
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design direct Z-scheme BiOBr/g-C3N4 heterojunction with enhanced visible photocatalytic activity for organic pollutants elimination
Hongfei Li,
Aiqiong Ma *,
Dian Zhang,
Yunqin Gao and
Yonghao Dong
*,
Dian Zhang,
Yunqin Gao and
Yonghao Dong
College of Materials Science and Engineering, Xi'an University of Architecture and Technology, Xi'an, Shaanxi 710055, China. E-mail: maaiqiong@xauat.edu.cn
First published on 29th January 2020
Abstract
A rapid recombination of photo-generated electrons and holes, as well as a narrow visible light adsorption range are two intrinsic defects in graphitic carbon nitride (g-C3N4)-based photocatalysts. Inspired by natural photosynthesis, an artificially synthesized Z-scheme photocatalyst can efficaciously restrain the recombination of photogenerated electron–hole pairs and enhance the photoabsorption ability. Hence, to figure out the above problems, BiOBr/g-C3N4 composite photocatalysts with different mass ratios of BiOBr were successfully synthesized via a facile template-assisted hydrothermal method which enabled the BiOBr microspheres to in situ grow on the surface of g-C3N4 flakes. Furthermore, to explore the origin of the enhanced photocatalytic activity of BiOBr/g-C3N4 composites, the microstructure, photoabsorption ability and electrochemical property of BiOBr/g-C3N4 composites were investigated by X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), UV-vis diffuse reflectance spectroscopy (DRS), electrochemical impedance spectroscopy (EIS) and photocurrent (PC) response measurements. As a result, the introduction of BiOBr on g-C3N4 to constitute a direct Z-scheme heterojunction system can effectively broaden the light absorption range and promote the separation of photo-generated electron–hole pairs. Hence, compared with pure g-C3N4 and BiOBr, the resultant BiOBr/g-C3N4 composites exhibit the remarkable activity of photodegradated rhodamine B (RhB) and tetracycline hydrochloride (TC-HCl) under visible light irradiation. Simultaneously, the optimal BiOBr content of the BiOBr/g-C3N4 composites was obtained. The BiOBr/g-C3N4 composites exhibit an excellent photostability and reusability after four recycling runs for degradation RhB. Moreover, the active-group-trapping experiment confirmed that ·OH, ·O2− and h+ were the primary active groups in the degradation process. Based on the above research results, a rational direct Z-scheme heterojunction system is contrastively analyzed and proposed to account for the photocatalytic degradation process of BiOBr/g-C3N4 composites.
1. Introduction
With the development of dye and pharmaceuticals industry, organic pollutants of industrial emission have given rise to severe water pollution which could constitute a bad threat to the living environment of humanity. Thereinto, aromatic compounds with diffusely conjugated chromophores, such as RhB and TC-HCl, possess a physicochemical stability in water environment such that the traditional treatment measures hardly prove effective. Therefore, plenty of new technologies which can effectively decompose organic pollutants through redox reactions have emerged continuously.1–3 Semiconductor photocatalytic degradation technology has attracted a wide research interest,4,5 due to its unique advantages of the utilization visible light to excite semiconductor materials to produce highly efficient active groups.6,7 Nevertheless, most semiconductor photocatalysts with a wide band gap can merely utilize the ultraviolet light approximately, which accounts for only 4% of the solar energy.8,9 Although CdS,10 BiVO4,11 Ag3PO4,12 etc. with relatively narrow band gaps possess a visible light photocatalytic activity, they usually suffer from a rapid recombination between excited electrons and holes during light irradiation.13 As a consequence, it is attractive and significant to design and develop photocatalytic materials with the applicable band gaps, wide visible-light adsorption ranges and highly photocatalytic activity.14Graphitic carbon nitride (g-C3N4), a familiar metal-free semiconductor photocatalyst, has been frequently investigated because of its numerous excellent physicochemical traits, such as outstanding thermology, electricity and photology properties.15 Such performances result mainly from its stratiform structure of strong covalent C–N bonds in each layer and weak van der Waals force between layers.16,17 The g-C3N4 with a narrow band gap (2.7 eV) possesses an appropriate conduction band (CB) position (ECB = −1.07 eV), which is more negative than the reduction potential of E0(O2/·O2−) (about −0.33 eV).18 Therefore, the photo-generated electrons on the CB of g-C3N4 are capable of reacting with O2 to generate superoxide radicals (·O2−) which can subsequently participate in the process of degradation of organic pollutants. Nevertheless, on account of the weak van der Waals interactions between adjacent CN layers in the g-C3N4 crystal structure, g-C3N4 has high recombination odds of photo-generated electron–hole pairs which lessens the quantum efficiency of the photocatalytic degradation procedures.19,20 Therefore, for further improving the inherent shortcomings of g-C3N4 and enhancing its photocatalytic efficiency, several research works have been carried out and showed that combination with metal-oxide semiconductors to construct composite photocatalyst can remarkably enhance the absorption ability of sunlight and prolong the lifetime of the photo-generated carrier.21
Recently, BiOBr, as one of the highly active bismuth oxyhalide (BiOX, X = Cl, Br, I) photocatalytic materials, has received intensive attention in the photocatalytic field. It exhibits a high photocatalytic activity on account of its suitable band gap (2.81 eV) and wide visible light adsorption range (>410 nm).22 It is noteworthy that its unique lamellar crystal structure, just as the tetragonal matlockite structure, can afford adequate space to polarize the relevant atoms and orbitals, resulting in the generation of an internal electric field between the [Bi2O2]2+ layer and the Br− layer.23 On the basis of previous reports, BiOBr was used to combine with g-C3N4 to form an indirect Z-scheme heterojunction and showed excellent photocatalytic activity. In 2019, Zhang Mingming et al. prepared an indirect Z-scheme BiOBr/CDs/g-C3N4 photocatalyst by a facile hydrothermal method and the hybrids can degrade 17.5% of tetracycline (TC) within 180 min for which the reaction rates were 31.92 and 9.76 times those of pristine BiOBr and g-C3N4, respectively.24 Hence, this indirect Z-scheme-type composite shows remarkable potential as an efficient photocatalyst for the utilization of solar energy to decompose contaminants. To further improve the photocatalytic performance of such a Z-scheme BiOBr/g-C3N4 system, a high-efficiency direct Z-scheme type photocatalyst is proposed in which the photo-induced electrons can reduce unnecessary loss during transfer process by direct contact between BiOBr and g-C3N4. Furthermore, for the past few years, BiOBr has been synthesized via different methods, such as solvothermal, chemical precipitation, hydrothermal and so on. Among the different synthesis methods, a BiOBr microsphere structure obtained by a hydrothermal method reveals a remarkably photocatalytic activity thanks to its abundant porous structures providing BiOBr with sufficient active sites for the elimination of organic pollutants. To improve the microstructure on the basis of BiOBr microspheres, a facile template-assisted hydrothermal method was selected for the synthesis of BiOBr microspheres. Among the precursor solutions, ethylene glycol was used for prompting BiOBr nanosheets to self-assemble into microspheres. Simultaneously, the bio-template L-lysine with the polar functional groups of amino and hydroxyl, as a special surfactant, was used for regulating the microstructure of BiOBr microspheres so as to increase the efficacious active sites on the BiOBr surface.25–27 To the best of our knowledge, only a few researchers have focused on direct Z-scheme BiOBr/g-C3N4 composites synthesized by a template-assisted hydrothermal method and the number of efficient direct Z-scheme photocatalysts is still finite.
