
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransistor properties of salen-type metal complexes†
Kyohei Koyama,
Kodai Iijima,
Dongho Yoo‡
 and
Takehiko Mori*
and
Takehiko Mori*
Department of Materials Science and Engineering, Tokyo Institute of Technology, Ookayama 2-12-1, Meguro-ku, 152-8552, Japan. E-mail: mori.t.ae@m.titech.ac.jp
First published on 11th August 2020
Abstract
Schiff base complexes derived from salicylaldehyde and ethylene-, propylene-, and trans-1,2-cyclohexane-diamines exhibit p-channel transistor properties. The Cu complexes are open-shell compounds, but the oxidation and the hole transport occur at the highest occupied molecular orbital, where the singly occupied molecular orbital (SOMO) does not participate in conduction. Although Ni complexes tend to show larger mobilities than Cu complexes owing to the molecular planarity, the presence of SOMO is not harmful to the transistor properties.
Introduction
Owing to the potential application to plastic electronics, a great deal of attention has been paid to organic semiconductors used in organic light-emitting diodes, transistors, and photovoltaics. Among organic semiconductors, metal complexes are promising materials but not sufficiently explored.1 Square planar dithiolene complexes coordinated by four sulfur atoms (M–S4) are strong electron acceptors with the lowest unoccupied molecular orbital (LUMO) level located around −4.2 eV, which show air-stable n-channel transistor properties.2 Metal complexes coordinated by four nitrogen atoms (M–N4) are exemplified by phthalocyanine,3 which is a well known p-channel material. Several other M–N4 systems show p-channel transistor properties.4 A metal complex derived from 1,2-phenylenediamine is a strong electron donor,5,6 whose highest occupied molecular orbital (HOMO) level is located at as high as −4.44 eV. However, owing to the small energy gaps, the phenylenediamine complexes exhibit ambipolar properties on an inert interface.6,7 These results demonstrate that metal complexes realize very strong electron acceptor and donor abilities which are never attainable by ordinary organic materials, and at the same time very small energy gaps. Recently, infinite-sheet complexes of M–S4 and M–N4 are attracting attention as analogues of graphene; these complexes exhibit high electrical conductivity,8 and even superconductivity.9Inspired by these findings, we have investigated M–N2O2 complexes with more ordinary redox properties.10 Schiff base complexes derived from salicylaldehyde and ethylene diamine are known as salen complexes (Scheme 1), which are moderate electron donors with the HOMO levels located around −5.3 eV. Electron and hole mobilities of 10−5 cm2 V−1 s−1 have been reported in the time-of-flight measurement of an alkylthiophene-substituted salen complex.11 High hole mobility of 1.5 cm2 V−1 s−1 has been reported in a thiophene-based Schiff base of 1,2-phenylenediamine.12 Hole mobility in the 10−7 cm2 V−1 s−1 order has been also reported in a Schiff base with tetrathiafulvalene moieties.13 In M–N2S2 complexes including thiosalen, we have recently found ambipolar transistor properties associated with energy gaps as small as 0.5–0.6 eV.14 However, transistor properties are not investigated in the most fundamental salen complexes. In particular, all these Ni complexes are closed shell compounds, but open-shell Cu compounds have not been examined as transistors.
 | ||
| Scheme 1 Structures of salen-type metal complexes. | ||
In the present work, we have investigated transistor properties of ethylenediamine (salen),15–21 propylenediamine (salpn),22 and trans-1,2-cyclohexanediamine (salcyh) complexes (Scheme 1).23–26 Changing metal atoms, we can compare transistor properties of open- and closed-shell materials. A copolymer of salpn complex and thiophene has been reported to show conductivity of 83 mS cm−1.27 We have also examined conducting properties of a homopolymer containing Schiff base (P1).
Results and discussion
Electronic properties
The salen, salpn and salcyh complexes were prepared according to literature.15–26,28 Since these complexes do not decompose up to high temperatures,18,21 the materials were purified by sublimation, and the thin films were formed by vacuum evaporation.The monomer ligand, 4,4′-dihydroxy-1,1′-biphenyl-3,3′-dicarboaldehyde, was prepared from 4,4′-dihydroxy-1,1′-biphenyl,29 which was successively reacted with octadecyl amine and metal to afford the metal polymer (P1).
