
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe miscibility and spatial distribution of the components in electrospun polymer–protein mats†
Elizaveta Pavlova ab,
Igor Nikishinc,
Alexandra Bogdanovaab,
Dmitry Klinovab and
Dmitry Bagrov*ac
ab,
Igor Nikishinc,
Alexandra Bogdanovaab,
Dmitry Klinovab and
Dmitry Bagrov*ac
aFederal Research Clinical Center of Physical-Chemical Medicine of the Federal Medical and Biological Agency of Russia, 1a Malaya Pirogovskaya Street, 119435, Moscow, Russian Federation
bMoscow Institute of Physics and Technology, 9 Institutsky Per., Dolgoprudny, 141700, Moscow Region, Russian Federation
cLomonosov Moscow State University, Faculty of Biology, Department of Bioengineering, 1-12 Leninskie Gory, 119234, Moscow, Russian Federation. E-mail: bagrov@mail.bio.msu.ru
First published on 29th January 2020
Abstract
Biodegradable blended electrospun mats are promising for biomedical applications such as wound dressing, tissue engineering, and drug delivery. Electrospun mats based on polyesters can be modified by the addition of other polymers or proteins to accelerate the degradation, improve mechanical properties or biocompatibility. However, relatively little is known about the distribution of the components throughout the blended mats. In the present work, we prepared polylactide (PLA), bovine serum albumin (BSA), and the blended PLA–BSA electrospun mats. We demonstrated that PLA and BSA are miscible in a common solvent HFIP (1,1,1,3,3,3-hexafluoro-2-propanol) at concentrations below 3%, but become immiscible as concentration increases. We used three methods (fluorescence microscopy, EDX, and Raman microspectroscopy) to validate that PLA and BSA can be blended in a single electrospun fiber despite the phase separation in the blend. The homogeneity of the blend influences on the homogeneity of the distribution of PLA and BSA components throughout the electrospun mat, as measured by Raman microspectroscopy. When the blended electrospun mats were incubated in water, they demonstrated the prolonged release of BSA. The presented results show a step-by-step approach for manufacturing blended electrospun mats made of immiscible components, which involves the analysis of component miscibility, the mat morphology, and composition. This approach can be used for the rational design of multicomponent electrospun mats.
Introduction
Electrospinning is a popular method to obtain non-woven mats consisting of ultrafine fibers by applying high voltage to a polymer solution or melt.1 Electrospun mats are used as filters, sorbents, substrates for cell culturing, materials for wound dressing, and tissue engineering.2–4 Combining several components in a single mat allows one to finely tune its physical, chemical, and biological properties.5 Electrospun mats from protein-based blends are especially valuable for biomedical applications because proteins make mats more hydrophilic, biocompatible and bioactive.6 From the technological point of view, it is far easier to mix the two components in a common solvent than to use the alternative methods of multicomponent electrospinning, such as coaxial electrospinning7 or emulsion electrospinning.8 The electrospun polymer–protein mats can be used for prolonged release of proteins and peptides.9–11 However, most polymer–polymer and polymer–protein pairs are poorly miscible and undergo phase separation,12 which can alter the structure, morphology, and properties of the mats. How does the inhomogeneity of the electrospun blend manifest itself in the morphology of the blended mat? How are the different components distributed throughout the electrospun fibers – are they combined in a single fiber or form the independent networks? In other words, how homogeneous are the blended electrospun mats?Only a few works address these questions taking the poor miscibility of the components into account. In 2001 Mikhail Bognitzki and colleagues13 proposed three possible ways of two-component fibers structure, having regard to the size of the different phases. First, if the size of phases is large enough (more than fiber diameter), they form independent fibers. Second, if the size of the phases is close to the fiber diameter, the fibers consist of alternating domains. Third, if the phase's size is smaller than the fiber diameter, the two components could be spread within one fiber and form complex interpenetrating domains. The third case led to the formation of rough fiber morphology, and removal of one component resulted in holes on the fiber surface. In 2017 Annalisa Aluigi and colleagues14 used Fourier-transform infrared spectroscopy to demonstrate that homogeneous electrospun mats could be prepared from an immiscible blend (keratin and polyamide). The authors emphasized that the homogeneity resulted from the fast solvent evaporation, which helped to avoid phase separation. In 2019 our group15 also confirmed the presence of both components within the electrospun polymer–protein blend (gelatin and PLA) fiber using Raman spectroscopy. The immiscible polymer–protein blends sometimes yield special morphologies of the electrospun mats, including bimodal networks.16
In the present work, we investigated the relationship between the composition of blends used for electrospinning, the morphology and the composition of the resulting fibers. Our experimental system consisted of polylactide (PLA) and bovine serum albumin (BSA) with 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as the solvent. We used the PLA–BSA system, because PLA is a widely used biocompatible and biodegradable polyester, and albumin is a convenient model protein, which can be subjected to electrospinning and used for manufacturing scaffolds.17–19
Materials and methods
Materials
The following materials were used: PLA (REC, Russia), BSA (Sigma-Aldrich, USA), 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, P&M-Invest, Russia).The ternary phase diagram
In order to show the correlation between the composition of the PLA–BSA–HFIP blend and the compatibility of its components, we built the ternary phase diagram. Each point of the diagram reflected the composition of the PLA–BSA–HFIP mixture (mass concentration of the components) and different states of the solution: transparent, opalescent, or phase-separated. Transparent solutions had a ratio of components at which the components were compatible. Likewise, the opalescent blends contained partly compatible components (metastable state), and the blends with phase separation contained incompatible components. The blends were prepared by HFIP adding to dry mixes of BSA and PLA weighted using Ohaus (USA) lab balance. Prior to the examination, the mixtures were kept at a temperature of 25 °C at least 8 hours.Electrospinning
Electrospinning of PLA and BSA solutions and PLA–BSA blends in HFIP was carried out using the Nanofiber Electrospinning Unit (China) at 30 kV accelerating voltage and 1 ml h−1 solution feed rate. An inner diameter of a needle with a polymer solution or blend was 0.7 mm; the distance between the needle and the counter electrode was 20 cm. The electrospun mats were obtained either on aluminum foil or on a polypropylene frame positioned between the needle and the counter electrode.Fluorescence microscopy
BSA was labeled with rhodamine B (Rho B, Sigma-Aldrich, USA) in 0.1 M Na2CO3 solution at pH 9.3 for 1 hour at room temperature and then was purified by Sephadex G25 gel-chromatography followed by mild centrifugation at 700g. BSA-RhoB was dissolved at a concentration of 100 mg ml−1 in HFIP. Solutions (100 mg ml−1 in HFIP) of BSA-RhoB, BSA, and PLA were mixed in proportion 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:5 (BSA-RhoB:BSA:PLA). Then the blend was electrospun and fibers were collected on a glass slide (ApexLab, Russia). The fluorescent electrospun fibers were investigated using the fluorescent microscope Nikon Eclipse Ti (Nikon, Japan) with a Plan Fluor 60×/0.70 objective (Nikon, Japan) with a set of filters providing excitation for RhoB at 528–553 nm and emission at 590–650 nm. The images were captured with camera ORCA-Flash4.0 (Hamamatsu, Japan) with 500 ms exposure time.