In this study, a direct Z-scheme BiOBr microsphere/g-C3N4 flake heterojunction photocatalysts was successfully synthesized via a facile template-assisted hydrothermal method which enabled BiOBr microspheres to in situ grow on the surface of g-C3N4 flakes (Fig. 1). Through exploring the microstructure, optical and electrochemical properties of the composites, the BiOBr/g-C3N4 composites exhibit a higher photocatalytic activity proved by the degradation of RhB and TC-HCl, which could be attributed chiefly to the formation of a direct Z-scheme heterojunction system. Ultimately, a reasonable photocatalytic mechanism of the BiOBr/g-C3N4 composites for the degradation of RhB is contrastively analyzed and proposed by free radical trapping experiments.
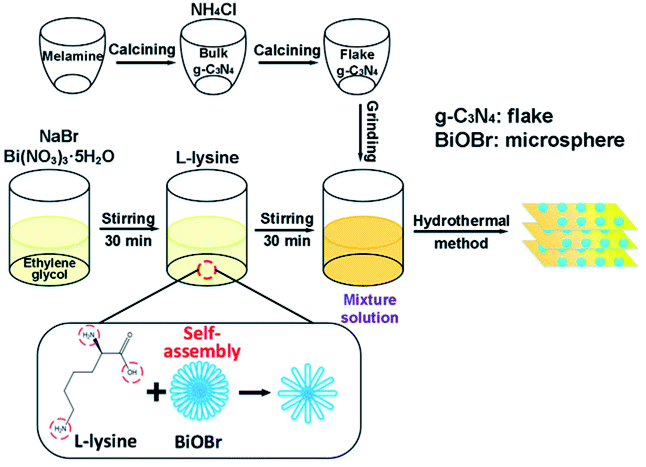 | ||
| Fig. 1 Abridged summarization of the synthesis process of the BiOBr/g-C3N4 composites. | ||
2. Experimental
2.1. Chemicals
All the chemicals were used as received without further purification. Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O) was acquired from Tianjin Kemiou Chemical Reagent Co. (Tianjing, China). Sodium bromide (NaBr) was acquired from Wuhan Tiantai Chemical Co. (Wuhan, China). Ethylene glycol ((CH2OH)2), sodium hydroxide (NaOH), ethyl alcohol absolute (C2H6O) and ammonium chloride (NH4Cl) were purchased from Tianjin Tianli Chemical Reagent Co. (Tianjing, China). Tetracycline hydrochloride (TC-HCl, C22H24N2O8·HCl) was purchased from Shanghai Macklin Biochemical Technology Co. (Shanghai, China).2.2. Synthesis of g-C3N4 flakes
Fig. 1 illustrates the synthesis procedure of the BiOBr/g-C3N4 composites and the particular processes were as follows: 10 g melamine was sintered at 550 °C with a heating rate of 3 °C min−1 in a muffle furnace for 4 h. The bulk-g-C3N4 was obtained after cooling down to room temperature. After 4.5 g of NH4Cl particles were dissolved in 50 mL of deionized water, 6 g of the obtained bulk-g-C3N4 was added in solution under vigorous stirring and sonicated for 2 h. Thereafter, the suspension was dried at 80 °C in a drying oven overnight. Afterwards, the obtained dry g-C3N4 was sintered at 550 °C once again with a heating rate of 3 °C min−1 in a muffle furnace for 4 h. The flakes of g-C3N4 were obtained after cooling down to room temperature.2.3. Synthesis of BiOBr/g-C3N4 nanocomposites
In a peculiar template-assisted hydrothermal process (Fig. 1), 0.16 mmol Bi(NO3)3·5H2O and 0.16 mmol NaBr were dissolved into 50 mL of ethylene glycol solution and magnetically stirred for 30 min to form a clear solution. Then, 0.16 mmol L-lysine was dispersed into the above mixture solution and stirred for 30 min, followed by adding 0.95 g of as-prepared g-C3N4 flakes under vigorous stirring and sonicated for 2 h. Subsequently, the pH value of the mixture was adjusted to 5 by adding NaOH solution under constant stirring. After the suspension was transferred to a Teflon-lined stainless-steel autoclave and maintained at 120 °C for 12 h, the resultant suspension was respectively washed with deionized water and ethyl alcohol three times. The wet products were dried at 80 °C to obtain the BiOBr/g-C3N4 composite with a BiOBr mass ratio of 5%. BiOBr/g-C3N4 nanocomposites with different mass ratios of BiOBr (5%, 10%, 15%, 20% and 25%) were synthesized similarly by tuning the dosage of BiOBr. The corresponding BiOBr/g-C3N4 samples are abbreviated as B-5/CN, B-10/CN, B-15/CN, B-20/CN and B-25/CN. For comparison, pure BiOBr and g-C3N4 photocatalysts were prepared under the same conditions.2.4. Characterization
The samples were characterized by X-ray diffraction (XRD) (D/max-2500, Rigaku), field emission scanning electron microscopy (FESEM) (Gemini SEM 300, Zeiss) and transmission electron microscope (TEM) (H-7650, Hitachi). Optical diffuse reflectance spectra (DRS) were measured by using UV-vis DRS (CARY 5000, Agilent).The electrochemical impedance spectroscopy (EIS) and photocurrent (PC) responses measurements were performed by using an electrochemical workstation (CHI660E, CH Instruments Ins.) with a standard three-electrode configuration at room temperature. The as-prepared sample, a Pt wire and Ag/AgCl were used as the working electrode, counter electrode and reference electrode, respectively. The working electrode was prepared by a fluorinated tin oxide (FTO) sheet glass with an active area of 1 × 1 cm2 containing 10 mg of photocatalyst and ultimately calcined at 105 °C for 2 h. The EIS experiment was executed in an aqueous 0.5 M Na2SO4 solution under dark conditions. For the PC measurement, a 500 W Xe lamp and Na2SO4 (0.5 M) aqueous solution served as the light source and electrolyte, respectively.