Cyclic voltammograms of these complexes show oxidation waves around 0.3–0.6 V (Fig. 1(a) and (b)). These complexes are moderate electron donors whose HOMO levels are located around −5.2 to −5.6 eV (Table 1).11,26 The Cu and Co complexes are open shell materials,19,23 but the observed oxidation waves seem to come from the HOMO (Fig. 2). The calculated singly occupied molecular orbital (SOMO) levels are located much above the HOMO (Fig. 2). Since these complexes are strongly dimerized in the crystals, we have carried out the calculations based on the dimers. The SOMO levels split to ±0.3 eV (Fig. S2†), but the resulting occupied levels (−3.8 to −4.2 eV) are still far from the observed EHOMO in Table 1. The Cu complexes show slightly large oxidation potentials and deep HOMO levels in comparison with other complexes (Table 1). This is in complete agreement with the calculated HOMO levels (Fig. 2). From these observations, the oxidation waves are attributed to the HOMO levels. Co(salen) exhibits another redox wave around 0 V (Fig. 1(a)), potentially related to the partially occupied HOMO level.32
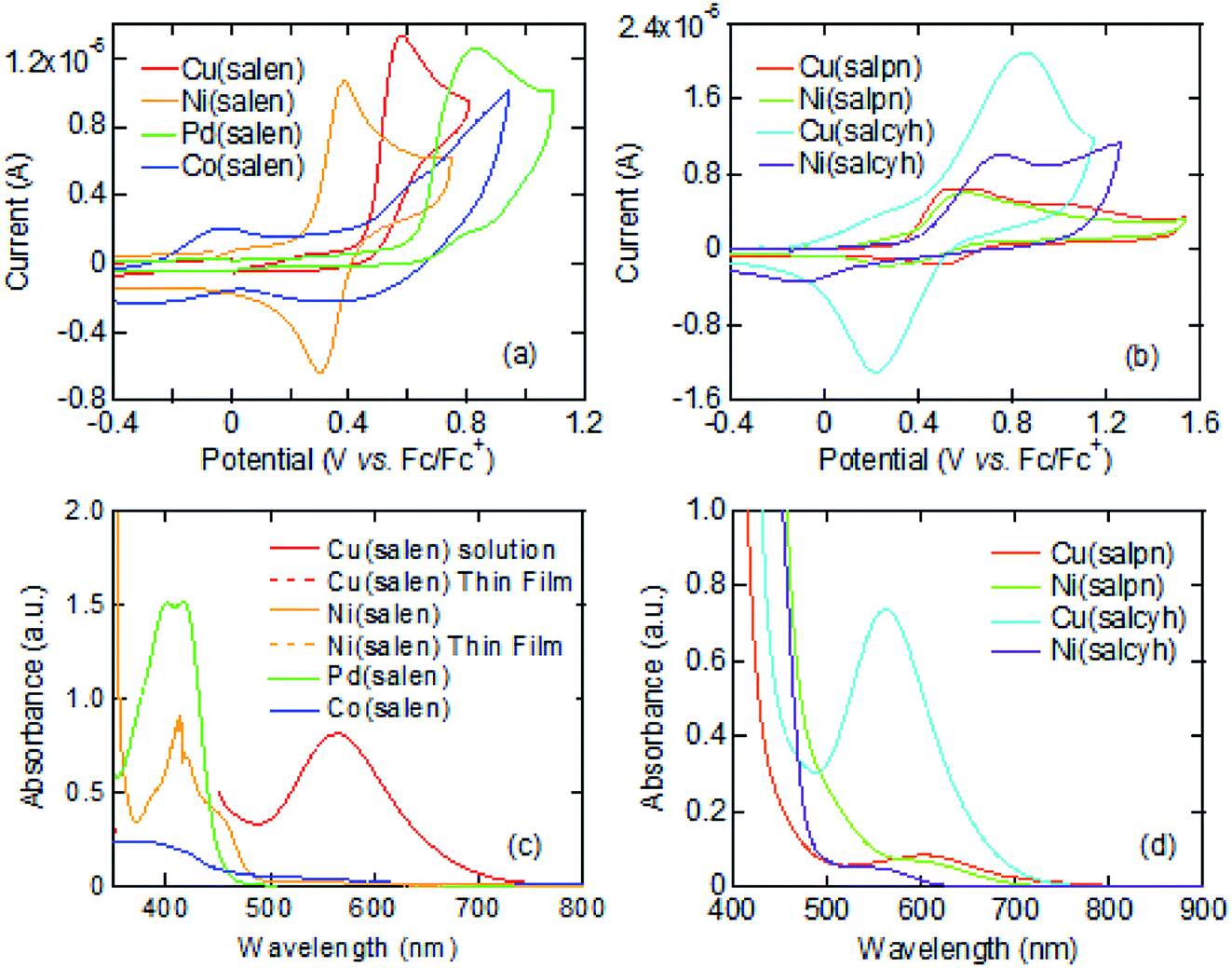 | ||
| Fig. 1 (a and b) Cyclic voltammograms, and (c and d) absorption spectra. | ||
| Complex | E11/2 (V) | EHOMOa (eV) | λedgeb (nm) | Optical gapb (eV) | ELUMOc (eV) |
|---|---|---|---|---|---|
| a The HOMO levels were estimated from the first oxidation potentials by assuming the reference energy level of ferrocene/ferrocenium to be 4.8 eV from the vacuum level.30 The values in the parentheses were calculated by using the ADF software with the B3LYP* functional and TZP basis set.31b The values in the parentheses were from the dd transitions.c The LUMO levels were obtained from the HOMO levels and the optical gaps. The values in the parentheses were related to the SOMO levels. | |||||
| Cu(salen) | 0.48 | −5.28 (−5.29) | 438 (676) | 2.83 (1.83) | −2.45 (−3.45) |
| Ni(salen) | 0.34 | −5.14 (−5.06) | 496 | 2.50 | −2.64 |
| Pd(salen) | 0.64 | −5.36 (−5.17) | 474 | 2.62 | −2.73 |
| Co(salen) | 0.47 | −5.27 (−5.13) | 470 | 2.64 | −2.63 |