:4:5 (BSA-RhoB:BSA:PLA). Then the blend was electrospun and fibers were collected on a glass slide (ApexLab, Russia). The fluorescent electrospun fibers were investigated using the fluorescent microscope Nikon Eclipse Ti (Nikon, Japan) with a Plan Fluor 60×/0.70 objective (Nikon, Japan) with a set of filters providing excitation for RhoB at 528–553 nm and emission at 590–650 nm. The images were captured with camera ORCA-Flash4.0 (Hamamatsu, Japan) with 500 ms exposure time.
Electron microscopy and EDX spectroscopy
Electrospun mats were covered by 10 nm gold–palladium alloy using Sputter Coater Q150T (Quorum Technologies, UK) and examined with a Zeiss Merlin microscope equipped with Gemini II Electron Optics (Zeiss, Oberkochen, Germany). SEM imaging was done at 1–3 kV accelerating voltage and 30–100 pA probe current.EDX was performed by SEM via Silicon Drift Detector (SDD) X-MaxN 150 (Oxford Instruments, Abingdon, Oxfordshire, UK) and AztecEnergy EDX Software (Version 3.0). For EDX analysis electrospun fibers were collected onto silicon wafers; the probe current and accelerating voltage were set to 500 pA and 8 kV.
Raman spectroscopy
Raman measurements were performed at room temperature with a confocal Raman microscope INTEGRA Spectra (NT-MDT, Russia). The spectra were recorded with an electron-multiplying charge-coupled device (Newton, Andor; Abingdon, Oxfordshire, UK) cooled down to −65 °C. The recorded spectra covered the range between 600 and 3000 cm−1 (the diffraction grating with 600 lines per mm was used). For the measurements, a 532 nm laser was used; the intensity was adjusted to 2 mW; the spectra were recorded using 100×/0.9 objective.The electrospun fibers were collected at low surface density onto aluminum foil to facilitate the acquisition of Raman signals from the individual fibers. Raman spectra were processed with median filtering, baseline subtraction (via Origin 8.6 software, OriginLab, Northampton, Massachusetts, USA) to reduce the noise; then the peaks above the background were detected and measured.
BSA release kinetics from PLA–BSA blended electrospun mats
The release kinetics of BSA in water (at room temperature) from the electrospun mats (3% BSA + 3% PLA) was examined by measuring the concentration of BSA in supernatants by NanoDrop 2000c UV-vis spectrophotometer (Thermo Scientific, Waltham, MA, USA). The fraction of the released BSA was calculated as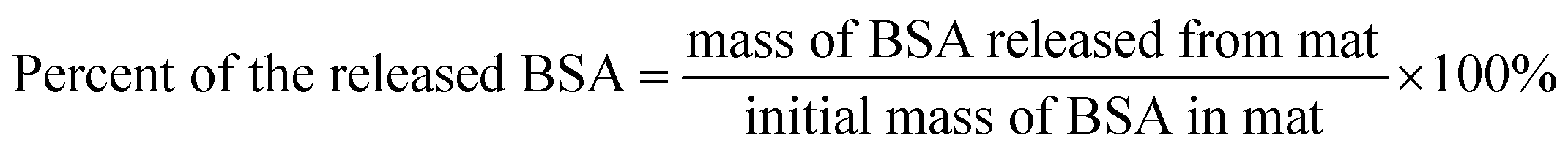
Results and discussion
Ternary phase diagram of the PLA–BSA–HFIP mixture
The polymers are poorly miscible,20 and the polyester–protein pair PLA–BSA is not an exclusion. We built the ternary phase diagram for PLA–BSA–HFIP system via the method of turbidity points13,21 (Fig. 1). The solutions (ternary mixtures) were homogenous only at relatively low concentrations (PLA and BSA less than 3% wt in total) or when one of the components was prevalent and the system behaved as a two-component mixture (PLA concentration less than 1% and BSA concentration less than 5%). Red dots on the phase diagram corresponded to opalescent blends, which maintained this metastable state at least 2 weeks. Remaining areas (black dots) of phase diagram reflected blends with clear phase separation. The diagram demonstrates a spinodal curve, which is typical for the ternary mixtures of two immiscible polymers and a common solvent.13,21,22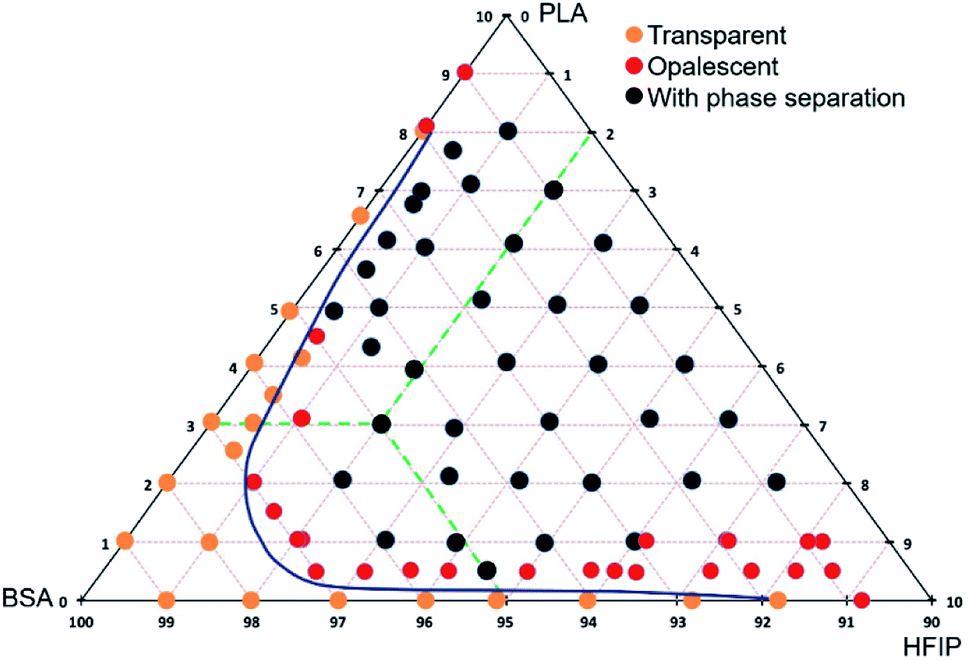 | ||
| Fig. 1 Ternary phase diagram of PLA–BSA–HFIP system. The mass concentration range for BSA and PLA is from 0% to 10%, green dashed lines show the concentration scheme. | ||
Morphology of single-component and blended electrospun mats
Several solutions and blends were electrospun at the same conditions, and the morphology of electrospun mats was investigated by SEM (Fig. 2, 3 and Table SI1†). We focused on the electrospinning of three types of solutions: PLA in HFIP, BSA in HFIP and PLA–BSA in HFIP at equal mass fractions of PLA and BSA.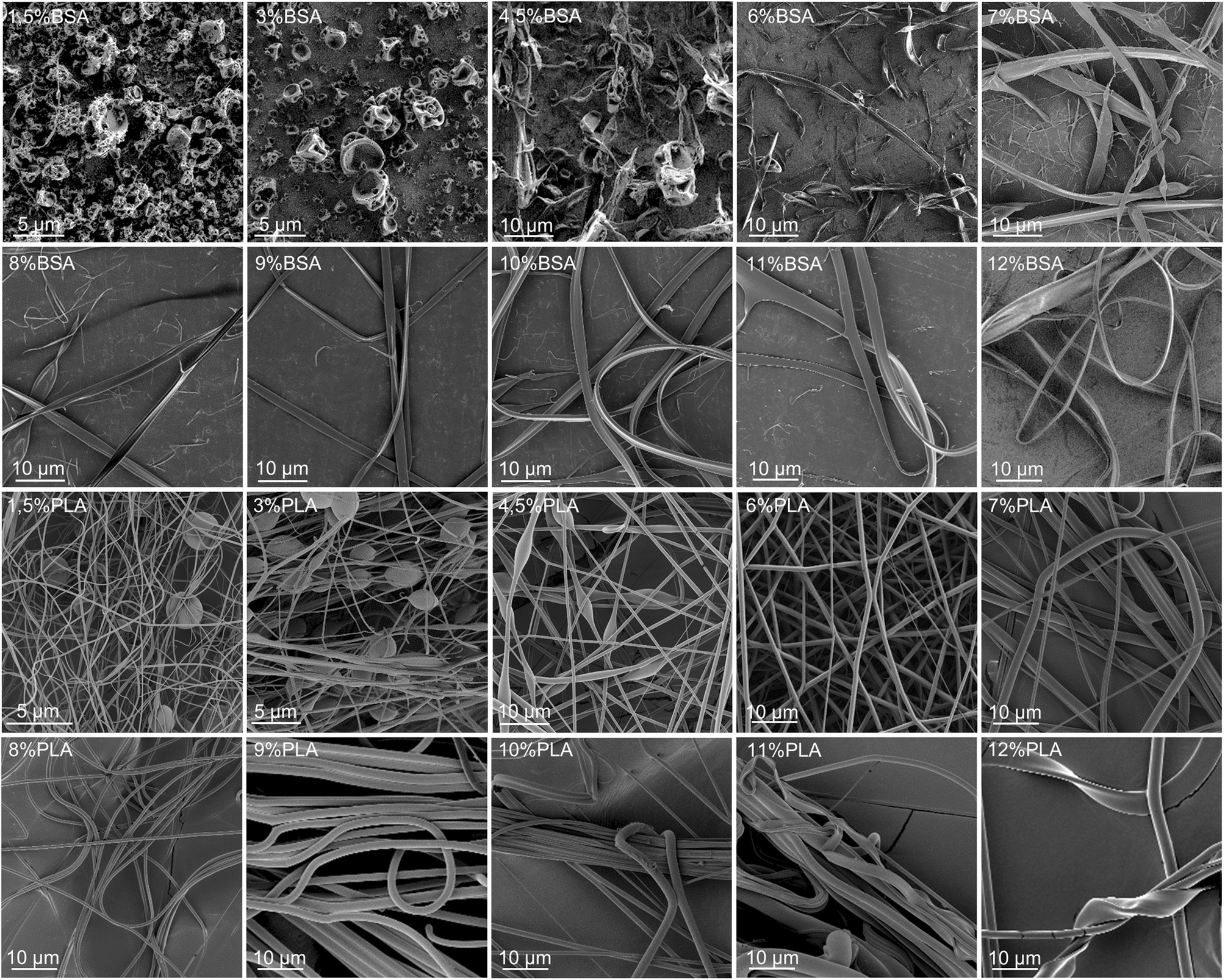 | ||
| Fig. 2 SEM images of BSA (two upper lines) and PLA (two lower lines) electrospun mats obtained from HFIP solutions at different mass concentrations (from 1.5% to 12%). | ||
 | ||
| Fig. 3 Electrospun mats made from PLA–BSA blends in HFIP. | ||