2.5. Photocatalytic activity
The photocatalytic activities of the as-prepared samples were evaluated by means of the degradation of the RhB solutions under a 500 W Xe-lamp with a cut off filter (λ ≥ 420 nm). Briefly, the process was performed through immersing 50 mg of the as-prepared samples into 50 mL of RhB (TC-HCl) at an initial concentration of 15 mg L−1 (20 mg L−1). Before lighting up the illuminant, the adsorption–desorption equilibrium was achieved by continuously stirring the suspensions in the dark for 40 min. At a certain time interval, 4 mL of the suspension was fetched and centrifuged to obtain a purified liquid. Finally, the RhB and TC-HCl concentrations were detected via the UV-vis spectrophotometer (UV-1200, Macy) at an absorption wavelength of 554 nm for RhB and 356 nm for TC-HCl. The stability of the B-20/CN composites was evaluated by four successive cycling experiments.3. Results and discussion
3.1. Crystal structures of the samples
Fig. 2 exhibits XRD patterns of all the synthesized samples. Pure g-C3N4 shows distinct diffraction peaks at approximately 27.5°, corresponding to the (002) planes of g-C3N4 (JCPDS no. 87-1526). The diffraction peaks at 2θ = 10.9°, 25.2°, 32.2°, 40.6°, 46.2°, and 57.2° correspond to the (001), (101), (110), (112), (200), and (212) crystal planes of BiOBr (JCPDS No. 09-0393). Moreover, no other diffraction peaks were detected, revealing the high purity and single phase of the as-obtained samples. However, no other characteristic peaks of BiOBr were detected in B-5/CN except diffraction peaks at 2θ = 25.2° and 32.2°, owing to the low BiOBr content in the B-5/CN composite. With the increase of BiOBr content, the characteristic peaks intensity of BiOBr also have increases of varying degree. Meanwhile, the characteristic peak intensities of g-C3N4 gradually weaken thanks to the decrease of g-C3N4 content. The strong diffraction peaks correspond with g-C3N4 and BiOBr, which indicates that the BiOBr/g-C3N4 composites were successfully synthesized through the facile template-assisted hydrothermal method.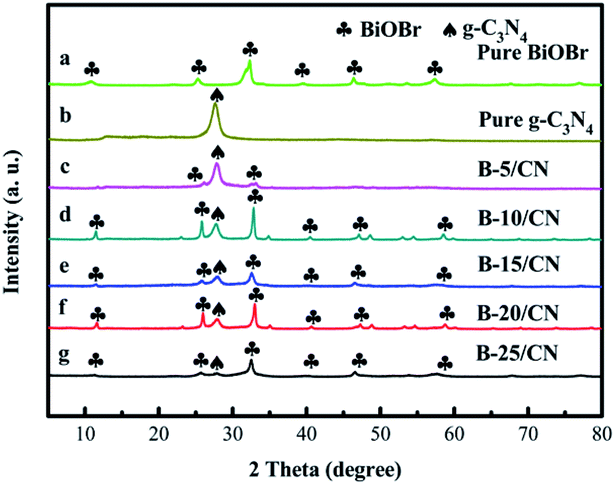 | ||
| Fig. 2 XRD patterns of (a) pure BiOBr, (b) pure g-C3N4, (c) B-5/CN, (d) B-10/CN, (e) B-15/CN, (f) B-20/CN and (g) B-25/CN. | ||
3.2. Morphology characteristics of the samples
The microstructures and morphology of the as-prepared samples were observed by FESEM and TEM, and the results are displayed in Fig. 3. As can be seen from the FESEM image of pure g-C3N4 in Fig. 3a, the pure g-C3N4 synthesized by simple polymerization and gas phase methods consists of thin flakes with an approximate thickness of 200 nm. The small thickness of g-C3N4 flakes is conducive to the separation of photo-generated carriers and the increase of built-in electric field intensity perpendicular to the lamellae.28 From Fig. 3b, BiOBr microspheres are hardly detected on the surface of g-C3N4 flakes on account of the lower BiOBr content of the B-5/CN sample of than others. As you can see from Fig. 3c–f, BiOBr microspheres are randomly dispersed on the surface of the g-C3N4 flakes and between the g-C3N4 flakes. With the increase of BiOBr content, the number of BiOBr microspheres on the surface of the g-C3N4 flakes gradually multiplies. Furthermore, from the FESEM image of B-20/CN in Fig. 3e, a mass of BiOBr microspheres existed between the g-C3N4 flakes which provided multiple cellular structures for the BiOBr/g-C3N4 composites. Such structures are convincing evidence that the B-20/CN sample possesses a better photoabsorption performance than the other samples in below UV–vis DRS spectra. | ||
| Fig. 3 FESEM images of (a) pure g-C3N4, (b) B-5/CN, (c) B-10/CN, (d) B-15/CN, (e) B-20/CN, (f) B-25/CN, TEM images of (g) B-20/CN, (h) template-free BiOBr and (i) BiOBr prepared by using bio-templates. | ||
A TEM image of B-20/CN is shown in Fig. 3g, providing a more distinct observation about the two components. The darker area with a spherical shape ought to be BiOBr and the lighter area should be g-C3N4, which further testifies the good dispersion of BiOBr microspheres on the g-C3N4 flakes and the direct contact between the two components. Homogeneous dispersion of the BiOBr microspheres constructs an effective physical junction between the BiOBr and g-C3N4 semiconductors that unquestionably will accelerate the generation and separation of photo-generated carriers due to the direct Z-scheme heterojunction structure of the composites.29,30 Furthermore, TEM images of template-free BiOBr and BiOBr prepared by using a bio-template are shown in Fig. 3h and i, reflecting the effect of a bio-template on the microsphere structures within the BiOBr photocatalyst. Obviously, BiOBr microspheres consist of vast BiOBr nanosheets via a self-assembly process. Meanwhile, the bio-template L-lysine25–27 with polar functional groups of amino and hydroxyl, as a special surfactant, can selectively adsorb on specific crystal faces and control the crystal growth and self-assembly process of BiOBr. Compared with template-free BiOBr (Fig. 3h), the internal structure of BiOBr microspheres (diameter: about 400 nm) prepared by using the bio-template (Fig. 3i) shows more loose structures and a further exposure degree of the BiOBr nanosheets in order to enhance the amount of active sites for the degradation of organic pollutants.