| Cu(salpn) | 0.53 | −5.33 (−5.18) | 495 (824) | 2.50 (1.50) | −2.83 (−3.78) |
| Ni(salpn) | 0.44 | −5.24 (−4.97) | 510 | 2.40 | −2.84 |
| Cu(salcyh) | 0.53 | −5.33 (−5.43) | 480 (737) | 2.58 (1.68) | −2.75 (−3.57) |
| Ni(salcyh) | 0.23 | −5.03 (−4.95) | 490 | 2.53 | −2.53 |
| P1 | 0.57 | −5.13 | 482 | 2.57 | −2.56 |
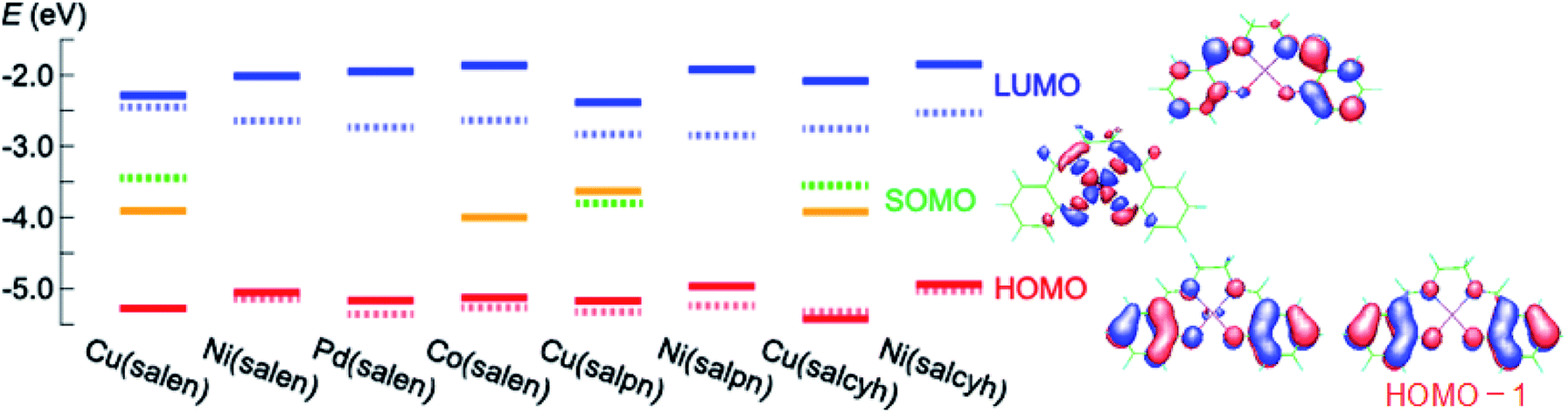 | ||
| Fig. 2 Calculated (solid) and experimentally determined (dashed) energy levels as well as the molecular orbitals. | ||
The absorption edges around 490 nm come from the ππ* transitions in the Ni and Pd complexes (Fig. 1(c)), indicating the HOMO–LUMO gaps of 2.5 eV (Table 1).16 This leads to the LUMO levels around −2.9 eV, which do not conflict with the calculated LUMO levels around −2.0 eV (Fig. 2). In addition, reduction waves are not observed, and electron conduction is unlikely.30 In analogy with the M–N2S2 complexes,14 this is partly because the two ligands are not conjugated.
The Cu complexes show an enhanced dd transition around 590 nm (Fig. 1(c) and (d)),26 but this is much reduced in the thin films probably due to the dimerization. This transition suggests the presence of additional levels around −3.5 to −3.7 eV coming from the d-like SOMO levels. These orbitals are σ-like for the ligands (Fig. 2), and the oxidation occurs on the HOMO rather than the SOMO. It is expected from the HOMO levels that these complexes show hole conduction.30
Transistor properties
These complexes were vacuum evaporated on a SiO2/Si substrate, and bottom-gate top-contact transistors were fabricated. These complexes show p-channel transistor properties as depicted in Fig. 3. From these characteristics, the transistor parameters are extracted as listed in Table 2. Cu(salen) shows maximum mobility of 4.3 × 10−3 cm2 V−1 s−1, but the Ni, Pd, and Co analogues show by more than one order lower mobilities.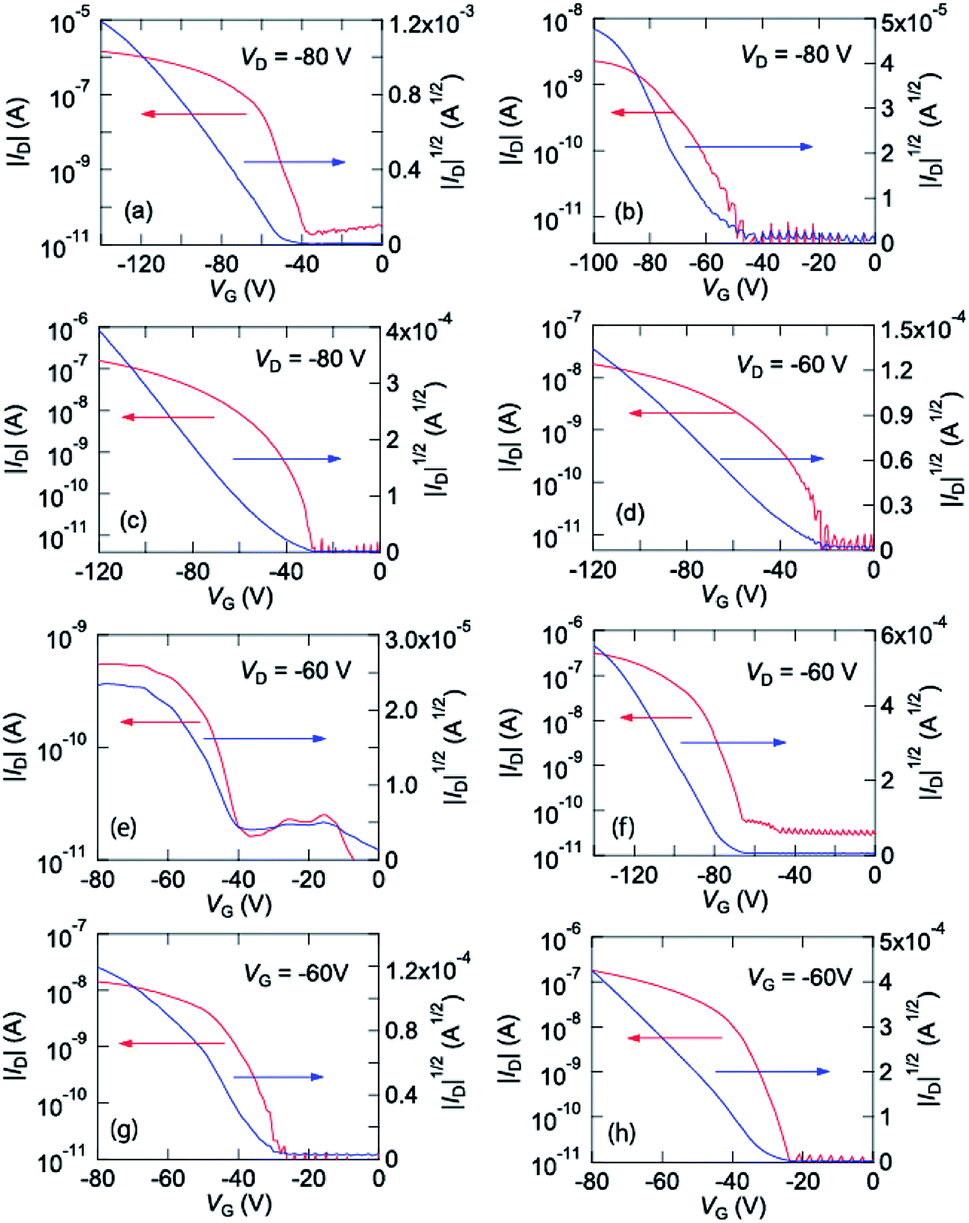 | ||
| Fig. 3 Transfer characteristics of (a) Cu(salen), (b) Ni(salen), (c) Pd(salen), (d) Co(salen), (e) Cu(salpn), (f) Ni(salpn), (g) Cu(salcyh), and (h) Ni(salcyh). | ||