At low concentrations, the solutions of BSA in HFIP formed single electrospun beads with the morphology of “deflated balls”. As BSA concentration increased, the number of balls decreased, and the flat ribbons appeared (Fig. 2). At concentrations below 4.5% BSA–HFIP system was in electrospray regime, rather than electrospinning due to low solution viscosity. Noteworthy, in electrospun BSA samples only a few cylindrical (round-sectioned) fibers were observed, and the ribbons were the dominant morphology. A similar ribbon-like morphology of BSA fibers was obtained from 14% BSA solution in TFE with β-mercaptoethanol addition.18 The electrospinning of a 7% albumin solution in HFIP also resulted in ribbons.23
Electrospinning of PLA solutions at low concentrations resulted in beaded fibers, which became smoother (beadless) and thicker as PLA concentration increased (Fig. 2). The same behavior was observed for polystyrene,24 polymethyl methacrylate,25 poly(ε-caprolactone),26 silk fibroin27 and PLA.27–29 After electrospinning of the 11–12% PLA solutions in HFIP, some ribbons were observed along with the cylindrical fibers.
The ribbon formation can be explained by buckling instability.30,31 Evaporation of the solvent from the surface of an electrospun jet leads to the formation of the core–shell structure with a glassy polymer shell. The further contraction of the jet and mechanical mismatch between the core and the shell can result in buckling of the shell, which is similar to the instability of a long elastic rod under compression. The instability requires a certain threshold compressive pressure, which depends on the mechanical properties and the sizes of the core and the shell.31 It yields a wrinkled fiber or a ribbon – if the wrinkle wavelength is limited by the fiber circumference.32
For a given polymer–solvent pair, the “fiber to ribbon” transition is facilitated by the increase in concentration, molecular weight, or applied voltage (Table 1). Firstly, an increase in viscosity (or molecular weight) raises the diameter of the jet during the electrospinning process,33,34 and leads to an increase in local charge density. Secondly, high applied voltage facilitates the ionization of polymer chains. As a result, an increase in the abovementioned parameters leads to an increase in the charge density per unit length of the polymer jet. We hypothesize that the charge density makes one of the most important contributions to glassy shell formation and, as a result, to flat ribbons obtaining. An increase in charge density leads to an increase of Coulomb repulsion between polymer molecules, and they tend to locate in the periphery, forming a shell.
| Material | Solvent | Factors promoting the “fiber to ribbon” transition | References |
|---|---|---|---|
| Gelatin | Formic acid | (1) Concentration increase | 35 |
| (2) Voltage increase | |||
| PVA | Water | (1) Molecular weight increase | 38 and 39 |
| (2) Concentration increase | |||
| Nylon 6 | Formic acid | (1) Concentration increase | 40 |
| Silk fibroin | Water | (1) Concentration increase | 37 |
| (2) Voltage increase | |||
| Zein | 70% ethanol | (1) Concentration increase | 41 |
| Polyacrylamide | Water with sodium bis(2-ethylhexyl) sulfosuccinate | (1) Concentration increase | 42 |
| Poly(vinyl pyrrolidone) | Ethanol | (1) Concentration increase | 43 |
Our results are in overall agreement with the previous data (Table 1). First, we observed more electrospun ribbons at higher concentrations of solution. Second, the electrospinning of BSA solutions in HFIP resulted mostly in ribbons. This is not surprising since many proteins tend to form ribbons rather than fibers – gelatin,35 BSA,18,36 human serum albumin,16 silk fibroin.37
The SEM images of electrospun PLA–BSA blends are shown in Fig. 3 (PLA:BSA = 1:1, total concentration of 1%, 2%, 3%, 6%, 9% and 12%). The first two blends (1% and 2%) belong to the homogeneous area of the ternary phase diagram; the 3% blend is at the spinodal curve, and the rest ones (6%, 9%, 12%) belong to the heterogeneous area with phase separation (Fig. 1). All the blends were stirred violently before loading into the syringe. Electrospinning of the first three blends (1%, 2%, and 3%) led to roughly similar mats consisting of fibers and beads, and the number of beads decreased with increasing concentration. Electrospinning of 6%, 9% and 12% blends resulted in fibers and ribbons formation without beads (Fig. 3). Thus, the concentration increase caused the disappearance of the beads and the transition from fibers to ribbons (Fig. 3).