3.3. UV-vis DRS and band gap analysis
The optical properties of the as-prepared samples with different BiOBr content were evaluated by UV-vis DRS. From Fig. 4a, the light absorption edges of g-C3N4 and BiOBr are approximately 465 and 428 nm, respectively. After g-C3N4 combines with BiOBr, the light absorption edge of the B-5/CN composite has red-shifted to 470 nm, which indicates that the BiOBr microspheres are likely to settle onto the surface of g-C3N4 flakes. It's worth noting that the photoabsorption performance of the blended samples seems stronger than those of pure g-C3N4 and BiOBr in the range of 470–495 nm. This phenomenon can be ascribed to the irregular cellular structure of BiOBr microspheres scattering and reflecting light, as well as the strong interfacial interaction between BiOBr and g-C3N4 in the BiOBr/g-C3N4 hetero-phase junction.31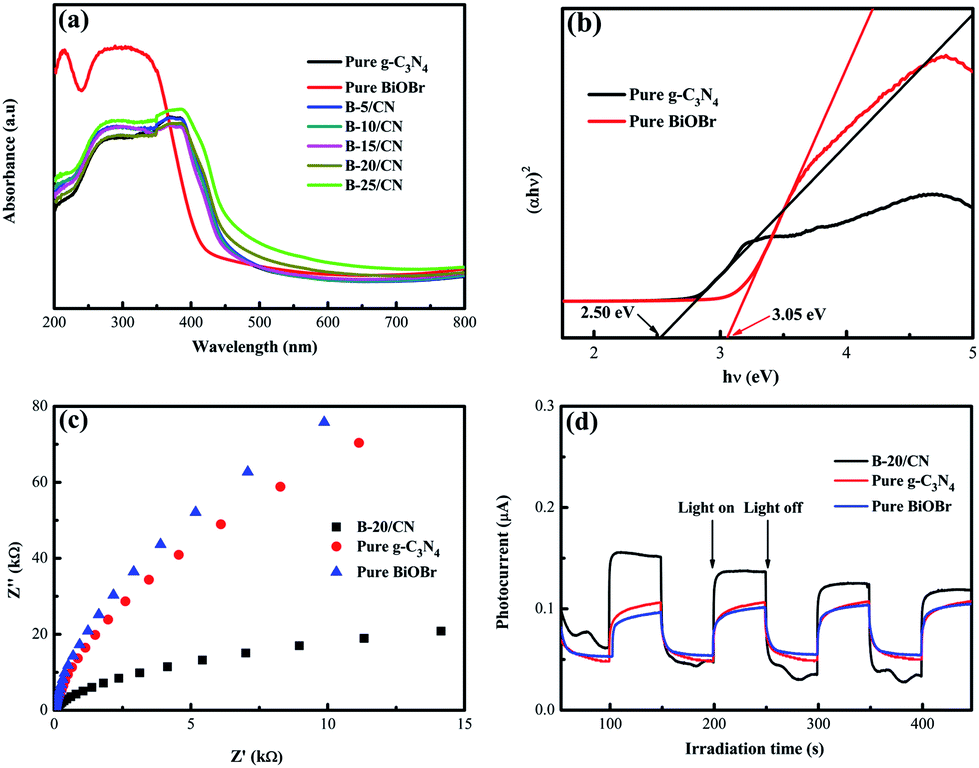 | ||
| Fig. 4 (a) DRS spectra; (b) plot of (αhν)2 vs. hν; (c) EIS Nyquist plot and (d) transient photocurrent response of the as-prepared samples with light on/off cycles. | ||
Fine approximations of the band gaps of BiOBr and g-C3N4 semiconductors are provided in Fig. 4b. From the spectra, the band gaps (Eg) of g-C3N4 and BiOBr are 2.50 and 3.05 eV, respectively. Furthermore, the conduction band (CB) and valence band (VB) edge redox potentials of g-C3N4 and BiOBr can be estimated on the basis of the extensively accepted equations:
| ECB = X − Ee − Eg/2 | (1) |
| EVB = ECB + Eg | (2) |
The X values for BiOBr and g-C3N4 are 6.18 eV and 4.73 eV, respectively.32,33 EVB and ECB stand for the VB and CB potentials, respectively, and Ee is the energy of free electrons on the hydrogen scale (about 4.5 eV).34 According to eqn (1) and (2), the ECB and EVB of g-C3N4 are −1.02 and +1.48 eV, respectively. Furthermore, the ECB and EVB of BiOBr are +0.16 and +3.21 eV, respectively.