| Complex | μmax (cm2 V−1 s−1) | μave (cm2 V−1 s−1) | Vth (V) | On/off ratio |
|---|---|---|---|---|
| Cu(salen) | 4.3 × 10−3 | 2.5 × 10−3 | −45 | 105 |
| Ni(salen) | 6.1 × 10−5 | 4.4 × 10−5 | −44 | 105 |
| Pd(salen) | 8.6 × 10−4 | 5.4 × 10−4 | −60 | 105 |
| Co(salen) | 6.0 × 10−5 | 4.3 × 10−5 | −32 | 105 |
| Cu(salpn) | 5.1 × 10−5 | 2.9 × 10−5 | −37 | 103 |
| Ni(salpn) | 1.7 × 10−3 | 1.0 × 10−3 | −83 | 105 |
| Cu(salcyh) | 2.6 × 10−4 | 1.3 × 10−4 | −36 | 103 |
| Ni(salcyh) | 2.0 × 10−3 | 1.4 × 10−3 | −28 | 104 |
Ni(salpn) and Ni(salcyn) exhibit comparable mobilities to Cu(salen), whereas Cu(salpn) and Cu(salcyn) show lower mobilities. In these two series, the closed-shell Ni complexes show higher mobilities than the open-shell Cu complexes.
Transfer integrals
Dimerized crystal structures have been reported in Cu(salen), Ni(salen), and Pd(salen) (Fig. 4),33–36 where each dimer is arranged in an alternately tilted manner. Cu(salpn) and Ni(salpn) make loosely packed stacking structures.22,37 Cu(salcyh) and Ni(salcyh) also have dimerized structures.25,38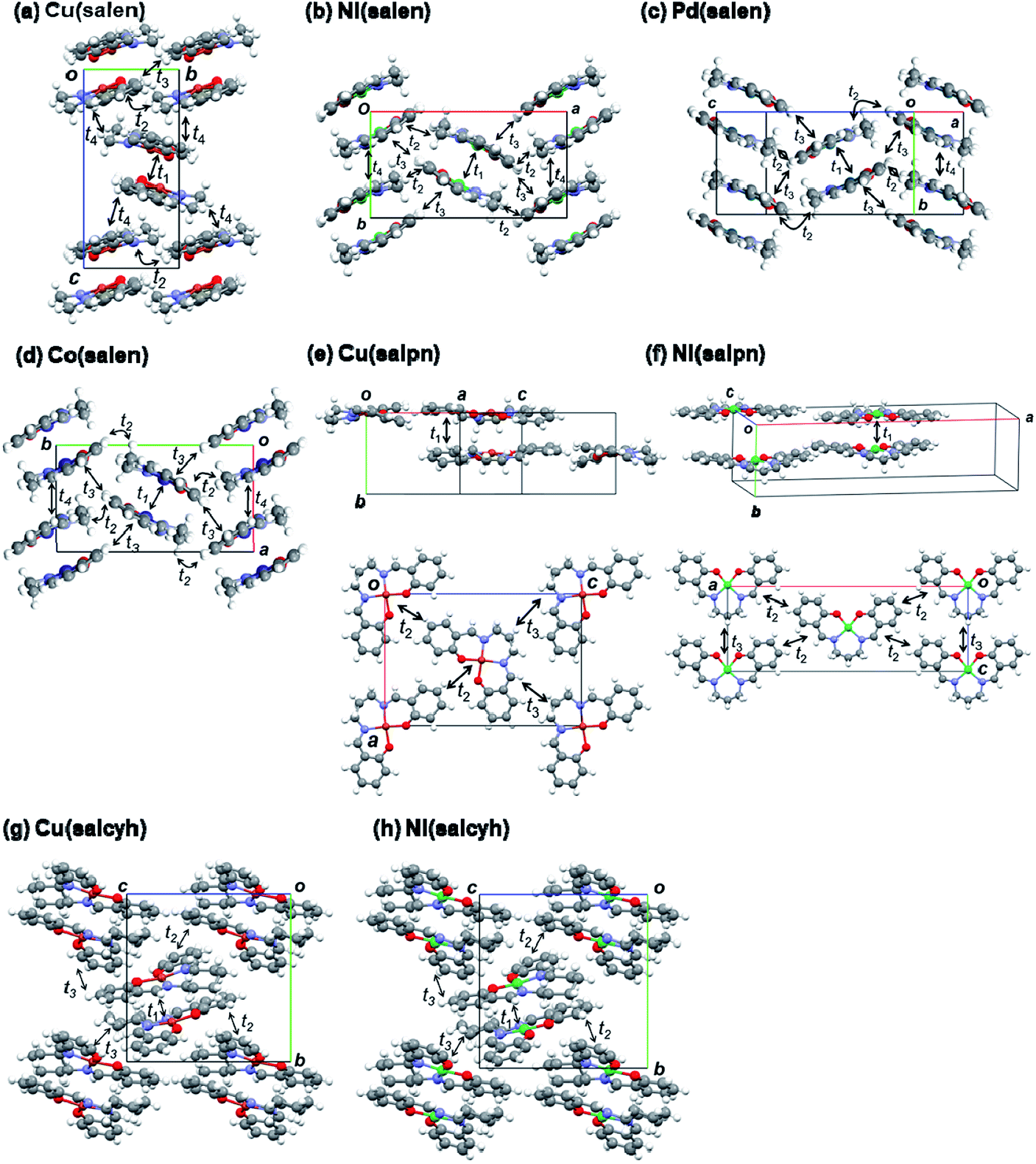 | ||
| Fig. 4 Crystal structures and transfer integrals of (a) Cu(salen),33 (b) Ni(salen),34 (c) Pd(salen),35 (d) Co(salen),36 (e) Cu(salpn),22,37 (f) Ni(salpn),37 (g) Cu(salcyh),25 and (h) Ni(salcyh).38 | ||
In order to interpret the observed significant difference of the transistor performance, transfer integrals of the HOMO are investigated.39,40 As shown in Fig. 4 and Table 3, the salen complexes form strongly dimerized herringbone structures, but the structures are not isostructural. Cu(salen) has a dihedral angle of ∼140° (Fig. 4(a)), and the unit cell is elongated along the “stacking” (c) axis.23 Parallel dimers are aligned along the horizontal (b) axis. Ni(salen) has a similar dihedral angle of ∼140° (Fig. 2(b)), but the cell is elongated along the transverse (a) axis.34 Parallel dimers are aligned along the vertical (b) axis. Pd(salen) (Fig. 4(c)) is similar to Ni(salen),35 though the latter has a double layer structure along the c axis.