Thus, we have shown the following features of the PLA–BSA–HFIP system:
(1) HFIP is a good solvent for both BSA and PLA, but the ternary system PLA–BSA–HFIP demonstrates a spinodal decomposition behavior.
(2) Regardless of the doping solution composition and regardless of the phase separation, we observed that
(a) At a low concentration of the doping solution, the electrospun mats involved beads.
(b) At a high concentration of the doping solution, the electrospun mats involved ribbons.
(3) BSA demonstrated a higher tendency to form electrospun ribbons than PLA.
Fig. 3 (PLA–BSA mats) demonstrates the same trends as shown in Fig. 2 for the single-component mats. Thus, we did not observe any drastic change in morphology of the electrospun mats when the solution composition shifted from the homogeneous area of the diagram through the spinodal curve and into the “phase separation” region. How are the PLA and BSA components distributed in the electrospun fibers? To answer this question, we investigated the electrospun PLA–BSA fibers using fluorescent microscopy, EDX analysis, and Raman microspectroscopy. Hereafter we focused on the 6% blend, because it is prone to phase separation, and it allowed us to observe the ribbons and fibers simultaneously.
Tracing the fibers composition
In order to investigate the composition of individual fibers within the electrospun PLA–BSA mat we used three methods: fluorescent microscopy, EDX and Raman microspectroscopy. All the three methods proved that the two components of the dry mat (PLA and BSA) could be combined in a single fiber.Fluorescent microscopy showed that BSA-RhoB was detected in all the fibers obtained by electrospinning of a freshly prepared PLA–BSA–HFIP blend (Fig. 4). Although the fraction of the labeled BSA was only 10% of all the mat weight, it was enough to label all the fibers.44,45
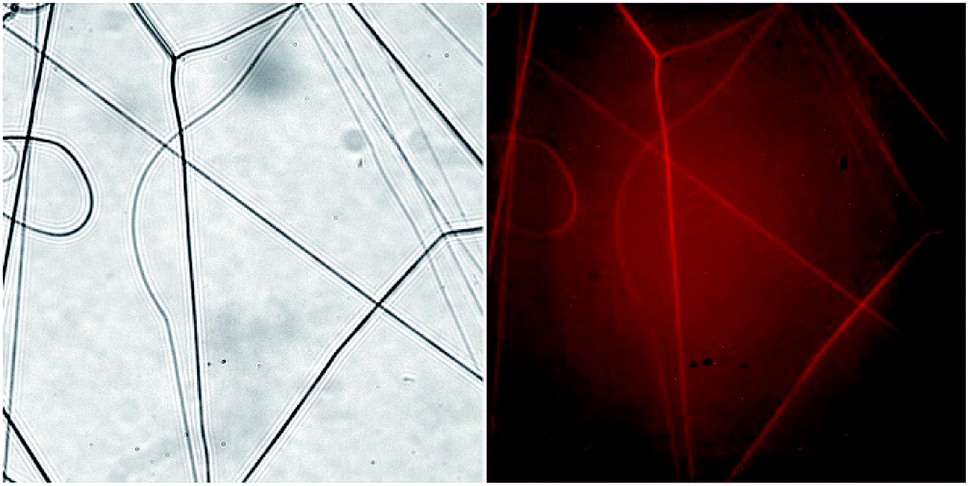 | ||
| Fig. 4 Light microscopy of BSA-RhoB:BSA:PLA (1:4:5) electrospun fibers. Left – transmitted light, right – fluorescence image. Images size is 222 μm × 222 μm. | ||
A similar result was obtained by EDX analysis. We plotted the elemental maps of carbon, oxygen, and nitrogen and focused on the localization of nitrogen, since it is present in BSA and absent in PLA. We compared three samples: fibers obtained by electrospinning of the 6% blend (3% PLA and 3% BSA) and two control samples obtained by electrospinning of the single-component PLA and BSA 6% solutions (Fig. 5). The nitrogen elemental maps were relatively noisy because the characteristic peak of nitrogen (Kα 392 eV) is difficult to detect in the EDX spectra between the peaks of carbon (Kα 277 eV) and oxygen (Kα 525 eV). However, we managed to detect nitrogen both in fibers and ribbons produced by electrospinning of the 6% blend (3% PLA and 3% BSA). When the control samples were examined, nitrogen was observed in the BSA sample and was not detected in the PLA sample.
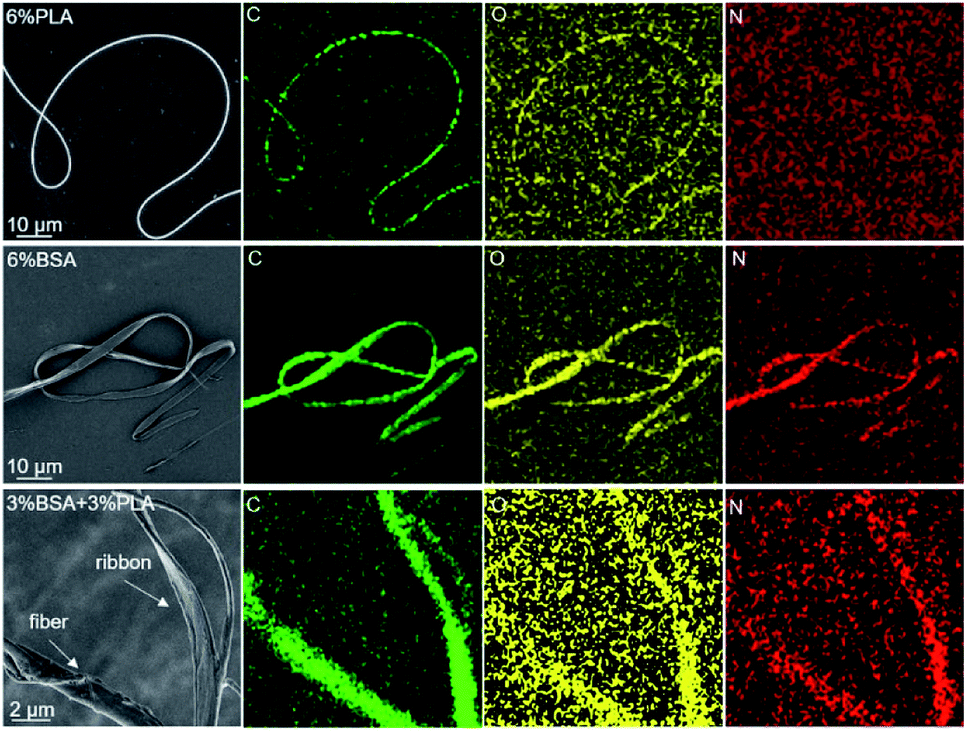 | ||
| Fig. 5 EDX analysis of the electrospun fibers. The top row fibers obtained from 6% PLA solution, the middle ribbons obtained from 6% BSA solution, the bottom row – from 6% blend (3% PLA and 3% BSA). | ||
The fluorescent microscopy and EDX analysis prove that both fibers and ribbons are bi-component, but these methods cannot provide us with information about components ratios of different types of blended fibers. Therefore, our further investigation was performed by Raman spectroscopy.