3.4. EIS analysis and photocurrent response measurements
EIS analysis and photocurrent response measurements were applied to testify the high efficiency of direct Z-scheme BiOBr/g-C3N4 hybrids in facilitating the segregation of photogenerated electron–hole pairs. The EIS spectra of pure BiOBr, pure g-C3N4 and the B-20/CN composite are shown in Fig. 4c. The diameter of the EIS Nyquist plot of B-20/CN is smaller than those of pure BiOBr and g-C3N4. Given that the diameters on the EIS spectra reveal the reaction rate at the surface of the electrode, the plots in Fig. 4c display that B-20/CN possesses a lower resistance than pure BiOBr and g-C3N4. It's worth noting that lower resistance is conducive to accelerating the interfacial charge transfer and separating photo-generated electron–hole pairs.35 Fig. 4d reveals the photoelectric conversion ability for pure g-C3N4, pure BiOBr and the B-20/CN electrodes. As is well-known, during the electron transfer from the CB of samples to the electrodes, the photocurrent is simultaneously generated. Generally speaking, the sample with a higher photocurrent signifies the presence of a longer lifetime of the photo-generated electrons and holes which enhances the photocatalytic activity.36 As is shown in Fig. 4d, high-frequency and consistent photocurrent responses are visibly observed for each switch-on and switch-off event in the electrodes. The photocurrent of the B-20/CN electrode is higher than those of the pure g-C3N4 and BiOBr electrodes. This conclusion is in accordance with the EIS analyses, and explicitly indicates that the introduction of BiOBr into g-C3N4 can effectively promote the separation of photogenerated electron–hole pairs.3.5. Photocatalytic activity
The photocatalytic activity of all the semiconductor photocatalysts was measured by degrading RhB under visible light irradiation. Before irradiation, the adsorption–desorption equilibrium between the RhB solution and photocatalyst was attained by magnetic stirring for 40 min under dark conditions. From Fig. 5a, it can be observed that the direct photolysis of RhB is negligible without the addition of a photocatalyst. Pure g-C3N4 possesses a relatively lower photocatalyst activity than those of the other BiOBr/g-C3N4 composites, and only 53.2% of RhB was degraded within 120 min. In addition, the pure BiOBr has a relatively higher adsorptive property and photocatalyst activity than those of pure g-C3N4 due to the special structure of the microspheres, and 59.9% of RhB was degraded within 120 min. With an incremental BiOBr content, the photocatalytic efficiency of the composites was gradually improved due to the formation of direct Z-scheme heterostructures and the resulting promotion of light absorption properties in the visible light range.37 The B-20/CN composite with an optimal photocatalytic efficiency can decompose 97.1% of RhB. Nonetheless, while the mass ratio of BiOBr was higher than 20%, the photocatalytic efficiency of B-25/CN decreased by 11.5% for the degradation of RhB. This phenomenon can be attributed to the excessive BiOBr covering the active sites of g-C3N4, leading to a low photocatalytic performance.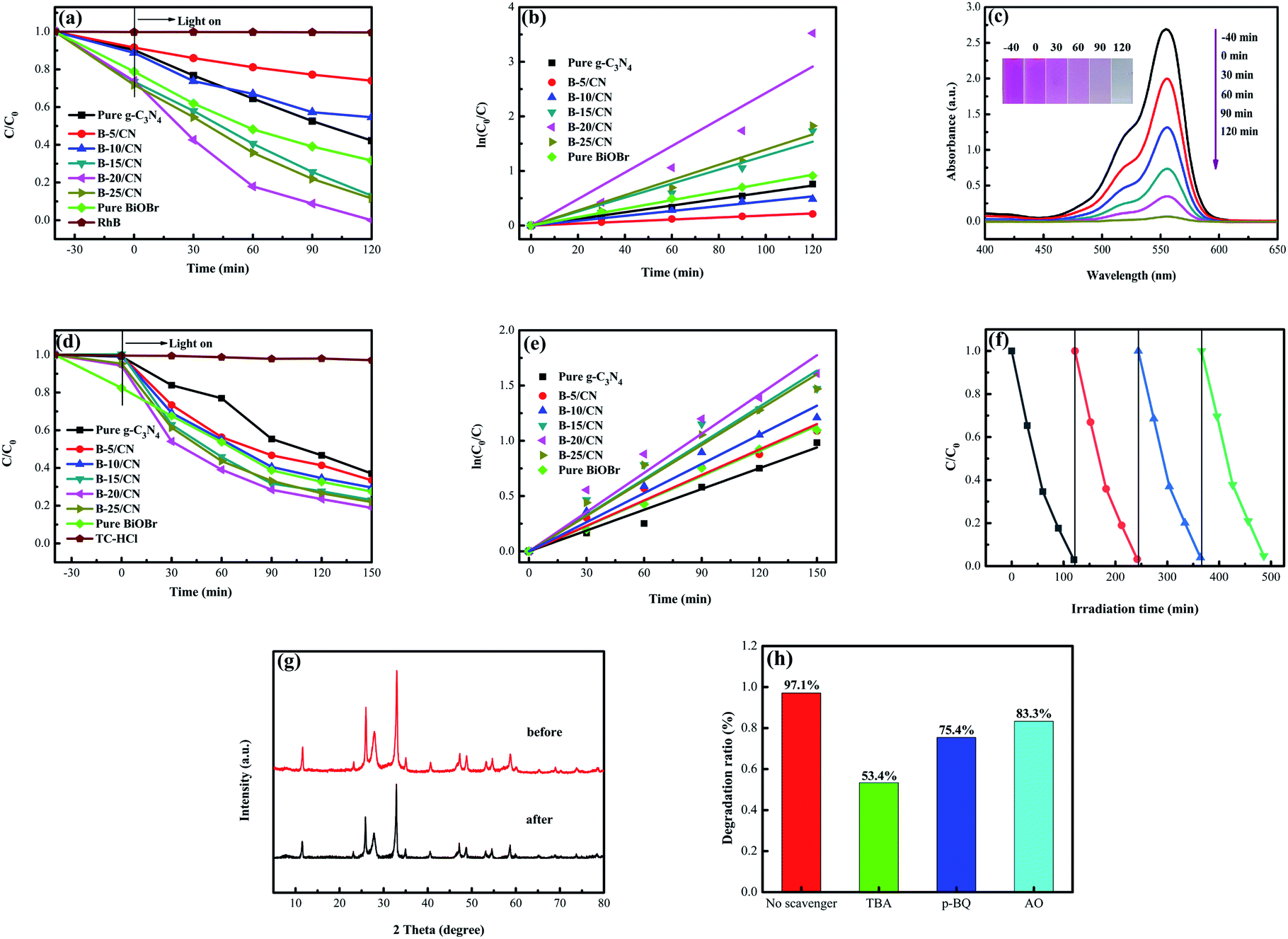 | ||
| Fig. 5 (a) Photocatalytic activity for the degradation of RhB; (b) the relative pseudo-first-order kinetic plots; (c) UV-vis spectral curves of RhB degradation over B-20/CN; (d) photocatalytic activity for the degradation of TC-HCl; (e) the relative pseudo-first-order kinetic plots of the as-prepared samples; (f) four recycling runs of B-20/CN for the degradation of RhB; (g) the XRD patterns of B-20/CN samples before and after the photocatalytic degradation experiments for RhB and (h) the effects of various scavengers on the degradation efficiency of B-20/CN toward RhB. | ||
Moreover, aiming to intensively research the kinetics curves of the photodegradation of RhB, the experimental data were applied to a pseudo-first-order model:
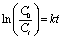 | (3) |
| Samples | Degradation rate (Vis) | |
|---|---|---|
| RhB (min−1) | TC-HCl (min−1) | |
| Pure g-C3N4 | 0.0061 | 0.0063 |
| Pure BiOBr | 0.0077 | 0.0075 |
| B-5/CN | 0.0018 | 0.0077 |
| B-10/CN | 0.0044 | 0.0088 |
| B-15/CN | 0.0128 | 0.0109 |
| B-20/CN | 0.0242 | 0.0118 |
| B-25/CN | 0.0139 | 0.0106 |