| Complex | t1 | t2 | t3 | t4 | Angle (°) |
|---|---|---|---|---|---|
| Cu(salen) | 237 | −9 | 1 | 40 | 358.7 |
| Ni(salen) | 191 | −9 | 15 | 21 | 358.6 |
| Pd(salen) | 212 | −10 | −22 | −19 | 360.0 |
| Co(salen) | −34 | 3 | −27 | −87 | 360.0 |
| Cu(salpn) | −94 | −6 | −6 | 365.8 | |
| Ni(salpn) | −110 | 3 | −9 | 359.4 | |
| Cu(salcyh) | 189 | −13 | −4 | 361.7 | |
| Ni(salcyh) | 102 | −10 | −25 | 360.1 |
The intradimer transfer t1 is as large as 200 meV (Table 3), and the other interdimer transfers are nearly by one order of magnitude smaller than t1. 2t1 affords the splitting of the two energy bands, and t2–t4 give the bandwidth. In Cu(salen), t4 makes a two-dimensional network (Fig. 4(a)), while t2–t4 are one half in Ni(salen) and Pd(salen). This may be related to the comparatively large mobility of Cu(salen). Co(salen) has only a highly one-dimensional interaction. Although not strictly isostructural, Ni(salen), Pd(salen), and Co(salen) have very similar molecular packing, and the two-dimensional network of Cu(salen) seems to be more advantageous to the charge transport.
The salpn complexes have uniform stacking structures with t1 ∼ 100 meV (Fig. 4(d) and (e)).36 Although the molecular planarity is not very good, the interplanar distance is short (3.45 and 3.36 Å) and the adjacent molecule is located exactly on the top of another molecule. Therefore, the transfer is smaller than t1 in the salen complexes (Table 3). Strong dimerization is not inherent in the present metal complexes, and the uniform chain does not seem to be too disadvantageous to the conduction.
The salcyh complexes have strongly dimerized structures (Fig. 4(g) and (h)).25,38 Nonetheless, Ni(salcyh) has larger interdimer interaction t3 than Cu(salcyh), and this may be the origin of the difference of the mobility.
In order to investigate planarity of the complexes, sums of four bond angles around the metal atoms are compared in Table 3.22,33–38 When the complex is planar, the angle is 360°. The deviation is particularly large in Cu(salpn),22 related to the reduced transistor performance in comparison with Ni(salpn). Cu(salcyh) shows larger non-planarity than Ni(salcyh) as well. By contrast, non-planarity is not serious in Cu(salen) with a shorter alkyl diamine. The transistor performance seems to be closely related to the molecular planarity.