For obtaining Raman spectra from individual nanofibers we used the procedure which was developed previously for the electrospun PLA–gelatin fibers.15 Individual blended fibers were deposited onto aluminum foil and mapped by light microscopy and elastic scattering signal. After that Raman spectra were collected form approximately 1 μm spots. Raman spectra of single component BSA and PLA fibers have specific characteristic peaks which are not overlapping (Fig. 6): ∼1655 cm−1 for BSA (the amide I peak) and ∼1765 cm−1 for PLA (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching).15
O stretching).15
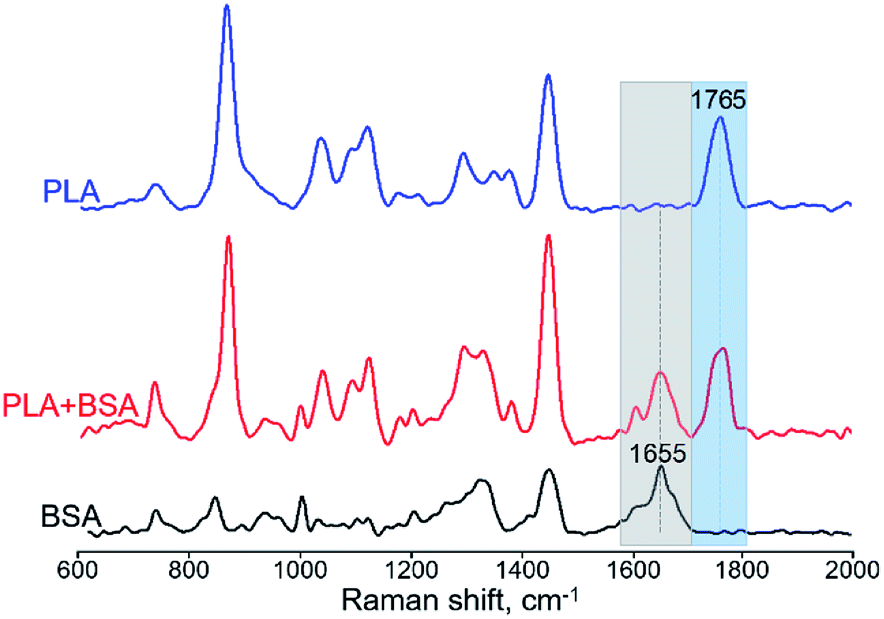 | ||
| Fig. 6 Raman spectra of BSA, PLA, and PLA–BSA electrospun fibers. Characteristic PLA and BSA peaks marked by blue (PLA) and grey (BSA) bands correspondingly. | ||
We followed the ratio of the characteristic peak intensities 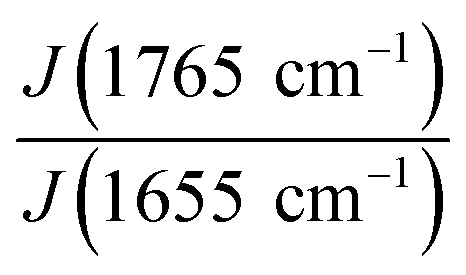 of single fibers obtained from 6% blend (3% PLA and 3% BSA) and noticed that the ratio was changing greatly from one fiber to another (Fig. 7), despite the violent stirring of the blend before electrospinning. For comparison, the ratio of the characteristic peak intensities had a relatively narrow spread when the fibers were obtained by electrospinning the homogeneous 2% blend (1% PLA and 1% BSA) (Fig. 7). The fibers were relatively uniform in composition when we used the homogeneous 2% blend than instead of the phase-separated 6% blend. However, despite the phase separation in the solution, the fibers could incorporate both components – PLA and BSA. This result is in agreement with the previous results on electrospinning of PLA–gelatin mixture.15 Fig. 7 shows that the median ratios of the characteristic peaks were almost the same for the fibers obtained from the 2% and the 6% blends (1.34 and 1.36). This corresponds to the constant mass ratio of the PLA and BSA components in the dry mats.
of single fibers obtained from 6% blend (3% PLA and 3% BSA) and noticed that the ratio was changing greatly from one fiber to another (Fig. 7), despite the violent stirring of the blend before electrospinning. For comparison, the ratio of the characteristic peak intensities had a relatively narrow spread when the fibers were obtained by electrospinning the homogeneous 2% blend (1% PLA and 1% BSA) (Fig. 7). The fibers were relatively uniform in composition when we used the homogeneous 2% blend than instead of the phase-separated 6% blend. However, despite the phase separation in the solution, the fibers could incorporate both components – PLA and BSA. This result is in agreement with the previous results on electrospinning of PLA–gelatin mixture.15 Fig. 7 shows that the median ratios of the characteristic peaks were almost the same for the fibers obtained from the 2% and the 6% blends (1.34 and 1.36). This corresponds to the constant mass ratio of the PLA and BSA components in the dry mats.
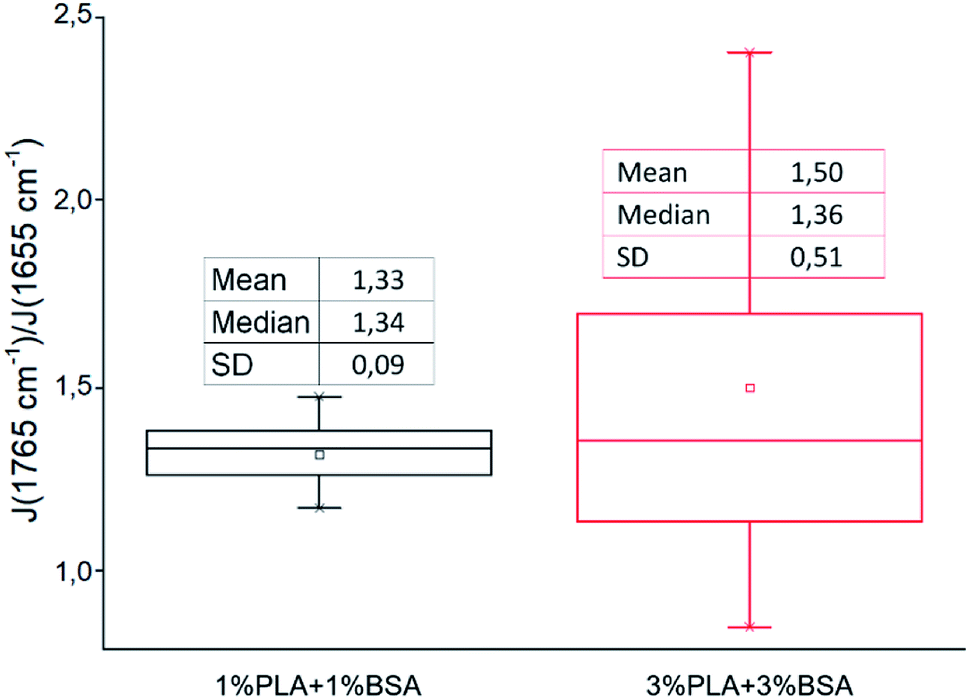 | ||
| Fig. 7 The ratio of characteristic peaks PLA and BSA from Raman spectra of blended mats. | ||
Since 6% blend is unstable and tends to be phase-separated, after 7 days of storage at room temperature it yielded two conjugated solutions (Fig. SI1†). We calculated their composition using spectrophotometry and estimation of the volumes. The upper transparent solution contained BSA at the concentration from 8 to 11% (N = 7 experiments). If we assume that the upper solution had only trace amount of PLA, then the lower solution contained approximately 4% PLA. However, the lower solution was not transparent, so the system did not reach the equilibrium state due to the high viscosity. Thus, the lower solution had also a small BSA fraction.