To shield against specific degradation of the photocatalysts, TC-HCl, regarded as an antibiotic residue, was applied to evaluate the photocatalytic efficiency of the as-prepared samples under visible light irradiation.39 From Fig. 5d, it can be observed that the direct photolysis of TC-HCl is negligible without the photocatalysts. Not surprisingly, the B-20/CN sample exhibits the highest photodegradation efficiency for TC-HCl, which can reach 81.1% within 150 min. With the increasing BiOBr content, the photocatalytic efficiency of the composites was gradually promoted compared with pure g-C3N4 and BiOBr. However, for the BiOBr content higher than 20%, a further augmentation of the BiOBr content leads to a decrease in the photocatalytic efficiency of TC-HCl degradation. The photodegradation process of TC-HCl accords with a pseudo-first order model (Fig. 5e). The degradation rate of B-20/CN is 1.87, 1.57, 1.53, 1.39, 1.08 and 1.11 times higher than those for pure g-C3N4, pure BiOBr, B-5/CN, B-10/CN, B-15/CN and B-25/CN, respectively (Table 1). The above conclusion testifies that the B-20/CN composite possesses a great potential to decompose antibiotics from the sewage of the pharmaceuticals industry.
As shown in Fig. 5f, in order to evaluate the stability of the as-prepared photocatalyst, B-20/CN was selected for a recycling test to degrade RhB under visible light irradiation. After four recycling runs, the degradation efficiency diminished from 97.1% to 95.3%, indicating that the B-20/CN composite exhibits an excellent photocatalysis stability. Furthermore, to further testify the stability of the B-20/CN photocatalyst, the XRD pattern after the photocatalytic degradation experiments for RhB was measured.35 The result (Fig. 5g) exhibits that the crystal structure did not change during the photocatalytic degradation of RhB. Hence, the B-20/CN composite has a remarkable stability and recyclability.
To illustrate the photocatalytic mechanism of the B-20/CN photocatalyst, the primary active groups generated in the photodegradation process were detected via an active-groups-trapping experiment. In this experiment, tertiary butanol (TBA), benzoquinone (p-BQ) and ammonium oxalate (AO) served as the scavengers for ·OH, ·O2− and h+, respectively.40 Fig. 5h demonstrates the effects of the three scavengers on the photodegradation process of RhB. As TBA, p-BQ and AO were added into the photocatalytic process, the photocatalytic degradation efficiency of B-20/CN was reduced from 97.1% to 53.4%, 75.4% and 83.3%, respectively. The results indicate that the ·OH, ·O2− and h+ play major roles in the degradation course of RhB.
3.6. Photocatalytic mechanism
According to the free radical trapping experiments, as well as optical and electrochemical analysis, a possible photocatalytic mechanism of BiOBr/g-C3N4 photocatalyst is elaborated in Fig. 6. Followed by BiOBr and g-C3N4 being stimulated under visible light irradiation, photoexcited electrons (e−) and holes (h+) are located in their VB and CB, respectively. For BiOBr/g-C3N4, the transfer process of the photo-generated of e− and h+ can be classified as a type II heterojunction structure (Fig. 6a).41 Chiefly, the VB potential of g-C3N4 (+1.48 eV) is more negative than that of BiOBr (+3.21 eV) and the CB potential of g-C3N4 (−1.02 eV) is more negative than that of BiOBr (+0.16 eV).42 Secondly, since the band gap of g-C3N4 (Eg = 2.50 eV) is narrower than that of BiOBr (Eg = 3.05 eV), the VB of g-C3N4 is likely to generate more carriers than BiOBr under visible light irradiation.43 For the above two reasons, the e− from the CB of g-C3N4 can transmit to the CB of BiOBr and the h+ from VB of BiOBr can simultaneously transfer to the VB of g-C3N4. As a result, the e− and h+ may respectively concentrate on the CB of BiOBr and the VB of g-C3N4, which effectually restrains the recombination of e− and h+. Nevertheless, during the migration of e−/h+ to more positive/negative energy levels, the reducing capacity of the photo-generated electrons and oxidizing capacity of the photo-generated holes will be subject to inevitable loss.44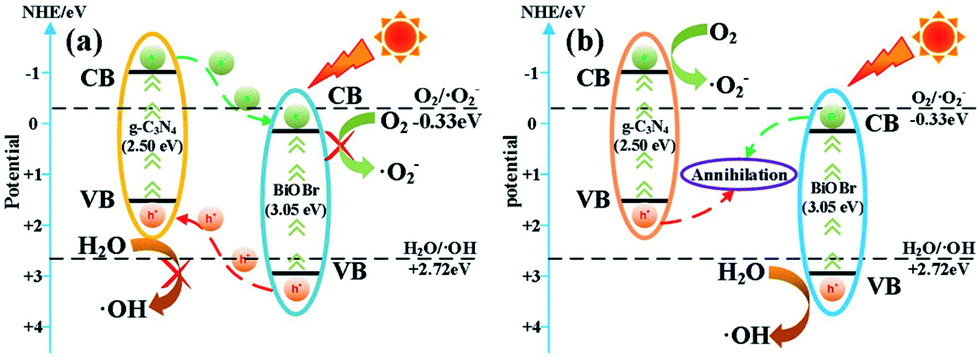 | ||
| Fig. 6 Schematic of charge carriers transfer in (a) BiOBr/g-C3N4 heterojunction and (b) direct Z-scheme BiOBr/g-C3N4 heterojunction. | ||
