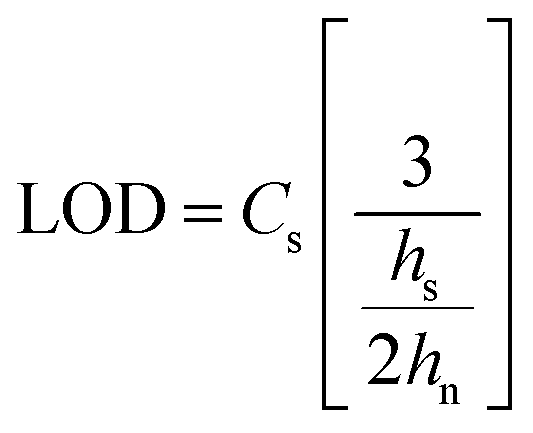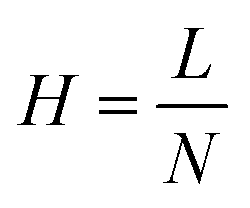DOI:
10.1039/C9AY02305D
(Paper)
Anal. Methods, 2020,
12, 85-90
HPLC detection of organic gunshot residues collected with silicone wristbands
Received
25th October 2019
, Accepted 25th November 2019
First published on 26th November 2019
Abstract
Gunshot Residue (GSR) is a common subject in criminal analysis due to the frequency of firearm-related crimes. There have been multiple studies aimed at determining the fastest and most efficient techniques to collect and detect GSR. Scanning Electron Microscopy remains a popular technique in the analysis of key Inorganic Gunshot Residues (IGSR) such as barium, lead, and antimony. However, introduction of heavy-metal-free ammunition creates the need for a different approach. This research focuses on developing a robust HPLC procedure for separation and detection of five Organic Gunshot Residues (OGSR), dimethylphthalate (DMP), diphenylamine (DPA), 2-nitrodiphenylamine (2-NDPA), 4-nitrodiphenylamine (4-NDPA), and ethyl centralite (EC) collected through passive sampling with silicone wristbands. By calculating figures of merit including resolution, retention factor, selectivity factor, theoretical plate number and height, and limit of detection for different HPLC conditions, it was found that water/acetonitrile (52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48) is the most efficient mobile phase using a Poroshell 120 EC-C18 column. The HPLC data showed that silicone wristbands are capable of adsorbing the selected OGSR compounds in laboratory settings. Compounds appeared to adsorb better individually than as a mix. This paper details the careful method development for the analysis of OGSR extracted from silicone bracelet. It provides researchers with the foundation required to move forward with real-world forensic samples by validating the important process of sample collection, extraction, and analysis.
:48) is the most efficient mobile phase using a Poroshell 120 EC-C18 column. The HPLC data showed that silicone wristbands are capable of adsorbing the selected OGSR compounds in laboratory settings. Compounds appeared to adsorb better individually than as a mix. This paper details the careful method development for the analysis of OGSR extracted from silicone bracelet. It provides researchers with the foundation required to move forward with real-world forensic samples by validating the important process of sample collection, extraction, and analysis.