We electrospun the upper and lower conjugated solutions separately, imaged the mats using SEM and performed Raman microanalysis from individual fibers (or ribbons). The upper one yielded ribbons (Fig. 8a), as expected for a concentrated BSA solution (Fig. 2), and only BSA peak was observed by Raman spectroscopy from ribbons (Fig. 8b). The bottom solution yielded round-sectioned fibers without ribbons (Fig. 8c). However, Raman spectroscopy indicated the presence of BSA (Fig. 8d) and the ratio of the characteristic peak intensities was 2.0 ± 0.6 (mean ± SD).
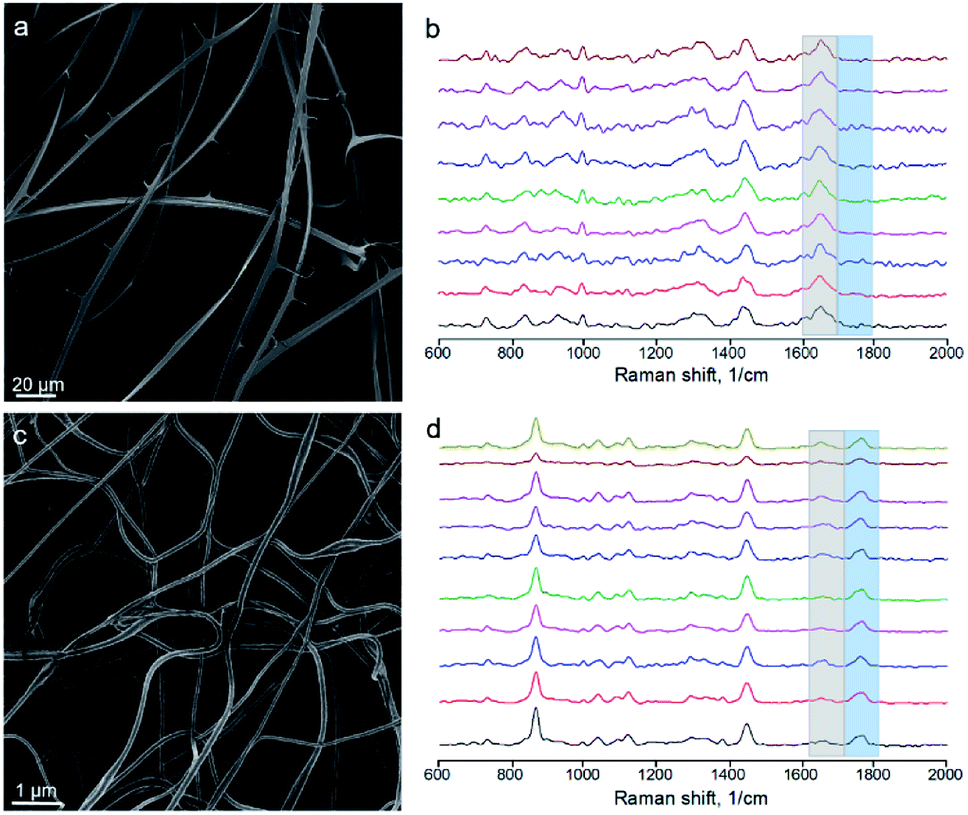 | ||
| Fig. 8 Electrospinning of the conjugated solutions. (a) Ribbons formed by the electrospinning of the upper solution (the one enriched with BSA). (b) Raman spectra from independent ribbons of mat (a). (c) Round-sectioned fibers formed by electrospinning of the bottom solution (the one enriched with PLA). (d) Raman spectra from independent fibers of mat (c). | ||
Thus, we analyzed the distribution of PLA and BSA throughout the fibers. If the 6% blend (3% PLA and 3% BSA) was stirred violently before electrospinning, each fiber consisted of both PLA and BSA. If the conjugated solutions were isolated after phase separation and electrospun, they yielded different morphologies. Electrospinning of the BSA-enriched solution resulted in ribbons, and the PLA-enriched one yielded round sectioned-fibers.
BSA release from the blended mats
Electrospun mats can be used for encapsulation and prolonged release of bioactive substances, including drugs, peptides, and therapeutic proteins.46–48 Incorporation of proteins into electrospun mats can be tricky because most proteins undergo denaturation during electrospinning due to the strong electric field and the presence of organic solvent.49 However, some proteins withstand both denaturation factors and retain their functionality after electrospinning. These proteins include insulin,50 chondroitinase,9 lysozyme,51,52 chymotrypsin,53,54 and others.BSA is a commonly used model protein. The blended mats described in the current study provide its prolonged release, as shown in Fig. 9. We incubated the blended mats (3% PLA + 3% BSA) in the water at room temperature during ten days, calculated concentration of the released BSA by UV spectrophotometry, and estimated the mass percentage of released BSA. Approximately 50% of BSA dissolved from the electrospun mat for ten days. A similar BSA release profile was obtained for electrospun polystyrene–BSA mats prepared by emulsion electrospinning55 and for the blended poly(hydroxybutyrate-co-hydroxyvalerate)–BSA mats.36
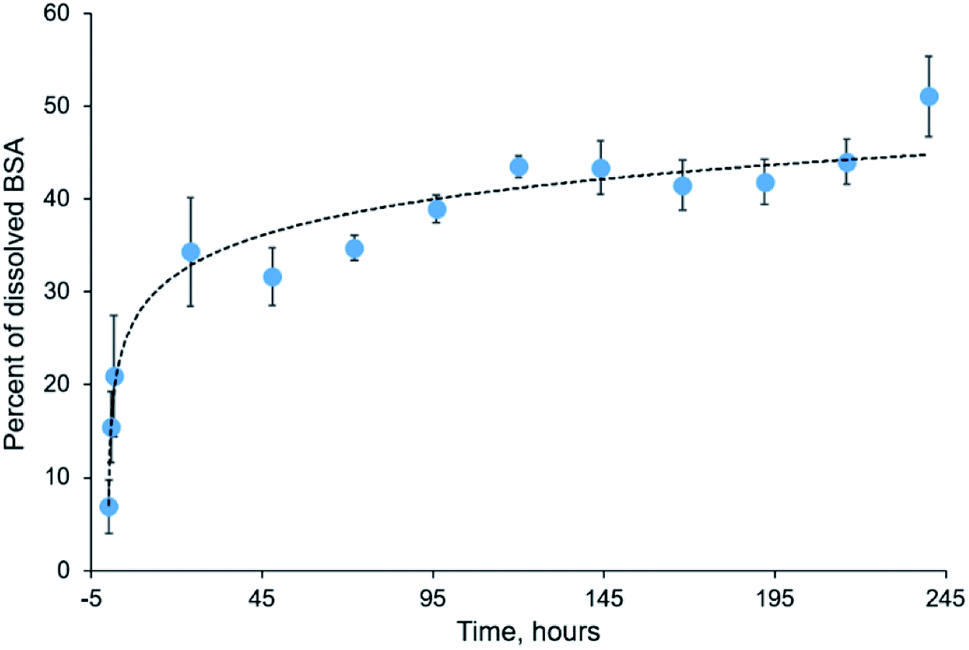 | ||
| Fig. 9 Kinetics of BSA release from electrospun blended mat during ten days. | ||
Although preparation of blended PLA–BSA mats was accompanied by the dissolution of BSA in HFIP which can promote the formation of alpha-helices in proteins and peptides and cause protein denaturation,56,57 we expect the denaturation effect to be relatively low since BSA has a high content of alpha-helices. Previously we demonstrated that HFIP-treated BSA could bind with the anti-BSA antibodies.58
Conclusion
Incorporation of a protein is a popular way of bringing bio-functionality to electrospun mats. Here we analyzed the structure of electrospun blended mats made of PLA and BSA mixed in a common solvent – HFIP. We can draw the following observations and conclusions:(1) We plotted the ternary phase diagram of the PLA–BSA–HFIP system and observed spinodal decomposition behavior, which is typical for the ternary mixtures of two immiscible polymers and a common solvent.