Obviously, in the direct Z-scheme BiOBr/g-C3N4 heterojunction structure (Fig. 6b), the contact surface between BiOBr and g-C3N4 serves as a charge transmission bridge to facilitate annihilation of electron–hole pairs with a weaker oxidation–reduction ability, which efficiently figures out the above deficiency.45,46 According to energy band theory, the photo-generated e− in the CB of BiOBr can quickly transfer to the contact surface through the Schottky barrier47,48 and recombine with h+ on the VB of g-C3N4, leaving the e−/h+ with a stronger oxidation-reduction ability in the CB/VB of g-C3N4/BiOBr. Moreover, the CB potential of g-C3N4 is more negative than E0(O2/·O2−), which means the e− on the CB of g-C3N4 can more easily reduce O2 to ·O2−.49,50 Meantime, the VB potential of BiOBr is more positive than E0(H2O/·OH), so the h+ in the VB of BiOBr is capable of oxidizing H2O to ·OH. The active groups, such as ·O2− and ·OH, are able to directly react with RhB to produce CO2, H2O or intermediate products.51,52 The procedure can be depicted as follows:
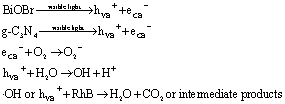
The deduction is well in accord with the above conclusion of the radical trapping experiments that the ·O2−, ·OH and h+ are the primary oxidation active groups for the degradation of RhB.
4. Conclusions
In summary, a direct Z-scheme BiOBr/g-C3N4 heterojunction system has been successfully constructed via a facile template-assisted hydrothermal method. Compared with pure g-C3N4 and BiOBr, the BiOBr/g-C3N4 composite with a BiOBr mass ratio of 20% (B-20/CN) exhibits a more remarkable photocatalytic performance for the degradation of RhB and TC-HCl. In such a direct Z-scheme system, the intimate synergistic interactions between BiOBr and g-C3N4, the significant extension of the light absorption edge, as well as the segregation of the strong oxidation–reduction capacity of e− and h+ lead to the remarkable photocatalytic activity of BiOBr/g-C3N4 composites. The trapping experiments manifest that the ·OH, ·O2− and h+ are the main active groups for the degradation of RhB. Moreover, the as-prepared B-20/CN composite exhibits excellent photostability and reusability after 4 runs under the same conditions. This work is supposed to open up new insights into the structural design of novel direct Z-scheme photocatalysts with a high photoactivity and further employment in the fields of the environment and energy.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was supported by the Natural Science Foundation of Shaanxi Province, China [grant no. 2020318029].References
- Z. Y. Ruan, G. H. Liu, J. X. Shu, C. G. Ren and Z. H. Wang, RSC Adv., 2019, 9, 5858–5864 RSC.
- S. D. Balgude, Y. A. Sethi, B. B. Kale, D. P. Amalnerkar and P. V. Adhyapak, RSC Adv., 2019, 9, 10289–10296 RSC.
- M. Sabet, S. Saeednia, M. Hatefi-Ardakani and R. Sheykhisarem, Nano-Struct. Nano-Objects, 2018, 13, 21–29 CrossRef CAS.
- N. Tian, H. W. Huang, Y. He, Y. X. Guo, T. R. Zhang and Y. H. Zhang, Dalton Trans., 2015, 44, 4297–4307 RSC.
- L. Cheng, Q. J. Xiang, Y. L. Liao and H. W. Zhang, Energy Environ. Sci., 2018, 11, 1362–1391 RSC.
- X. Li, J. G. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC.
- Z. F. Huang, L. Pan, J. J. Zou, X. W. Zhang and L. Wang, Nanoscale, 2014, 6, 14044–14063 RSC.
- L. L. Zhang, H. C. Zhang, H. Huang, Y. Liu and Z. H. Kang, New J. Chem., 2012, 36, 1541–1544 RSC.
- C. Wang, X. M. Wang, B. Q. Xu, J. C. Zhao, B. X. Mai, P. A. Peng, G. Y. Sheng and J. M. Fu, J. Photochem. Photobiol., A, 2004, 168, 47–52 CrossRef CAS.
- D. W. Jing and L. J. Guo, J. Phys. Chem. B, 2006, 110, 11139–11145 CrossRef CAS PubMed.
- M. C. Long, W. M. Cai, J. Cai, B. X. Zhou, X. Y. Chai and Y. H. Wu, J. Phys. Chem. B, 2006, 110, 20211–20216 CrossRef CAS PubMed.
- A. Petala, D. Spyrou, Z. Frontistis, D. Mantzavinos and D. I. Kondarides, Catal. Today, 2019, 328, 223–229 CrossRef CAS.
- Y. Zhang, J. Di, W. Tong, X. L. Chen, J. Z. Zhao, P. H. Ding, S. Yin, J. X. Xia and H. M. Li, Res. Chem. Intermed., 2019, 45, 437–451 CrossRef CAS.
- F. Cao, J. M. Wang, Y. N. Wang, J. Zhou, S. Li, G. W. Qin and W. Q. Fan, Nanoscale Adv., 2019, 1, 1124–1129 RSC.
- D. L. Huang, Z. H. Li, G. M. Zeng, C. Y. Zhou, W. J. Xue, X. M. Gong, X. L. Yan, S. Chen, W. J. Wang and M. Cheng, Appl. Catal., B, 2019, 240, 153–173 CrossRef CAS.