Thin-film properties
Evaporated films of the salen complexes show sharp X-ray diffraction (XRD) peaks together with the higher-order peaks (Fig. 5(a)). The d-values are about 13 Å (Table 4), which are in good agreement with half of the crystallographic a (Cu) and c (Ni, Pd, and Co) axes.33–36 We have observed several higher-order peaks. The molecules are standing perpendicular to the substrate keeping the crystallographic bc and ab planes parallel to the substrate.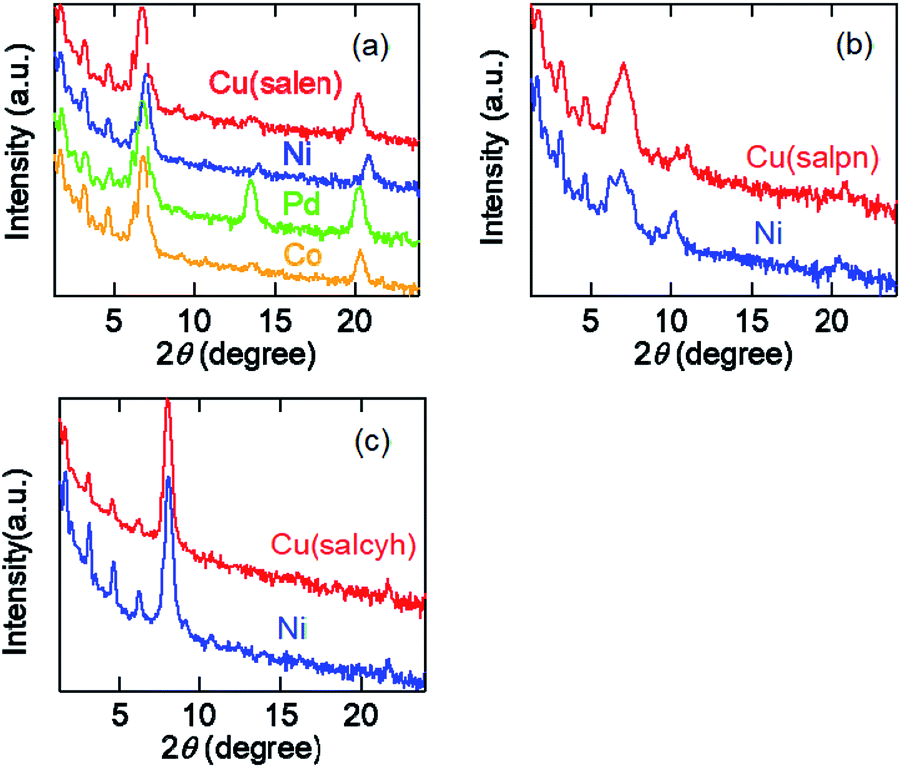 | ||
| Fig. 5 (a) XRD patterns of evaporated films of the salen complexes. (b) XRD patterns of evaporated films of the salpn complexes. (c) XRD patterns of evaporated films of the salcyh complexes. | ||
| Complex | 2θ (°) | d (Å) | Complex | 2θ (°) | d (Å) |
|---|---|---|---|---|---|
| Cu(salen) | 6.696 | 13.20 | Cu(salpn) | 6.979 | 12.67 |
| Ni(salen) | 6.956 | 12.71 | Ni(salpn) | 6.878 | 12.85 |
| Pd(salen) | 6.723 | 13.15 | Cu(salcyh) | 8.020 | 11.02 |
| Co(salen) | 6.761 | 13.07 | Ni(salcyh) | 8.062 | 10.97 |
The salpn complexes show XRD peaks at d = 12.7–12.8 Å (Fig. 3(b) and Table 4). These values correspond to the crystallographic b (Cu) and half of the a axes (Ni),37 and the molecules are standing perpendicular to the substrate again. However, the comparatively broad peaks indicate that the crystallinity is not as good as the salen complexes.
Cu(salcyn) and Ni(salcyn) show sharp XRD peaks around 11.0 Å (Fig. 3(c) and Table 4). These values correspond to the crystallographic a axes. These molecules are standing perpendicular to the substrate, where the bc plane is parallel to the substrate.
Atomic force microscopy (AFM) images are shown in Fig. 6. These compounds form crystalline films; exceptions are Pd(salen) and Co(salen) (Fig. S4†). Cu(salen) shows large plate-like crystals, whereas other complexes exhibit needle-like crystals. The plate-like crystals are related to the two-dimensional electronic structure and the comparatively high mobility. Ni(salen) and Cu(salcyh) have rough surface, and probably this is associated with the relatively low mobility.
 | ||
| Fig. 6 Atomic force microscopy (AFM) images of (a) Cu(salen), (b) Ni(salen), (c) Cu(salpn), (d) Ni(salpn), (e) Cu(salcyh) and (f) Ni(salcyh). | ||
Polymer
Since an electrochemically prepared copolymer of VO(salen) and thiophene has been reported to show conductivity of 83 mS cm−1,13 a homopolymer P1 is chemically prepared. In contrast to the electrochemical polymers, the pristine polymer is entirely undoped. Since P1 does not show transistor properties, P1 is exposed to iodine vapor, where the conductivity increases from 1.0 × 10−7 to 3.7 × 10−4 S cm−1 (Fig. 7). The increase is saturated after about 30 min. When the sample is taken out from the iodine vapor, the conductivity decreases slightly, but is maintained around 0.8 × 10−4 S cm−1 for a long time (Fig. S6†). These results demonstrate efficient chemical hole doping is possible in P1.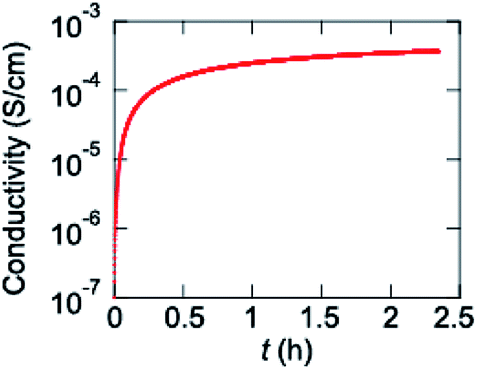 | ||
| Fig. 7 Conductivity of P1 upon iodine doping. | ||
Conclusions
We have demonstrated that salen-type metal complexes exhibit p-type transistor properties. Although Cu and Co complexes are open shell compounds,19,23 the oxidation occurs at the same HOMO as the other metal complexes, and the hole transport is mediated by the HOMO. The SOMO does not participate in the oxidation and conduction due to the d-like character. Owing to the difference of the molecular packing and thin-film morphology, Cu(salen) shows larger mobility than other salen complexes, whereas the Ni salpn and salcyh complexes show larger mobilities than the Cu complexes. The latter is due to the molecular planarity of the Ni complexes, but the former example proves the presence of SOMO is not harmful to the transistor properties.Although almost all organic semiconductors investigated so far are closed-shell materials,4 copper phthalocyanine (CuPc) is an open-shell compound.41 CuPc is an extensively investigated material,3 but the SOMO is localized on the metal atom as well,40 and the oxidation occurs at the HOMO.42 Mobilities larger than 1 cm2 V−1 s−1 have been reported in the single-crystal transistors,3e,f but the thin-film mobility is typically 0.02 cm2 V−1 s−1,3b which is not largely different from the present materials. CuPc sometimes shows ambipolar transistor properties, but this has been attributed to the narrow-gap nature.3f,7
In mixed-stack charge-transfer complexes of tetracyanoquino-dimethane (TCNQ),43 the donor HOMO does not participate in the hole conduction, but hybridization of the donor next HOMO with the TCNQ LUMO leads to the electron-only transport. Similar to the present metal complexes, this is another example in which the most “frontier” orbital does not participate in the conduction, and a lower orbital mediates the charge transport owing to the larger intermolecular orbital overlap.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the JSPS KAKENHI Grant Number 18H02044, and the Takahashi Industrial and Economic Research Foundation (08-003-022). The authors are grateful to Tokyo Institute of Technology Center for Advanced Materials Analysis for XRD measurement.References
- (a) T. Mori, Chem. Lett., 2011, 40, 428 CrossRef CAS; (b) T. Mori, J. Phys.: Condens. Matter, 2008, 20, 184010 CrossRef.