(2) Regardless of the doping solution composition and regardless of the phase separation, we observed that (1) at a low concentration of the doping solution, the electrospun mats involved beads, and (2) at a high concentration of the doping solution, the electrospun mats involved ribbons.
(3) BSA demonstrated a higher tendency to form electrospun ribbons than PLA. BSA solution in HFIP showed a strong tendency to form ribbons regardless of how the solution was obtained – by the simple dissolution of BSA in HFIP or by phase separation in a ternary blend.
(4) If the phase-separated ternary blend (3% PLA, 3% BSA, 94% HFIP) was stirred violently before electrospinning, both components of the dry mats (PLA and BSA) were present in each of the fibers. This conclusion was confirmed by three analytical methods – fluorescence microscopy, EDX, and Raman microspectroscopy.
(5) Raman spectra proved that when a homogeneous blend (1% PLA, 1% BSA, 98% HFIP) was used for electrospinning, the ratio of PLA and BSA component were spread more uniformly throughout the mat, than in case of a heterogeneous blend (3% PLA, 3% BSA, 96% HFIP).
The observations related to the immiscible ternary blends can be explained if we regard the blend as a set of domains enriched either by the polymer (PLA) or by the protein (BSA), described by Hilliard–Cahn waves59 or later theories.60,61 If the size of the domains is small, they are mixed during electrospinning, and the polymer and protein components co-localize in a single fiber or ribbon. As the phase separation occurs and the domains grow, the probability of ribbon formation increases. However, when the two conjugated solutions form, only the one enriched with protein yields ribbons.
The data obtained in the current work have several aspects which contribute to the significance
(1) We demonstrated the rational way to control the morphology of an electrospun mat prepared from a blend consisting of two polymers and a solvent. The mat preparation should start with plotting the ternary phase diagram.13 If we use the homogeneous blend for electrospinning, the two components of the dry mat will be distributed more uniformly than in the case of the heterogeneous blend.
(2) When an immiscible blend is used for electrospinning, it follows the same general tendencies (relation between the blend concentration and mat morphology) as a single-component polymer solution.
(3) Raman spectroscopy seems a suitable tool to assess the chemical composition of individual electrospun fibers. Previously it was used to examine molecular orientation in individual fibers,62 and the core–shell structure.63
(4) The prolonged protein release from blended mats can be useful for biomedical applications, including wound dressing, drug delivery, and tissue engineering.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Our research was supported by the Russian Science Foundation (grant no 19-74-00037). We thank Dr Levchishin S. Yu (D. Mendeleev University of Chemical Technology of Russia) for fruitful discussion.Notes and references
- G. C. Rutledge and S. V. Fridrikh, Adv. Drug Delivery Rev., 2007, 59, 1384–1391 CrossRef CAS PubMed.
- J. Ding, J. Zhang, J. Li, D. Li, C. Xiao, H. Xiao, H. Yang, X. Zhuang and X. Chen, Prog. Polym. Sci., 2019, 90, 1–34 CrossRef CAS.
- T. Hemamalini and V. R. Giri Dev, Int. J. Biol. Macromol., 2018, 106, 712–718 CrossRef CAS.
- J. Xie, M. R. MacEwan, A. G. Schwartz and Y. Xia, Nanoscale, 2010, 2, 35–44 RSC.
- S. N. Hanumantharao and S. Rao, Fibers, 2019, 7, 66 CrossRef.
- D. B. Khadka and D. T. Haynie, Nanomedicine, 2012, 8, 1242–1262 CrossRef CAS PubMed.
- A. K. Moghe and B. S. Gupta, Polym. Rev., 2008, 48, 353–377 CrossRef CAS.
- N. Nikmaram, S. Roohinejad, S. Hashemi, M. Koubaa, F. J. Barba, A. Abbaspourrad and R. Greiner, RSC Adv., 2017, 7, 28951–28964 RSC.
- T. Xia, B. Huang, S. Ni, L. Gao, J. Wang, J. Wang, A. Chen, S. Zhu, B. Wang, G. Li, S. Zhu and X. Li, Biomed. Pharmacother., 2017, 86, 354–362 CrossRef CAS.
- J. F. C. Viana, J. Carrijo, C. G. Freitas, A. Paul, J. Alcaraz, C. C. Lacorte, L. Migliolo, C. A. Andrade, R. Falcão, N. C. Santos, S. Gonçalves, A. J. Otero-González, A. Khademhosseini, S. C. Dias and O. L. Franco, Nanoscale, 2015, 7, 6238–6246 RSC.
- Y. H. Hsu, C.-T. Lin, Y.-H. Yu, Y.-C. Chou, S.-J. Liu and E.-C. Chan, Int. J. Nanomed., 2016, 11, 3927–3937 CrossRef CAS PubMed.
- I. Khan, M. Mansha and M. A. J. Mazumder, Polym. Blends, 2018, 1–38 Search PubMed.
- M. Bognitzki, T. Frese, M. Steinhart, A. Greiner, J. H. Wendorff, A. Schaper and M. Hellwig, Polym. Eng. Sci., 2001, 41, 982–989 CrossRef CAS.
- A. Aluigi, A. Varesano, C. Vineis and A. Del Rio, Mater. Des., 2017, 127, 144–153 CrossRef CAS.
- D. V. Bagrov, I. I. Nikishin, E. R. Pavlova and D. V. Klinov, AIP Conf. Proc., 2019, 2064, 040001 CrossRef.
- A. P. Tiwari, M. K. Joshi, J. I. Kim, A. R. Unnithan, J. Lee, C. H. Park and C. S. Kim, J. Colloid Interface Sci., 2016, 476, 29–34 CrossRef CAS PubMed.
- B. H. Noszczyk, T. Kowalczyk, M. Łyżniak, K. Zembrzycki, G. Mikułowski, J. Wysocki, J. Kawiak and Z. Pojda, Biofabrication, 2015, 7, 015011 CrossRef CAS PubMed.
- S. Fleischer, A. Shapira, O. Regev, N. Nseir, E. Zussman and T. Dvir, Biotechnol. Bioeng., 2014, 111, 1246–1257 CrossRef CAS.
- C.-C. Hsu, A. Serio, N. Amdursky, C. Besnard and M. M. Stevens, ACS Appl. Mater. Interfaces, 2018, 10, 5305–5317 CrossRef CAS.
- L. H. Sperling, in Introduction to Physical Polymer Science, John Wiley & Sons, Inc, 4th edn, 2006, pp. 687–756 Search PubMed.