- S. Zhang, P. C. Gu, R. Ma, C. T. Luo, T. Wen, G. X. Zhao, W. C. Cheng and X. K. Wang, Catal. Today, 2019, 335, 65–77 CrossRef CAS.
- R. G. He, K. Y. Cheng, Z. Y. Wei, S. Y. Zhang and D. F. Xu, Appl. Surf. Sci., 2019, 465, 964–972 CrossRef CAS.
- Z. F. Jiang, W. M. Wan, H. M. Li, S. Q. Yuan, H. J. Zhao and P. K. Wong, Adv. Mater., 2018, 30, 1706108 CrossRef PubMed.
- A. Mishra, A. Mehta, S. Basu, S. Shetti and P. Nagaraj, Carbon, 2019, 149, 693–721 CrossRef CAS.
- M. J. Lima, A. M. T. Silva, C. G. Silva and J. L. Faria, J. Catal., 2017, 353, 44–53 CrossRef CAS.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- J. Fu, Y. L. Tian, B. B. Chang, F. N. Xi and X. P. Dong, J. Mater. Res., 2012, 22, 21159–21166 CAS.
- L. D. Cao, D. K. Ma, Z. L. Zhou, C. L. Xu, C. Cao, P. Y. Zhao and Q. L. Huang, Chem. Eng. J., 2019, 368, 212–222 CrossRef CAS.
- M. M. Zhang, C. Lai, B. S. Li, D. L. Huang, G. M. Zeng, P. Xu, L. Qin, S. Y. Liu, X. G. Liu, H. Yi, M. F. Li, C. C. Chu and Z. Chen, J. Catal., 2019, 369, 469–481 CrossRef CAS.
- P. Alam, P. Fagerlund, P. Hägerstrand, J. Töyrylä, S. Amini, M. Tadayon, A. Miserez, V. Kumar, M. Pahlevan and M. Toivakka, Compos. Appl. Sci. Manuf., 2015, 75, 84–88 CrossRef CAS.
- J. Zhang, J. H. Hu, D. T. Wu, J. F. Ma, Y. X. Tao, Y. Qin and Y. Kong, Int. J. Biol. Macromol., 2019, 129, 786–791 CrossRef CAS PubMed.
- D. G. He, X. X. He, K. M. Wang and Y. X. Zhao, Chin. Chem. Lett., 2013, 24, 99–102 CrossRef CAS.
- J. Li, Y. Yu and L. Z. Zhang, Nanoscale, 2014, 6, 8473–8488 RSC.
- J. J. Liu, B. Cheng and J. G. Yu, Phys. Chem. Chem. Phys., 2016, 18, 31175–31183 RSC.
- Y. Z. Hong, Y. H. Jiang, C. S. Li, W. Q. Fan, X. Yan, M. Yan and W. D. Shi, Appl. Catal., B, 2016, 180, 663–673 CrossRef CAS.
- M. S. Zhu, S. Y. Kim, L. Mao, M. Fujitsuka, J. Y. Zhang, X. C. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- Y. X. Chen, X. B. Ji, S. Vadivel and B. Paul, Ceram. Int., 2018, 44, 23320–23323 CrossRef CAS.
- J. F. Zhang, J. W. Fu, Z. L. Wang, B. Cheng, K. Dai and W. K. Ho, J. Alloys Compd., 2018, 766, 841–850 CrossRef CAS.
- Y. Bai, P. Q. Wang, J. Y. Liu and X. J. Liu, RSC Adv., 2014, 4, 19456–19461 RSC.
- Z. J. Xie, Y. P. Feng, F. L. Wang, D. N. Chen, Q. X. Zhang, Y. Q. Zeng, W. Y. Lv and G. G. Liu, Appl. Catal., B, 2018, 229, 96–104 CrossRef CAS.
- Y. M. He, L. H. Zhang, M. H. Fan, X. X. Wang, M. L. Walbridge, Q. Y. Nong, Y. Wu and L. H. Zhao, Sol. Energy Mater. Sol. Cells, 2015, 137, 175–184 CrossRef CAS.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J. M. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- Q. Yang, Z. Chen, X. R. Yang, D. T. Zhou, X. X. Qian, J. J. Zhang and D. Zhang, Mater. Lett., 2018, 212, 41–44 CrossRef CAS.
- R. G. He, J. Q. Zhou, H. Q. Fu, S. Y. Zhang and C. J. Jiang, Appl. Surf. Sci., 2018, 430, 273–282 CrossRef CAS.
- J. X. Low, J. G. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- R. Dingle, H. L. Störmer, A. C. Gossard and W. Wiegmann, Appl. Phys. Lett., 1978, 33, 665–667 CrossRef CAS.
- F. L. Liu, C. Huang, C. X. Liu, R. Shi and Y. Chen, Chem.–Eur. J., 2019 DOI:10.1002/chem.201904594.
- X. Q. Liu and L. Cai, Appl. Surf. Sci., 2018, 445, 242–254 CrossRef CAS.
- J. Yang, H. T. Zhang, B. B. Chen, H. Tang, C. S. Li and Z. Z. Zhang, RSC Adv., 2015, 5, 64254–64260 RSC.
- J. G. Yu, S. H. Wang, J. X. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- T. T. Jiang, J. L. Li, Y. Gao, L. Li, T. Lu and L. K. Pan, J. Colloid Interface Sci., 2017, 490, 812–818 CrossRef CAS PubMed.
- J. F. Zhang, Y. F. Hu, X. L. Jiang, S. F. Chen, S. G. Meng and X. L. Fu, J. Hazard. Mater., 2014, 280, 713–722 CrossRef CAS PubMed.
- P. F. Xia, B. C. Zhu, B. Cheng, J. G. Yu and J. S. Xu, ACS Sustain. Chem. Eng., 2017, 6, 965–973 CrossRef.
- W. K. Jo and T. S. Natarajan, Chem. Eng. J., 2015, 281, 549–565 CrossRef CAS.
- Q. L. Xu, L. Y. Zhang, J. G. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Mater. Today, 2018, 21, 1042–1063 CrossRef CAS.
- H. J. Li, W. G. Tu, Y. Zhou and Z. G. Zou, Adv. Sci., 2016, 3, 1500389 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |