
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReliability and relevance evaluations of REACH data†
Ellen
Ingre-Khans
 *a,
Marlene
Ågerstrand
a,
Anna
Beronius
b and
Christina
Rudén
a
*a,
Marlene
Ågerstrand
a,
Anna
Beronius
b and
Christina
Rudén
a
aDepartment of Environmental Science and Analytical Chemistry, Stockholm University, 106 91 Stockholm, Sweden. E-mail: ellen.ingre-khans@aces.su.se; Tel: +46 (0)8 6747337
bInstitute of Environmental Medicine, Karolinska Institutet, 171 77 Stockholm, Sweden
First published on 15th October 2018
Abstract
Regulatory authorities rely on hazard and risk assessments performed under REACH for identifying chemicals of concern and to take action. Therefore, these assessments must be systematic and transparent. This study investigates how registrants evaluate and report data evaluations under REACH and the procedures established by the European Chemicals Agency (ECHA) to support these data evaluations. Data on the endpoint repeated dose toxicity were retrieved from the REACH registration database for 60 substances. An analysis of these data shows that the system for registrants to evaluate data and report these evaluations is neither systematic nor transparent. First, the current framework focuses on reliability, but overlooks the equally important aspect of relevance, as well as how reliability and relevance are combined for determining the adequacy of individual studies. Reliability and relevance aspects are also confused in the ECHA guidance for read-across. Second, justifications for reliability evaluations were mainly based on studies complying with GLP and test guidelines, following the Klimisch method. This may result in GLP and guideline studies being considered reliable by default and discounting non-GLP and non-test guideline data. Third, the reported rationales for reliability were frequently vague, confusing and lacking information necessary for transparency. Fourth, insufficient documentation of a study was sometimes used as a reason for judging data unreliable. Poor reporting merely affects the possibility to evaluate reliability and should be distinguished from methodological deficiencies. Consequently, ECHA is urged to improve the procedures and guidance for registrants to evaluate data under REACH to achieve systematic and transparent risk assessments.
1. Introduction
Data on the properties and hazards of chemicals submitted under the European Chemicals Regulation REACH1 form the backbone for identifying chemicals of concern that are subject to regulation. Consequently, such data must be reliable and relevant for identifying and characterising potential hazards and risks.REACH requires manufacturers to register data for industrial chemicals produced within, or imported into the EU in volumes of ≥1 metric tonne per year before the chemical can be put on the European market. The data should show that the substance can be used in a safe way, i.e. that the risk is “adequately controlled”. The manufacturers and importers of chemicals, i.e. the registrants, should use all available and relevant information for identifying the hazardous properties of their substances (Art 12, REACH). Different information requirements apply, depending on the annual volume produced or imported (Annexes VI–XI to REACH). The required data are reported and submitted electronically in a registration dossier to the European Chemicals Agency (ECHA) through the software programme IUCLID. IUCLID provides a standard format for reporting, evaluating and submitting data on chemicals. Thus, original studies, referred to as “full study reports” under the REACH legislation, are summarised in study summaries.2Full study reports are comprehensive reports that are either generated by a test house, e.g. a contract laboratory, or data published in the literature, such as academic studies, (Art 3(27), REACH). The registration dossiers are processed by the ECHA and disseminated on their website.3
The (eco)toxicological data registered by industry must be evaluated for their adequacy, i.e. their appropriateness for the purpose of hazard and risk assessment.4 The adequacy of studies is reported under REACH as key, supporting, weight of evidence or disregarded study.2 If several studies are available for an endpoint, most weight is given to the most reliable and relevant studies, i.e. the study(ies) designated as key. Reliability refers to the inherent scientific quality of the study, whereas relevance refers to the study's appropriateness for identifying and characterising a certain hazard and/or risk.4
Several frameworks have been developed for evaluating reliability and to some extent relevance of (eco)toxicological studies for risk assessment.5,6 The Klimisch method is a common approach for evaluating data in regulatory settings and is also the method recommended under REACH.4 In the Klimisch method, studies are assigned to one of four reliability categories: (1) reliable without restriction, (2) reliable with restriction, (3) not reliable and (4) not assignable.4,7 Registrants should assign studies included in the registration dossier to one of the four reliability categories and provide a rationale for the reliability category.2
However, the choice of method used for evaluating data has been shown to influence the outcome of the assessment.8,9 For example, the Klimisch method has been criticised for lacking certain criteria as well as guidance for evaluating reliability and relevance and consequently requiring more expert judgment compared to other methods.8,9 Expert judgement is an inherent part of the risk assessment process and evaluations of data may as a result vary between experts due to different expertise and experience.10,11 It is therefore important that interpretation and evaluations of data are systematic and transparent and possible for third parties to scrutinise.