- N. Posharnowa, A. Schneider, M. Wünsch, V. Kuleznew and B. A. Wolf, J. Chem. Phys., 2001, 115, 9536–9546 CrossRef CAS.
- J. A. Gonzalez-Leon and A. M. Mayes, Macromolecules, 2003, 36, 2508–2515 CrossRef CAS.
- W. Nuansing, D. Frauchiger, F. Huth, A. Rebollo, R. Hillenbrand and A. M. Bittner, Faraday Discuss., 2013, 166, 209–221 RSC.
- S. Megelski, J. S. Stephens, D. Bruce Chase, J. F. Rabolt, D. B. Chase and J. F. Rabolt, Macromolecules, 2002, 35, 8456–8466 CrossRef CAS.
- H.-S. Bae, A. Haider, K. M. K. Selim, D.-Y. Kang, E.-J. Kim and I.-K. Kang, J. Polym. Res., 2013, 20, 158 CrossRef.
- M. Chen, P. K. Patra, S. B. Warner and S. Bhowmick, Tissue Eng., 2007, 13, 579–587 CrossRef CAS PubMed.
- S. Shahverdi, M. Hajimiri, M. A. Esfandiari, B. Larijani, F. Atyabi, A. Rajabiani, A. R. Dehpour, A. A. Gharehaghaji and R. Dinarvand, Int. J. Pharm., 2014, 473, 345–355 CrossRef CAS.
- R. Casasola, N. L. Thomas and S. Georgiadou, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1483–1498 CrossRef CAS.
- R. Casasola, N. L. Thomas, A. Trybala and S. Georgiadou, Polymer, 2014, 55, 4728–4737 CrossRef CAS.
- L. Wang, C. Pai, M. C. Boyce and G. C. Rutledge, Appl. Phys. Lett., 2009, 94, 151916 CrossRef.
- G. Herrmann and M. J. Forrestal, AIAA J., 1965, 3, 1710–1715 CrossRef.
- S. Koombhongse, W. Liu and D. H. Reneker, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2598–2606 CrossRef CAS.
- Y. Hong, in Advances in Polyurethane Biomaterials, 2016 Search PubMed.
- P. Filip and P. Peer, Processes, 2019, 7, 948 CrossRef.
- F. Topuz and T. Uyar, Mater. Sci. Eng., C, 2017, 80, 371–378 CrossRef CAS PubMed.
- E. R. Pavlova, D. V Bagrov, M. N. Kopitsyna, D. A. Shchelokov, A. P. Bonartsev, I. I. Zharkova, T. K. Mahina, V. L. Myshkina, G. A. Bonartseva, K. V. Shaitan and D. V. Klinov, J. Appl. Polym. Sci., 2017, 45090 CrossRef.
- H. Wang, Y. Zhang, H. Shao and X. Hu, J. Mater. Sci., 2005, 40, 5359–5363 CrossRef CAS.
- J. Tao and S. Shivkumar, Mater. Lett., 2007, 61, 2325–2328 CrossRef CAS.
- A. Koski, K. Yim and S. Shivkumar, Mater. Lett., 2004, 58, 493–497 CrossRef CAS.
- S. Y. Tsou, H. S. Lin and C. Wang, Polymer, 2011, 52, 3127–3136 CrossRef CAS.
- L. Moradkhannejhad, M. Abdouss, N. Nikfarjam, S. Mazinani and V. Heydari, J. Mater. Sci.: Mater. Med., 2018, 29, 165 CrossRef PubMed.
- Y. Y. Zhao, Q. B. Yang, X. F. Lu, C. Wang and Y. Wei, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 2190–2195 CrossRef CAS.
- S. Chuangchote, T. Sagawa and S. Yoshikawa, J. Appl. Polym. Sci., 2009, 114, 2777–2791 CrossRef CAS.
- J. Dai, J. Jin, S. Yang and G. Li, Mater. Res. Express, 2017, 1–21 Search PubMed.
- C. Tang, A. E. Ozcam, B. Stout and S. A. Khan, Biomacromolecules, 2012, 13, 1269–1278 CrossRef CAS.
- E. J. Torres-Martinez, J. M. Cornejo Bravo, A. Serrano Medina, G. L. Pérez González and L. J. Villarreal Gómez, Curr. Drug Delivery, 2018, 15, 1360–1374 CrossRef CAS PubMed.
- Y. Sun, S. Cheng, W. Lu, Y. Wang, P. Zhang and Q. Yao, RSC Adv., 2019, 9, 25712–25729 RSC.
- P. Wen, Y. Wen, M. H. Zong, R. J. Linhardt and H. Wu, J. Agric. Food Chem., 2017, 65, 9161–9179 CrossRef CAS PubMed.
- D. I. Zeugolis, S. T. Khew, E. S. Y. Yew, A. K. Ekaputra, Y. W. Tong, L.-Y. L. Yung, D. W. Hutmacher, C. Sheppard and M. Raghunath, Biomaterials, 2008, 29, 2293–2305 CrossRef CAS PubMed.
- K. Stephansen, M. García-Díaz, F. Jessen, I. S. Chronakis and H. M. Nielsen, Int. J. Pharm., 2015, 495, 58–66 CrossRef CAS.
- S. K. Tiwari and S. Venkatraman, Polym. Int., 2012, 61, 1549–1555 CrossRef CAS.
- T. G. Kim, D. S. Lee and T. G. Park, Int. J. Pharm., 2007, 338, 276–283 CrossRef CAS PubMed.
- S. K. Murase, L. J. Del Valle, S. Kobauri, R. Katsarava and J. Puiggalí, Polym. Degrad. Stab., 2015, 119, 275–287 CrossRef CAS.
- A. Díaz, L. J. Del Valle, D. Tugushi, R. Katsarava and J. Puiggalí, Mater. Sci. Eng., C, 2015, 46, 450–462 CrossRef.
- X. Wang, Y. Yuan, X. Huang and T. Yue, J. Appl. Polym. Sci., 2015, 132, 41811 Search PubMed.
- K. Sode, S. Ochiai, N. Kobayashi and E. Usuzaka, Int. J. Biol. Sci., 2007, 3, 1–7 CAS.
- N. Hirota, K. Mizuno and Y. Goto, Protein Sci., 1997, 6, 416–421 CrossRef CAS PubMed.
- O. V. Morozova, E. R. Pavlova, D. V. Bagrov, N. A. Barinov, K. A. Prusakov, E. I. Isaeva, V. V. Podgorsky, D. V. Basmanov and D. V. Klinov, Int. J. Nanomed., 2018, 13, 6637–6646 CrossRef CAS PubMed.
- J. W. Cahn and J. E. Hilliard, J. Chem. Phys., 1958, 28, 258–267 CrossRef CAS.
- R.-J. Roe, J. Chem. Phys., 1975, 62, 490 CrossRef CAS.
- E. Helfand and Y. Tagami, J. Chem. Phys., 1972, 56, 3592–3601 CrossRef CAS.
- L. M. Bellan and H. G. Craighead, Polymer, 2008, 49, 3125–3129 CrossRef CAS.
- L. Sfakis, A. Sharikova, D. Tuschel, F. X. Costa, M. Larsen, A. Khmaladze and J. Castracane, Biomed. Opt. Express, 2017, 8, 1025–1035 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10910b |
| This journal is © The Royal Society of Chemistry 2020 |