Introduction
Gunshot Residue (GSR) is frequently analysed after firearm-related-crimes to aid criminal investigations. The reliability and specificity of the method used to detect GSR is a crucial factor that needs to be considered before presenting evidence in court.1 Several recent studies have been aimed at determining the fastest and most efficient techniques to detect Inorganic Gunshot Residue (IGSR).2–12 For example, Fedick and Bain focused their research on developing an in situ procedure for collecting GSR from the hands of shooters and other surfaces using swab touch spray mass spectroscopy.13 Ilker also chose shooters' skin as the target for collecting GSR using Scanning Electron Microscopy (SEM).10 In fact, SEM combined with energy dispersion X-ray analysis has been a widely used technique in identifying IGSR.10–12 The introduction of heavy metal-free ammunition eliminates the possibility of studying the key IGSR elements, such as lead and antimony.2,3,14 Moreover, the identification of IGSR particles from lead-free and heavy-metal free ammunition appears to be problematic due to the similarity of these particles to the ones from unrelated sources,15 such as fireworks and vehicle brake linings.16 Such issues create the need for an additional technique in order to support and strengthen the found results. A complementary organic analysis along with the metallic analysis would ensure the identity of the GSR.17 Although, early research of volatile GSR compounds was directed towards health and environmental issues,18 it has rapidly extended to forensic analysis as well. Organic Gunshot Residue (OGSR) has been analysed using a wide range of analytical techniques, such as liquid chromatography, gas chromatography, micellar electrokinetic capillary electrophoresis, time-of-flight secondary ion mass spectrometry and others.15 Liquid chromatography appears to be a better separation technique than gas chromatography due to the lack of thermal stability of the explosives.1 In addition, there is a variety of collection techniques for deposited GSR, like tape lifting, swabbing, vacuum lifting, glue lifting and film lifting.19 However, there does not appear to be adequate research on the use of passive sampling devices (PSDs) to collect OGSR. PSDs would not require further collection methods of OGSR that could potentially result in sample loss. Active sampling techniques such as swabbing can result in unacceptable losses of analyte.20 Passive sampling devices such as silicone wristbands may provide improved sample recovery. They have been used for personal monitoring, such as water vapour, sulphur dioxide, and organic contaminants due to silicone's known ability to adsorb a wide range of compounds from volatile benzene to more hydrophobic compounds containing hydroxyl or carboxyl groups. O'Connell et al. report the qualitative identification of 47 compounds including PAHs, consumer products, pesticides, phthalates and industrial compounds.21 By being worn on the wrist under normal daily activity, silicone wristbands adsorb chemicals from any media they contact.22 Due to the fact that many OGSR compounds can be found in environmental and occupational materials, it is important to choose characteristic OGSR materials in order for them to hold forensic relevance.23 The OGSR compounds should meet three criteria: they should have a known origin, have a strong association with the ammunition, and have a limited occupational and environmental prevalence.23 Compounds that have been found to have a strong association with OGSR include ethyl centralite, methyl centralite, nitroglycerin, nitroguanidine, 2,4-dinitrotoluene, akardite II, 2-nitrodiphenylamine, 4-nitrodiphenylamine and diphenylamine.23 Diphenylamine, specifically, has proven to be a successful target due to its uniqueness as a stabilizer in smokeless gunpowders.1 High performance liquid chromatography (HPLC) for identification of DMP also appears to be rapid and reliable.17 The target OGSR compounds in this research are ethyl centralite (EC), dimethyl phthalate (DMP), diphenylamine (DPA), 2-nitrodiphenylamine (2-NDPA) and 4-nitrodiphenylamine (4-NDPA). The main goal of this research is to develop and validate methodology for the analysis of OGSR extracted from PSDs using HPLC as the analytical method.
Experimental
Standard preparation
DMP (99%), DPA (99%), 2-NDPA (98%), and EC (99%) were purchased from Fisher Scientific; 4-NDPA (99%) was purchased from Sigma-Aldrich. All laboratory glassware and tools were rinsed with (1:1) methanol/acetonitrile before use. Stock solutions of 1000 ppm for each of the five analytes (DMP, DPA, 2-NDPA, 4-NDPA, EC) were prepared using methanol/acetonitrile (1:1) as the solvent. A mix stock solution of 1000 ppm of all five analytes was also prepared. Diluted standards of the mix stock solution were prepared in concentrations of 10, 50, and 100 ppm by pipetting calculated amounts of the solution into appropriate volumetric flasks and diluting them with the prepared solvent. Once the dilutions were made, they were pipetted into labelled HPLC vials (10 ppm, 50 ppm, and 100 ppm) for analysis.
Wristbands and wristband cleaning
Wristbands were obtained from the corresponding author. These silicone wristbands were bright green in colour and offered as a free giveaway at a campus event. Prior to testing, foreign compounds on the wristbands that might interfere with the future analytes were removed using published cleaning procedures.21 Refer to Table 1 for a summary of the cleaning procedure. The silicone wristbands were cut into approximately one gram pieces. The procedure used 300 mL of mixed solvent for the bracelet pieces per exchange. A mixture of ethyl acetate/hexane (1:1) was used for the first three exchanges, and ethyl acetate/methanol (1:1) was used for the last two. Each exchange occurred for 30 minutes at 60 rotations per minute using a magnetic stir plate. After cleaning, the cleaned wristbands were dried in the oven at 70 °C (≤24 hours). Throughout the research, dried wristbands were stored in a glass beaker covered with a watch glass.
Table 1 Wristband cleaning procedure
| Exchange number |
Solvent |
Volume (mL) |
Rotations/minute |
Time (min) |
| 1 |
Ethyl acetate/hexane (1:1) |
300 |
60 |
30 |
| 2 |
Ethyl acetate/hexane (1:1) |
300 |
60 |
30 |
| 3 |
Ethyl acetate/hexane (1:1) |
300 |
60 |
30 |
| 4 |
Ethyl acetate/methanol (1:1) |
300 |
60 |
30 |
| 5 |
Ethyl acetate/methanol (1:1) |
300 |
60 |
30 |
Wristband samples and extraction
Three different sample adsorption protocols were developed for assessing bracelet extraction efficiencies: open vessel soaking, closed container soaking, and direct pipetting. These were chosen to test the feasibility of using the silicone as PSD for OGSR. Each of the sample types were completed in triplicate. For the open vessel soaking, individual bracelet pieces (∼1.0 g) were placed into six different 50 mL beakers. The bracelet piece was treated with exactly 20.00 mL of 1000 ppm solution (DMP, DPA, 2-NDPA, 4-NDPA, EC, or mix). The beakers were covered with watch glasses and were left to soak for 7 days. For the closed container soaking, individual bracelet pieces were placed into six different 50 mL glass jars (Fisher). As with the open vessel, the bracelet piece was treated with exactly 20.00 mL of 1000 ppm solution (DMP, DPA, 2-NDPA, 4-NDPA, EC, or mix) was pipetted into each jar. The jars were covered with lids and left to soak for 7 days. The direct pipetting technique was only completed with DMP, because it is the only liquid compound of the five target OGSR. Three bracelet pieces were placed on three watch glasses. Aliquots of 10 mL, 50 mL, and 100 mL of DMP were pipetted directly onto each bracelet piece. The samples were covered with watch glasses and left to soak for 7 days.
After 7 days, all the bracelet pieces were placed into clean 50 mL beakers and dried in the oven for one hour at 100 °C. After cooling, 20 mL of acetonitrile/methanol (1:1) was added to each individual beaker for extraction. The samples were left to stir for one hour at 60 rotations per minute. After extraction, the solutions were quantitatively transferred to 25 mL volumetric flasks, diluted to the mark, and mixed. The final extraction solutions were stored in HPLC vials for future analysis.
Combustion simulation
Qualitative analysis of OGSR combustion simulations were performed to determine if residuals were detectable post-combustion or if OGSR was completely combusted to NOx, CO2 and H2O. Separate 1.0 g pellets of 2-NDPA, DPA, and EC were prepared using a press. Each pellet was placed into a Parr 101A Stainless Steel Bomb Calorimeter (Moline, IL) for combustion. Since DMP is a liquid, it could not be pressed into a pellet. In addition, 4-NDPA would not stay in a pressed pellet form. An ignition wire placed in contact with the sample to detonate and the bomb was sealed and pressurized to 15 atm oxygen. After ignition, it was allowed to cool, and the pressure valve was opened to release residual vapours. The stainless steel chamber was washed with methanol to collect the residuals for analysis. In addition to pure samples, a silicone wristband was included in the chamber with a pellet of DPA during detonation to test efficacy of the PSD. All samples obtained were analysed by HPLC.
HPLC analysis
The instrument used throughout this research was an Agilent 1220 Infinity Series LC. All solvents used for these analyses were HPLC-grade. Limit of detection (LOD) was obtained for all five OGSR prior to the extraction analysis. Standards of 1 ppm of the five OGSR were prepared using methanol/acetonitrile (1:1) as the solvent. These standards were analysed by UV-vis to determine the maximum wavelength of absorbance (Table 2). All standards, extractions, and combustion samples were analysed by the HPLC method listed in Table 3.
Table 2 HPLC diode array detector wavelengths
| Compound |
Wavelength (nm) |
| DMP |
230 |
| DPA |
280 |
| 2-NDPA |
260 |
| 4-NDPA |
395 |
| EC |
250 |
Table 3 HPLC parameters
| Column type |
Poroshell 120 EC-C18, 4.5 × 150 mm, 4 micron |
| Flow rate (mL min−1) |
1.5 |
| Mobile phase |
Water/acetonitrile (52/48) |
| Injection volume (μL) |
5 |
| Max. pressure (bar) |
600 |
| Stop time (min) |
11 |
| Temperature (°C)24 |
45 |
Results and discussion
Figures of merit
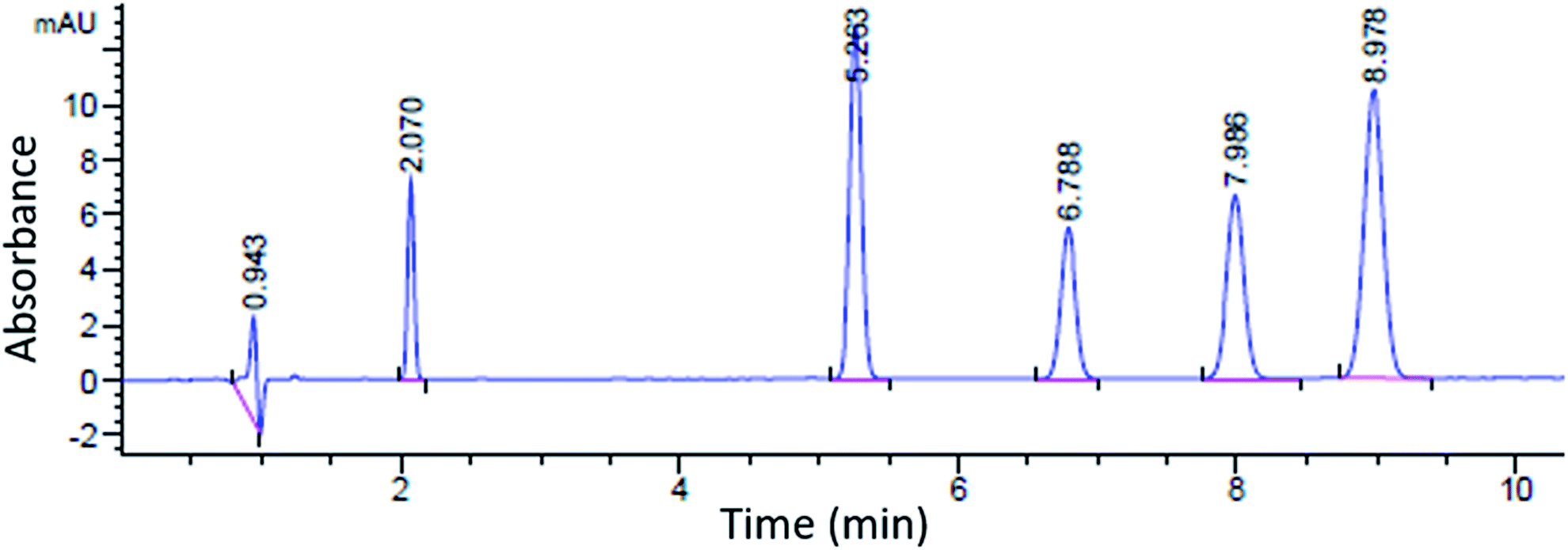
A sample chromatogram at 250 nm using the HPLC method above is presented in Fig. 1. Figures of merit including, Limit of Detection (LOD), retention factor, resolution, selectivity, theoretical plate number, and plate height were calculated and are presented in Tables 4 and 5. All figures of merit were calculated from the 250 nm chromatograph, except LOD where the best wavelength for each compound (see Table 2) was used. Limit of detection was calculated using eqn (1), where Cs is analyte's concentration, hs is the peak height of the analyte and hn is the largest deviation of the noise signal measured over a span of 10 peak widths from the retention time of the analyte (see Fig. 2)25. The LODs ranged between 0.023 ppm and 0.142 ppm. In terms of absolute LOD (by mass), this correlates to a range of 0.12 ng to 0.71 ng per injection. These are comparable to LODs found by previous researchers using HPLC-DAD. Specifically, Lopez-Lopez et al. determined absolute LODs of 0.5, 4.5, and 5 ng for DPA, 2-NDPA, and 4-NDPA respectively.24| | 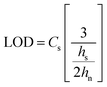 | (1) |
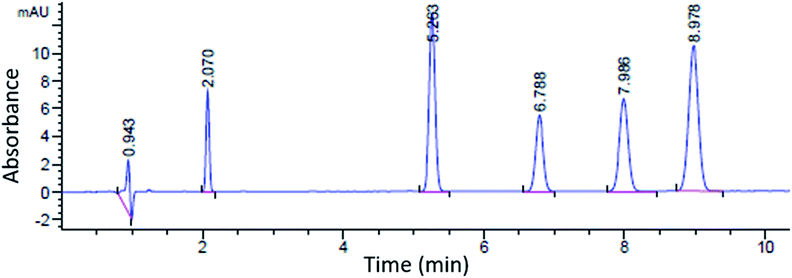 |
| | Fig. 1 Sample chromatogram at 250 nm; left to right: injection peak, DMP, 4-NDPA, DPA, EC, 2-NDPA. | |
Table 4 Limit of detection, retention time, and retention factor
| Compound |
LOD (ppm) |
Absolute LOD (ng) |
Retention time (min) |
Retention factor (k) |
| DMP |
0.023 |
0.12 |
2.07 |
1.20 |
| DPA |
0.026 |
0.13 |
6.79 |
4.58 |
| 2-NDPA |
0.082 |
0.41 |
8.98 |
6.20 |
| 4-NDPA |
0.036 |
0.18 |
5.26 |
7.47 |
| EC |
0.142 |
0.71 |
7.99 |
8.52 |
Table 5 Resolution and selectivity
| Compounds |
Resolution (Rs) |
Selectivity (α) |
| DMP & 4-NDPA |
44.7 |
3.82 |
| 4-NDPA & DPA |
14.7 |
1.35 |
| DPA & EC |
9.619 |
1.28 |
| EC & 2-NDPA |
6.944 |
1.14 |
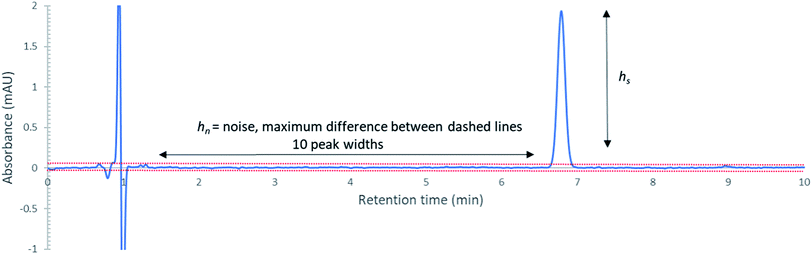 |
| | Fig. 2 Chromatogram of 1 ppm DPA at 280 nm for LOD determination. The first peak is the injection. | |
Retention factor (k) is used to compare the migration rates of different analytes. The preferable value for k is between 1 and 10, which results in better resolution and shorter run times. This was calculated using eqn (2), where tr is the retention time of the analyte and t0 is the hold-up time (the time it takes for the mobile phase to reach the detector).25,26 All k values of the studied analytes were found to be between 1 and 10.