The Klimisch method has also been criticised for giving more weight to studies performed according to internationally validated and standardised test guidelines and Good Laboratory Practice (GLP) over non-standard and non-GLP studies.8,12 Studies performed according to GLP and standardised test guidelines are typically financed or carried out by industry since such tests are generally required for regulatory purposes.13,14 In contrast, academic research studies are rarely performed under the rules of GLP and strictly following test guidelines, but may nevertheless contribute with important information to the hazard and risk assessment.15–19 Consequently, studies that could be valuable to the hazard and risk assessment may be assigned lower weight in the assessment, or even dismissed, when relying on the Klimisch method.
The aim of this study was to investigate the procedures and guidance for evaluating data and reporting data evaluations in REACH and examine how registrants evaluate and report their evaluations. The study focused in particular on how data were assigned to reliability categories and how the reliability categories were justified by the registrant. Since the ECHA guidance recommends the Klimisch method for evaluating reliability of data, it was of particular interest whether studies complying with GLP and test guideline compliance and based on industry reports were assigned a higher reliability category than non-GLP and non-standard studies. The overall purpose was to clarify both the practice of evaluating data under the REACH regulation and the transparency of such evaluations thereby contributing to the development of systematic and transparent risk assessment procedures.
2. Methods
Data for analysis were retrieved from the REACH database3 during August 2015 for 60 substances registered in the first registration deadline under REACH. Information was extracted from registration dossiers for the endpoint repeated dose toxicity (RDT). Fifty one of the 60 substances were selected from a list provided by the German Federal Institute of Risk Assessment (BfR) investigating compliance of REACH registration dossiers.20 From the list, the first 17 substances in each of the three categories good, less than good and inferior were selected to include dossiers with varying quality. A further32 nine substances were included that were undergoing the authorisation and/or restriction processes in 2015. Information was extracted from the lead dossiers, i.e. dossiers that have been submitted jointly by the manufacturers and importers of the substance, except for four substances for which the dossiers had been submitted by only one manufacturer or importer. In total, 349 study summaries based on study report or publication were included for the 60 substances.It should be noted that the set of 60 substances included in the study is not expected to be fully representative of all substances registered under REACH, and it was not within the scope of this investigation to extrapolate to all substances (or all endpoints) registered with the ECHA (amounting to ∼21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 substances in August 20183). Instead, this was an explorative study, aiming at investigating and describing the procedures and guidance for evaluating data and reporting data evaluations in the REACH system.
000 substances in August 20183). Instead, this was an explorative study, aiming at investigating and describing the procedures and guidance for evaluating data and reporting data evaluations in the REACH system.
Data for the analysis were gathered from the following fields in the study summary: adequacy of data, reliability, rationale for reliability incl. deficiencies, GLP compliance and qualifier for the test guideline as well as reference type. The fields in the IUCLID template that constitute the study summary are either pick-lists with predefined options or free text fields. All of the abovementioned fields comprise pick-lists except for the field rationale for reliability incl. deficiencies which also contains a free text field. The information extracted from the fields is described in detail below. The data from the REACH registration database were collected in a Microsoft Access database and further analysed qualitatively, and quantitatively in Microsoft Excel.
The information in the field adequacy of data reflects how the study summary is used in the hazard assessment (key, supporting, weight of evidence and disregarded study). The option disregarded study is used for studies that are flawed but show critical results.2,21,22 Study summaries with no assigned adequacy by the registrant are referred to as not specified in this study.
In the field reliability, registrants select one of the Klimisch reliability categories 1 to 4. Study summaries with no assigned reliability category were excluded from further analysis, in total 18 study summaries. Registrants' justifications of the reliability category are provided in the field rationale for reliability incl. deficiencies. The field has a pick-list of standard justifications as well as a supplementary free text field for additional information.21
Information on whether the study has been conducted according to GLP and standardised test guidelines is provided in the fields GLP compliance and qualifier. GLP compliance can be reported as yes, yes incl. certificate, no and no data. Standard phrases in the field qualifier include according to, equivalent or similar to and no guideline followed. In the analysis, the options according to and equivalent or similar to have been grouped into one category test guideline compliance. Options that indicate that no guideline has been followed or not reported in the study or by the registrant in the dossier have been grouped into one category non-test guideline/no data. Similarly, studies that do not follow GLP or for which no information has been provided on GLP compliance have also been grouped into one category non-GLP/no data.