- (a) E. C. P. Smits, T. D. Anthopoulos, S. Setayesh, E. van Veenendaal, R. Coehoorn, P. W. M. Blom, B. de Boer and D. M. de Leeuw, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 205316 CrossRef; (b) T. D. Anthopoulus, S. Setayesh, E. Smits, M. Colle, E. Cantatore, B. Boer, P. W. M. Blom and D. M. de Leeuw, Adv. Mater., 2006, 18, 1900 CrossRef; (c) J. Cho, B. Domercq, S. C. Jones, J. Yu, X. Zhang, Z. An, M. Bishop, S. Barlow, S. R. Marder and B. Kippelen, J. Mater. Chem., 2006, 17, 2642 RSC; (d) T. Taguchi, H. Wada, T. Kambayashi, B. Noda, M. Goto, T. Mori, K. Ishikawa and H. Takezoe, Chem. Phys. Lett., 2006, 421, 395 CrossRef CAS; (e) H. Wada, T. Taguchi, B. Noda, T. Kambayashi, T. Mori, K. Ishikawa and H. Takezoe, Org. Electron., 2007, 8, 759 CrossRef CAS.
- (a) L. Li, Q. Tang, H. Li and W. Hu, J. Phys. Chem. B, 2008, 112, 10405 CrossRef CAS PubMed; (b) Z. Bao, A. J. Lovinger and J. Lovinger, Appl. Phys. Lett., 1996, 69, 3066 CrossRef CAS; (c) Z. Bao, A. J. Lovinger and J. Brown, J. Am. Chem. Soc., 1998, 120, 207 CrossRef CAS; (d) R. Zeis, T. Siegrist and C. Kloc, Appl. Phys. Lett., 2005, 86, 022103 CrossRef; (e) L. Li, Q. Tang, H. Li, X. Yang, W. Hu, Y. Song, Z. Shuai, W. Xu, Y. Li and D. Zhu, Adv. Mater., 2007, 19, 2613 CrossRef CAS; (f) R. W. I. de Boer, A. F. Stassen, M. F. Cracium, C. L. Mulder, A. Molinari, S. Rogge and A. F. Morpurgo, Appl. Phys. Lett., 2005, 86, 262109 CrossRef; (g) A. T. Vartanyan, Zh. Fiz. Khim., 1948, 22, 769 CAS.
- (a) A. M. Whyte, Y. Shuku, G. S. nichol, M. M. Matsushita, K. Awaga and N. Robertson, J. Mater. Chem., 2012, 22, 17967 RSC; (b) M. Hunziker, B. Hilti and G. Rihs, Helv. Chim. Acta, 1981, 64, 82 CrossRef CAS.
- (a) S. Noro, H. Chang, T. Takenobu, Y. Murayama, T. Kanbara, T. Aoyama, T. Sassa, T. Wada, D. Tanaka, S. Kitagawa, Y. Iwasa, T. Akutagawa and T. Nakamura, J. Am. Chem. Soc., 2005, 127, 10012 CrossRef CAS PubMed; (b) S. Noro, T. Takenobu, Y. Iwasa, H. Chang, S. Kitagawa, T. Akutagawa and T. Nakamura, Adv. Mater., 2008, 20, 3399 CrossRef CAS.
- T. Kitamori, D. Yoo, K. Iijima, T. Kawamoto and T. Mori, ACS Appl. Electron. Mater., 2019, 1, 1633 CrossRef CAS.
- A. Opitz, M. Horlet, M. Kiwull, J. Wagner, M. Kraus and W. Brutting, Org. Electron., 2012, 13, 1614 CrossRef CAS.
- (a) T. Kambe, R. Sakamoto, T. Kusamoto, T. Pal, N. Fukui, K. Hoshiko, T. Shimojima, Z. Wang, T. Hirahara, K. Ishizaka, S. Hasegawa, F. Liu and H. Nishihara, J. Am. Chem. Soc., 2014, 136, 14357 CrossRef CAS PubMed; (b) D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dinca, J. Am. Chem. Soc., 2014, 136, 8859 CrossRef CAS PubMed.
- X. Huang, L. Liu, L. Yu, G. Chen, W. Xu and D. Zhu, Angew. Chem., Int. Ed., 2018, 57, 146 CrossRef CAS PubMed.
- J. A. McClevery, Prog. Inorg. Chem., 1968, 10, 49 Search PubMed.
- L. Qu, D. Wang, C. Zhong, Y. Zou, J. Li, D. Zou and J. Qin, Synth. Met., 2010, 160, 2299 CrossRef CAS.
- A. K. Asatkar, S. P. Senanayak, A. Bedi, S. Panda, K. S. Narayan and S. S. Zade, Chem. Commun., 2014, 50, 7036 RSC.
- (a) H. Nishikawa, A. Wachi, M. Chikamatsu and R. Azumi, Mol. Cryst. Liq. Cryst., 2016, 641, 81 CrossRef CAS; (b) A. Wachi, Y. Kudo, A. Kanesaka, H. Nishikawa, T. Shiga, H. Oshio, M. Chikamatsu and R. Azumi, Polyhedron, 2017, 136, 70 CrossRef CAS.