| | 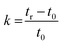 | (2) |
Resolution (Rs) is the column's ability to resolve two analytes into two different peaks. Achieving suitable resolution between analytes is the main objective in HPLC optimization. A resolution of 1.5 is typically sufficient for the baseline separation of adjacent peaks. Resolution was calculated using eqn (3) where tr is the retention time and w is the width at the base of the peak.25,26 All peaks reached resolution above 1.5, indicating sufficient separation.
| | 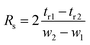 | (3) |
| | 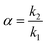 | (4) |
Selectivity (α) is the column's ability to discriminate between two different analytes. It is dependent on the chemistry between the analyte and the stationary phase. If the average number of theoretical plates is known, the selectivity necessary to get a resolution of 1.5 can be calculated. Selectivity is defined as the ratio of corresponding retention factors and was determined using eqn (4).25,26
Finally, efficiency is described as the degree of band broadening, which can be explained by the plate theory. It assumes that the column is divided into hypothetical plates and an analyte spends a certain time in each plate, which is sufficient to achieve equilibrium. Each plate has a finite height; therefore, the smaller the height, the greater the number of plates, and the better the separation. The number of theoretical plates (N) in the column and the plate height (H), where L is the length of the column, were calculate using eqn (5) and (6).25,26 The average N and H were calculated to be 17155 and 8.74 μm, respectively.
| | 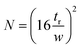 | (5) |
| | 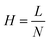 | (6) |
Extractions
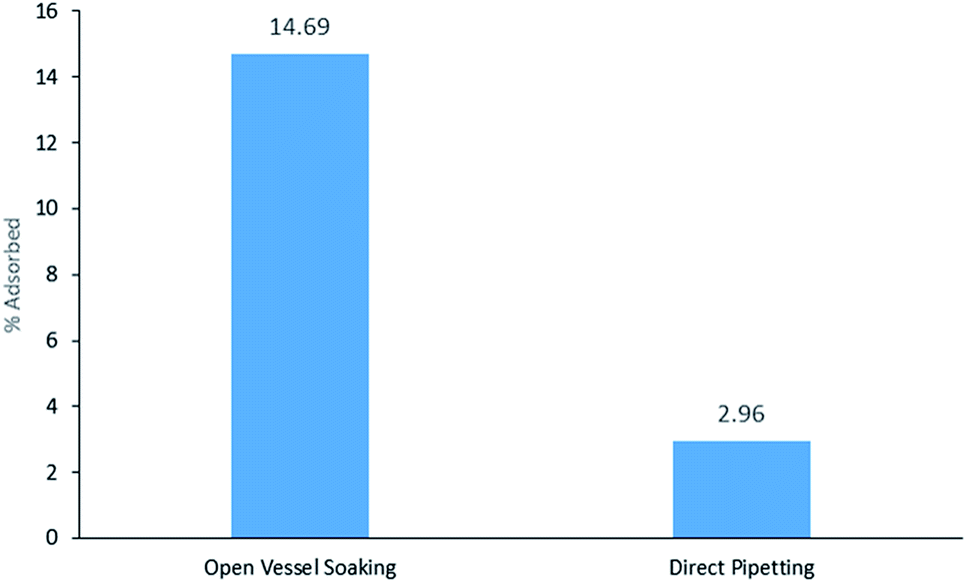
DMP was the only liquid sample and therefore, the three different extractions from the three DMP samples were compared: open vessel soaking, closed container soaking, and direct pipetting at three different volumes: 10, 50 and 100 μL. DMP was only detected above the LOD in the 100 μL direct pipetting sample. DMP was not detected from the closed container soaking on HPLC above the limit of detection. In Fig. 3, it can be seen that the open vessel soaking yields higher adsorption (14.69%) than direct pipetting (2.96%). The bracelet surface area that is exposed to sample is much higher in the open vessel soaking and this may account for the higher adsorption.
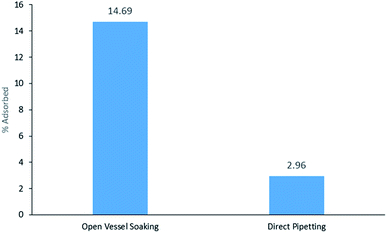 |
| | Fig. 3 % DMP adsorbed from 7 day open vessel soaking vs. direct pipetting (100 μL). | |
In order to compare how well silicone adsorbs each compound, the bracelet pieces were soaked in 1000 ppm solutions of individual compounds as well as the mix of all five analytes in both closed and open vessel soaking situations. The closed vessel samples were not detected above the LOD. The results obtained from open vessel samples are presented in Table 6. To fit the calibration curve, all solutions were diluted, which is a possible explanation to why individual 4-NDPA and EC, and DMP from the mix were not detected above the LODs. However, when comparing individual solutions of DPA and 2-NDPA to the ones from the mix, it can be seen that percentage adsorbed is higher in the individual solutions, than in a mix. However, the obtained results show that, although, they do not adsorb as well as they do individually, these compounds could still be detected using this technique. This research will be expanded in the future to apply this method to real forensic samples.
Table 6 Open vessel extractions
| Compound |
% individual analyte adsorbed |
% analyte adsorbed in mix |
|
Not detected above LOD.
|
| DMP |
14.69 ± 0.04 |
nda |
| DPA |
22.42 ± 0.02 |
13.23 ± 0.04 |
| 2-NDPA |
19.82 ± 0.05 |
16.85 ± 0.07 |
| 4-NDPA |
nda |
11.04 ± 0.04 |
| EC |
nda |
8.01 ± 0.02 |
Combustion simulations
The combustion residues were analysed by HPLC to mimic conditions in a firearm discharge. 2-NDPA was not detectable post combustion, but both DPA and EC were detected, suggesting incomplete combustion in the reaction vessel. More residues need to be tested in this format, but these results are encouraging. In addition to qualitative analysis of OGSR, this supports moving forward with samples from firearm discharge of commercially available ammunition by volunteer shooters, requiring human subject research application through the institutional review boards at both collaborating universities.
Conclusions
As a result of this research, silicone wristband personal sampling devices have shown promising results in proof-of-concept simulations. In addition, a suitable HPLC procedure for identification and separation of five target OGSR compounds (DMP, DPA, 2-NDPA, 4-NDPA, and EC) was developed with figures of merit comparable to previously published forensic research were achieved.24 It was found that these compounds adsorb better individually than as a mix. This may be due to adsorption affinity and will need to be tested further. In a real-life scenario, these analytes would be found as a mix. So far, only one extraction protocol has been used throughout this study. It is based on previous research using silicone wristbands as PSDs.21 In the future, other extraction methods, including sonication and varying the solvent type, could be studied. The direct pipetting method could be extended to the rest of the compounds; however, it would require solutions, rather than pure samples, as DMP is the only OGSR in the study that is a liquid at room temperature. The combustion simulation qualitatively confirmed the presence of OGSR post-detonation. It should be applied to quantitative analysis of multi-component OGSR mixtures to simulate commercially available ammunition. This study provides the needed baseline for future research involving the analysis of OGSR extracted from ammunition as well as OGSR extracted from silicone wristbands worn by actual shooters.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to thank the UTC Grote Foundation, the Westbrook Scholarship, the UTC Office for Undergraduate Research Search Award, and the UTC Chemistry and Physics Summer Undergraduate Research Program for supporting this research.
References
- D. B. Dahl, S. C. Slahck and P. F. Lott, Microchem. J., 1985, 31, 145–160 CrossRef CAS.
- L. A. Fambro, D. D. Vandenbos, M. B. Rosenberg and C. R. Dockery, Appl. Spectrosc., 2017, 71, 699–708 CrossRef CAS.
- L. A. Fambro, E. T. Miller, D. D. Vandenbos and C. R. Dockery, Anal. Methods, 2016, 8, 3132–3139 RSC.
- C. R. Dockery, M. B. Rosenberg, K. Kammerdiener, L. E. McAdams, N. A. Brutto, J. Turner, M. H. Chowdhury and M. K. Kiambuthi, J. Undergrad. Chem. Res., 2011, 10, 107–110 CAS.
- C. R. Dockery, J. Turner, M. B. Rosenberg, K. Kammerdiener and S. W. Mungai, Spectrosc. Lett., 2010, 43, 534–538 CrossRef CAS.
- M. B. Rosenberg and C. R. Dockery, Appl. Spectrosc., 2008, 62, 1238–1241 CrossRef CAS.
- C. R. Dockery and S. R. Groode, Appl. Opt., 2003, 42, 6153–6158 CrossRef CAS.
- S. R. Groode, C. R. Dockery, M. F. Bachmever, A. A. Niewland and S. L. Morgan, Trends Opt. Photonics, 2002, 81, 175–177 Search PubMed.
- P. W. Fedick and R. M. Bain, Forensic Chem., 2017, 5, 53–57 CrossRef CAS.
- I. Kara, Microsc. Res. Tech., 2017, 80, 1310–1314 CrossRef CAS.
- M. R. Rijnders, A. Stamouli and A. Bolck, J. Forensic Sci., 2010, 55, 616–623 CrossRef.
- F. Saverio Romolo and P. Margot, Forensic Sci. Int., 2001, 119, 195–211 CrossRef CAS.
- P. Fedick and R. Bain, Forensic Chem., 2017, 5, 53–57 CrossRef CAS.
- R. V. Taudte, C. Roux and A. Beavis, Forensic Sci. Int., 2017, 270, 55–60 CrossRef CAS.
- R. V. Taudte, C. Roux, D. Bishop, L. Blanes, P. Doble and A. Beavis, Anal. Methods, 2015, 7, 7447–7454 RSC.
- O. Dalby and J. W. Birkett, J. Chromatogr. A, 2010, 1217, 7183–7188 CrossRef CAS.
- L. S. Leggett and P. F. Lott, Microchem. J., 1989, 39, 76–85 CrossRef CAS.
- M. Gallidabino, F. S. Romolo and C. Weyermann, Anal. Bioanal. Chem., 2015, 407, 7123–7134 CrossRef CAS.
- E. Goudsmits, G. P. Sharples and J. W. Birkett, TrAC, Trends Anal. Chem., 2015, 74, 46–57 CrossRef CAS.
-
F. Tagliaro, Z. Deyl and I. Miksik, in HPLC in Enzymatic Analysis, ed. E. F. Rossomando, J. Wiley & Sons, Inc., New York, 1998, ch. 8, pp. 164–206 Search PubMed.
- S. G. O'Connell, L. D. Kincl and K. A. Anderson, Environ. Sci. Technol., 2014, 48, 3327–3335 CrossRef.
- A. J. Bergmann, P. E. North, L. Vasquez, H. Bello, M. d. C. G. Ruiz and K. A. Anderson, J. Exposure Sci. Environ. Epidemiol., 2017, 27, 560–568 CrossRef CAS.
- E. Goudsmits, G. P. Sharples and J. W. Birkett, Sci. Justice, 2016, 56, 421–425 CrossRef.
- M. Lopez-Lopez, J. C. Bravo, C. Garcia-Ruiz and M. Torre, Talanta, 2013, 103, 214–220 CrossRef CAS.
-
Y. Kazakevich and R. LoBrutto, HPLC for Pharmaceutical Scientists, Wiley-Interscience, Hoboken, N.J., 2007 Search PubMed.
-
L. R. Snyder, J. J. Kirkland and J. W. Dolan, Introduction to Modern Liquid Chromatography, John Wiley & Sons, Inc., Hoboken, New Jersey, 3rd edn, 2010 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b and
Gretchen E.
Potts
b and
Gretchen E.
Potts