEG-HA/CSO-SS-Hex/SPION/GA in vitro, the antitumor effects of the formulation in a 4T1 tumor model were evaluated. The TNBC tumor model was established by subcutaneous injection of 4T1 cells in BALB/c mice. The tumor-bearing mice were randomly divided into five groups (n = 5): saline, GA, CSO-SS-Hex/SPION/GA, mPEG-HA/CSO-SS-Hex/SPION/GA-MF, and mPEG-HA/CSO-SS-Hex/SPION/GA (6 mg kg−1 of GA), with an extra magnet attached to the tumor position. When the tumor volumes had grown to about 300 to 400 mm3, five groups of drugs were administered intravenously via tail vein to the mice on days 1, 3, 5, 7, 9 and 11, as shown in Fig. 4B. The tumors were harvested and weighed after 13 days of treatment.
The changes in tumor volume during the period of administration for the different groups are shown in Fig. 4C. The results show that the mPEG-HA/CSO-SS-Hex/SPION/GA group demonstrated the most obvious effect of inhibiting the growth of tumors compared to the other groups. The weights of the mice did not change significantly compared with the saline group (Fig. 4D). Fig. 4E and F show the significant antitumor effects of the mPEG-HA/CSO-SS-Hex/SPION/GA group compared to the other groups. The overall results prove that the mPEG-HA/CSO-SS-Hex/SPION/GA complex self-assembly has a good anti-tumor effect on TNBC.
3.6. Biosafety of mPEG-HA/CSO-SS-Hex/SPION/GA in vivo
In this study, the critical biomarkers BUN/CRE and ALT/AST of serum were measured to reflect renal and liver damage, respectively. Both the BUN/CRE and ALT/AST levels, as shown in Fig. 6A and B, showed that the GA group showed mild hepatorenal toxicity; however, the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly group was similar to the saline group, which indicates that the self-assembly has no significant renal or liver toxicity.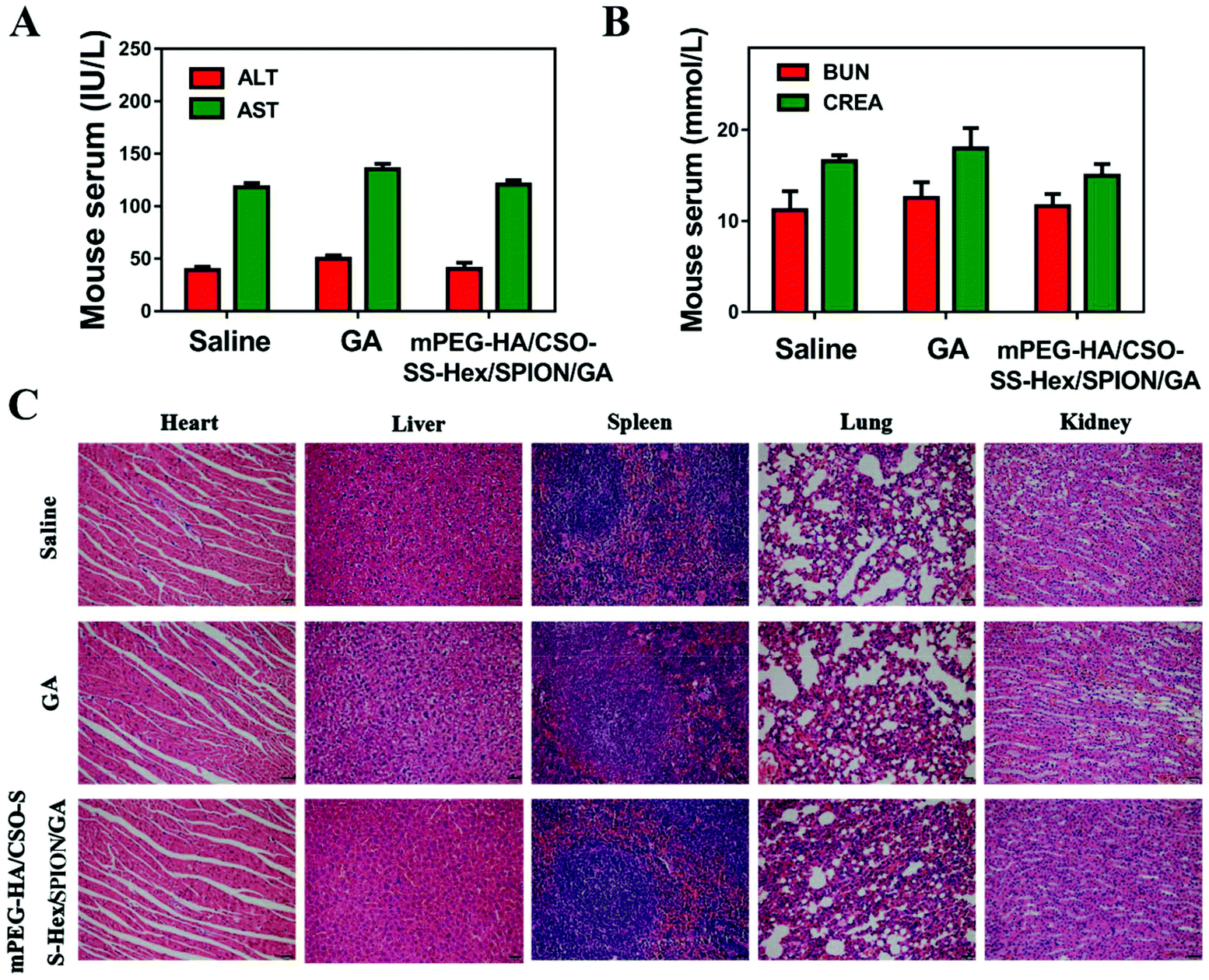 | ||
| Fig. 6 Biosafety of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly in vivo. (A) Serum levels of ALT and AST (liver function) at 48 h after the last treatment. (B) Serum levels of BUN and CRE (renal function) at 48 h after the last treatment. (C) H&E staining of female rat organs (hearts, livers, spleens, lungs, and kidneys) at the end of the experiments. | ||
H&E staining (Fig. 6C) was further used to investigate the potential toxicity of mPEG-HA/CSO-SS-Hex/SPION/GA in five organs, namely the heart, liver, spleen, lungs and kidneys. After treatment, the liver and kidney organs of the GA group showed slightly increased intercellular spaces; however, there was no obvious change in the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly compared to the saline group. The overall results confirmed that the caudal vein administration of the mPEG-HA/CSO-SS-Hex/SPION/GA self-assembly could decrease the toxicity of GA and showed good biocompatibility and tolerance in vivo.
4. Conclusions
In this study, we designed a double tumor targeting redox-responsive magnetic polymeric self-assembly (mPEG-HA/CSO-SS-Hex/SPION/GA) as a drug-carrying system for delivery of the natural compound GA; it showed a remarkable curative effect on TNBC. This complex self-assembly improved the GA uptake of TNBC cells in vitro and in vivo to produce better antitumor effects. The magnetic complex polymeric self-assembly quickly dis-assembled and subsequently released GA in the presence of GSH. Greater delivery efficiency was achieved with the magnetism-EPR-induced and HA-receptor-mediated endocytosis. Once the self-assembly was taken up into tumor cells, it could be rapidly decomposed, thus improving the drug release and anti-tumor efficacy. In conclusion, the in vivo and in vitro experiment results suggest that the tumor magnetic and CD44 receptor dual-targeting mPEG-HA/CSO-SS-Hex/SPION/GA redox-responsive polymeric self-assembly with excellent multi-functionality and bioavailability is an effective strategy for TNBC chemotherapy.Ethical statement
In this study, all animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of China Pharmaceutical University and approved by the Animal Ethics Committee of China Pharmaceutical University.Conflicts of interest
All authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81801819) and the “Double First-Class” University project (No. CPU2018GY34). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of China Pharmaceutical University and approved by the Animal Ethics Committee of China Pharmaceutical University.References
- C. M. Dawidczyk, C. Kim, J. H. Park, L. M. Russell, K. H. Lee, M. G. Pomper and P. C. Searson, J. Controlled Release, 2014, 187, 133–144 CrossRef CAS.
- Y. H. Choi and H.-K. Han, J. Pharm. Invest., 2018, 48, 43–60 CrossRef CAS.
- S. Yang, F. Zhu, Q. Wang, F. Liang, X. Qu, Z. Gan and Z. Yang, J. Mater. Chem. B, 2015, 3, 4043–4051 RSC.
- N. M. Dand, P. B. Patel, A. P. Ayre and V. J. Kadam, Chron. Young Sci., 2013, 4, 94 CrossRef.
- Q. Zhou, L. Zhang, T. Yang and H. Wu, Int. J. Nanomed., 2018, 13, 2921 CrossRef CAS.
- F. ud Din, W. Aman, I. Ullah, O. S. Qureshi, O. Mustapha, S. Shafique and A. Zeb, Int. J. Nanomed., 2017, 12, 7291 CrossRef CAS.
- T. Yang, W. Li, X. Duan, L. Zhu, L. Fan, Y. Qiao and H. Wu, PLoS One, 2016, 11, e0162607 CrossRef.
- J. H. Park, S. Lee, J.-H. Kim, K. Park, K. Kim and I. C. Kwon, Prog. Polym. Sci., 2008, 33, 113–137 CrossRef.
- A. Varela-Moreira, Y. Shi, M. H. Fens, T. Lammers, W. E. Hennink and R. M. Schiffelers, Mater. Chem. Front., 2017, 1, 1485–1501 RSC.
- X. X. Zhou, L. Jin, R. Q. Qi and T. Ma, R. Soc. Open Sci., 2018, 5, 171654 CrossRef PubMed.
- C. Hu, X. Cun, S. Ruan, R. Liu, W. Xiao, X. Yang, Y. Yang, C. Yang and H. Gao, Biomaterials, 2018, 168, 64–75 CrossRef CAS.
- R. Liu, W. Xiao, C. Hu, R. Xie and H. Gao, J. Controlled Release, 2018, 278, 127–139 CrossRef CAS.
- W. Yu, X. He, Z. Yang, X. Yang, W. Xiao, R. Liu, R. Xie, L. Qin and H. Gao, Biomaterials, 2019, 119309 CrossRef CAS.
- H. M. Coley, Cancer Treat. Rev., 2008, 34, 378 CrossRef CAS.
- A. M. Brewster, M. Chavez-Macgregor and P. Brown, Lancet Oncol., 2014, 15, 625–634 CrossRef.
- J. R. Palmer, E. Viscidi, M. A. Troester, C. C. Hong, P. Schedin, T. N. Bethea, E. V. Bandera, V. Borges, C. Mckinnon and C. A. Haiman, J. Natl. Cancer Inst., 2014, 106, dju237 CrossRef PubMed.
- K. Gelmon, R. Dent, J. R. Mackey, K. Laing, D. Mcleod and S. Verma, Ann. Oncol., 2012, 23, 2223 CrossRef CAS.
- L. A. Carey, E. C. Dees, L. Sawyer, L. Gatti, D. T. Moore, F. Collichio, D. W. Ollila, C. I. Sartor, M. L. Graham and C. M. Perou, Clin. Cancer Res., 2007, 13, 2329–2334 CrossRef CAS.
- N. C. Turner and J. S. Reis-Filho, Clin. Cancer Res., 2013, 19, 6380–6388 CrossRef CAS.
- H. Auterhoff, H. Frauendorf, W. Liesenklas and C. Schwandt, Arch. Pharm., 1962, 295/67, 833–846 CrossRef CAS.
- X. Wang, R. Deng, Y. Lu, Q. Xu, M. Yan, D. Ye and W. Chen, Basic Clin. Pharmacol. Toxicol., 2013, 112, 25–33 CrossRef CAS.
- R. Doddapaneni, K. Patel, I. H. Owaid and M. Singh, Drug Delivery, 2015, 23, 1–10 Search PubMed.
- Y. Yang, L. Yang, Q. D. You, F. F. Nie, H. Y. Gu, L. Zhao, X. T. Wang and Q. L. Guo, Cancer Lett., 2007, 256, 259 CrossRef CAS.
- Q. Qi, N. Lu, X. T. Wang, H. Gu, Y. Yang, W. Liu, C. Li, Q. You and Q. Guo, Biochem. Cell Biol., 2008, 86, 386 CrossRef CAS.
- H. Gu, S. Rao, J. Zhao, J. Wang, R. Mu, J. Rong, L. Tao, Q. Qi, Q. You and Q. Guo, J. Cancer Res. Clin. Oncol., 2009, 135, 1777–1782 CrossRef CAS.
- K. Hao, Acta Pharmacol. Sin., 2006, 27, 1253–1258 CrossRef.
- M. Sang, Z. Zhang, F. Liu, L. Hu, L. Li, L. Chen, F. Feng, W. Liu and W. Qu, J. Biomed. Nanotechnol., 2018, 14, 477–495 CrossRef CAS.
- A. K. Hauser, M. I. Mitov, E. F. Daley, R. C. Mcgarry, K. W. Anderson and J. Z. Hilt, Biomaterials, 2016, 105, 127–135 CrossRef CAS.
- Y. Kang, L. Lu, J. Lan, Y. Ding, J. Yang, Y. Zhang, Y. Zhao, T. Zhang and H. Rjy, Acta Biomater., 2018, 68, 137–153 CrossRef CAS.
- Y. W. Hu, Y. Z. Du, N. Liu, X. Liu, T. T. Meng, B. L. Cheng, J. B. He, J. You, H. Yuan and F. Q. Hu, J. Controlled Release, 2015, 206, 91–100 CrossRef CAS.
- Z. Yuan, L. Gui, J. Zheng, Y. Chen, S. Qu, Y. Shen, F. Wang, M. Er, Y. Gu and H. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 30994–31007 CrossRef CAS.
- L. Liu, H. Yi, H. He, H. Pan, L. Cai and Y. Ma, Biomaterials, 2017, 134, 166 CrossRef CAS.
- Z. Luo, C. Wang, H. Yi, P. Li, H. Pan, L. Liu, L. Cai and Y. Ma, Biomaterials, 2015, 38, 50–60 CrossRef CAS.
- S. Wang, M. Shao, Z. Zhong, A. Wang, J. Cao, Y. Lu, Y. Wang and J. Zhang, Drug Delivery, 2017, 24, 1791–1800 CrossRef CAS.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 265–273 Search PubMed.
- M. Zöller, Nat. Rev. Cancer, 2011, 11, 254–267 CrossRef.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS.
- J. Lee, T. Isobe and M. Senna, J. Colloid Interface Sci., 1996, 177, 490–494 CrossRef CAS.
- A. Dey, U. Koli, P. Dandekar and R. Jain, Polymer, 2016, 93, 44–52 CrossRef CAS.
- K. K. Sharma and E. Al, International conference on groundnut Aflatoxin management & Genomics, 2006, p. 88 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Zeta potential, drug loading capacity and encapsulation efficiency, cell viabilities, cell uptake, 1H NMR spectra, flow cytometry, organ fluorescence distribution images. See DOI: 10.1039/c9bm01171d |
| This journal is © The Royal Society of Chemistry 2020 |