- Y. Kato, K. Iijima, D. Yoo, T. Kawamoto and T. Mori, Chem. Lett., 2020, 49, 870 CrossRef CAS.
- (a) R. H. Holm, J. Am. Chem. Soc., 1960, 82, 5632 CrossRef CAS; (b) S. Di Bella, I. Fragala, I. Ledoux, M. A. Diaz-Garcia and T. J. Marks, J. Am. Chem. Soc., 1997, 119, 9550 CrossRef CAS.
- S. Zolezzi, A. Decinti and E. Spodine, Polyhedron, 1999, 18, 897 CrossRef CAS.
- A. Nakahara, H. Yamamoto and H. Matsumoto, Bull. Chem. Soc. Jpn., 1964, 37, 1137 CrossRef CAS.
- Y. Nakao, N. Nonagase and A. Nakahara, Bull. Chem. Soc. Jpn., 1969, 42, 452 CrossRef CAS.
- G. D. Simpson, G. O. Calisle and W. E. Hatfield, J. Inorg. Nucl. Chem., 1974, 36, 2257 CrossRef CAS.
- K. J. Miller, J. H. Baag and M. M. Abu-Omar, Inorg. Chem., 1999, 38, 4510 CrossRef CAS PubMed.
- N. Kumari, R. Prajapati and L. Mishra, Polyhedron, 2008, 27, 241 CrossRef CAS.
- (a) R.-G. Xiong, B.-L. Song, J.-L. Zuo and X.-Z. You, Polyhedron, 1996, 15, 903 CrossRef CAS; (b) L. C. Nathan, J. E. Koehne, J. M. Gilmore, K. A. Hannibal, W. E. Dewhirst and T. D. Mai, Polyhedron, 2003, 22, 887 CrossRef CAS.
- R. S. Downing and F. L. Urbach, J. Am. Chem. Soc., 1969, 91, 5977 CrossRef CAS.
- R. S. Downing and F. L. Urbach, J. Am. Chem. Soc., 1970, 92, 5861 CrossRef CAS.
- K. Bernardo, S. Leppard, A. Robert, G. Commenges, F. Dahan and B. Meunier, Inorg. Chem., 1996, 35, 387 CrossRef CAS PubMed.
- I. C. Santos, M. Vilas-Boas, M. F. M. Piedade, C. Freire, M. T. Duarte and B. de Castro, Polyhedron, 2000, 19, 655 CrossRef CAS.
- M. T. Nyuyen and B. J. Holliday, Chem. Commun., 2015, 51, 8610 RSC.
- (a) R. H. Bailes and M. Calvin, J. Am. Chem. Soc., 1947, 69, 1886 CrossRef CAS; (b) R. Warmuth and H. Elias, Inorg. Chem., 1991, 30, 5027 CrossRef CAS.
- Y. J. Lee, H. G. Heo and C. H. Oh, Tetrahedron, 2016, 72, 6113 CrossRef CAS.
- M. L. Tang, A. D. Reicharrdt, P. Wei and Z. Bao, J. Am. Chem. Soc., 2009, 131, 5264 CrossRef CAS.
- ADF2017.109, Scientific Computing & Modeling (SCM). Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- A. Bottcher, T. Takeuchi, K. I. Hardcastle, T. J. Maeda, D. Cwikel, M. Kapon and Z. Dori, Inorg. Chem., 1997, 36, 2498 CrossRef.
- (a) D. Hall and T. N. Waters, J. Chem. Soc., 1960, 2644 RSC; (b) L. M. Shkol'nikova, E. M. Yumal', E. A. Shugam and V. A. Voblikova, Zh. Strukt. Khim., 1970, 11, 886 Search PubMed; (c) A. G. Manfredotti and C. Guastini, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 863 CrossRef; (d) E. F. DiMauro and M. C. Kozlowski, Organometallics, 2002, 21, 1454 CrossRef CAS; (e) M. Kondo, K. Nabari, T. Horiba, Y. Irie, M. K. Kabir, R. P. Sarker, E. Shimizu, Y. Shimizu and Y. Fuwa, Inorg. Chem. Commun., 2003, 6, 154 CrossRef CAS.
- M. A. Siegler and M. Lutz, Cryst. Growth Des., 2009, 9, 1194 CrossRef CAS.
- N. Kumari, R. Prajapati and L. Mishra, Polyhedron, 2008, 27, 241 CrossRef CAS.
- W.-B. Yuan, H.-Y. Wang, J.-F. Du, S.-W. Chen and Q. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m3504 CrossRef CAS.
- M. G. B. Drew, R. N. Prasad and R. P. Sharma, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1755 CrossRef.
- A. Wojtczak, E. Szlyk, M. Jaskólski and E. Larsen, Acta Chem. Scand., 1997, 51, 274 CrossRef CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef CAS.
- T. Mori, A. Kobayashi, Y. Sasaki, H. Kobayashi, G. Saito and H. Inokuchi, Bull. Chem. Soc. Jpn., 1984, 57, 627 CrossRef CAS.
- F. Evangelista, V. Carravetta, G. Stefani, B. Jansik, M. Alagia, S. Stranges and A. Ruocco, J. Chem. Phys., 2007, 126, 124709 CrossRef PubMed.
- A. Wolberg and J. Manassen, J. Am. Chem. Soc., 1970, 92, 2982 CrossRef CAS PubMed.
- R. Sato, T. Kawamoto and T. Mori, J. Mater. Chem. C, 2019, 7, 567 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information for preparative details, dimer energy levels, devices fabrication, AFM images, and transistor characteristics. See DOI: 10.1039/d0ra05449f |
| ‡ Present address: Department of Chemical Engineering and Center for Advanced Soft Electronics, Pohang University of Science and Technology, 77 Cheongam-Ro, Nam-gu, Pohang 37673, Korea. |
| This journal is © The Royal Society of Chemistry 2020 |