Information on the bibliographic reference is provided in the section data source. For the purpose of this study, study summaries based on the reference types study report, other company data and publication were extracted from the field reference type. The reference types study report and other company data refer to information generated or funded by the industry. These categories were combined and are hereafter referred to as study report. Publication refers to reports published in the peer reviewed literature and includes academic and governmental research as well as industry study reports published as scientific papers. Study summaries referring to both study report and publication were excluded in order to analyse the two reference types separately. Information on author, year, title and bibliographic source were also collected to ensure that publications could be identified if needed. However, the data source for study reports is not disseminated on the ECHA website and therefore the number of unique studies could not be identified.23 Since several study summaries can be prepared from a single report, the number of study summaries is not equivalent to unique studies and consequently, information for the same study may have been counted multiple times in the analysis.
3. Results
In total, 349 study summaries based on the reference type study report or publication were included for 60 substances (ESI Tables a–d†). In this section, a general description of the study summaries is first provided (section 3.1) followed by an analysis of how the assigned reliability category relates to the adequacy of the studies (section 3.2). The reliability categories assigned to the studies are then related to compliance with GLP and standardised test guidelines as well as the type of reference (section 3.3). Finally, an overall description of the registrants’ rationales of reliability required for justifying the assigned reliability category is provided (section 3.4).3.1. General description36
Study summaries were mostly assigned to reliability category 1 “reliable without restriction” (31%) or 2 “reliable with restriction” (48%) by the registrants (Table 1). Only 10% and 11% of the study summaries were assigned to reliability categories 3 “not reliable” and 4 “not assignable”, respectively.| Adequacy | Reliability category 1 | Reliability category 2 | Reliability category 3 | Reliability category 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study report | Publication | % | Study report | Publication | % | Study report | Publication | % | Study report | Publication | % | |
| Key | 77 | 6 | 78 | 26 | 19 | 27 | 0 | 0 | 0 | 0 | 0 | 0 |
| Supporting | 22 | 1 | 21 | 57 | 52 | 65 | 2 | 5 | 20 | 8 | 3 | 27.5 |
| Weight of evidence | 1 | 0 | 1 | 0 | 3 | 2 | 0 | 0 | 0 | 0 | 1 | 2.5 |
| Disregarded study | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 14 | 0 | 0 | 0 |
| Not specified | 0 | 0 | 0 | 4 | 6 | 6 | 2 | 21 | 66 | 15 | 13 | 70 |
| Total (reference type) | 100 | 7 | 100 | 87 | 80 | 100 | 5 | 30 | 100 | 23 | 17 | 100 |
| Total (study summary) | 107 (31%) | 167 (48%) | 35 (10%) | 40 (11%) | ||||||||
It should be noted that 85 of the 349 study summaries (24%) were registered for one substance, DEHP (ESI Table 5†). This influenced the variation in the dataset, particularly in reliability categories 3 and 4, where DEHP constituted 21/35 (60%) and 18/40 (45%) of all the study summaries, respectively. In reliability category 4, which comprised 13 substances in total, 28/40 or 70% of the study summaries were registered for two of the substances (including DEHP). However, in general, 1–5 study summaries were registered per substance in reliability categories 1 (39 of 43 substances) and 2 (38 of 45 substances) (ESI Table e†).
3.2. Relationship between the assigned reliability category and adequacy
Registrants can assign studies to one of four adequacy categories, which indicates how the registrant uses the study in the hazard assessment. The most frequently used adequacy categories among the study summaries included here were key and supporting (278/349 or 80%). Study summaries assigned to reliability category 1 (reliable without restriction) were mainly used as key studies in the hazard assessment (83/107 or 78%) (Table 1), whereas study summaries assigned to reliability category 2 (reliable with restriction) were to a greater extent used as supporting information (109/167 or 65%). Only 27% of study summaries assigned to reliability category 2 were used as key evidence. Thus, study summaries were more likely to be used as key evidence if assigned to reliability category 1 than 2.No adequacy was reported for 17% (61/349) of the study summaries, i.e. the registrant had not specified how these studies were used in the hazard assessment. Most of these (51/61 or 84%) were assigned to reliability categories 3 and 4.
3.3. Relationship between the assigned reliability category and GLP and test guideline compliance
The following section presents to what extent study summaries assigned to the various reliability categories constituted GLP and test guideline studies and whether the study summaries were based on a study report or a publication. Since the Klimisch method is the recommended method to use for evaluating data under REACH, studies performed according to GLP and test guidelines were anticipated to be assigned to a higher reliability category and mainly constitute data from study reports.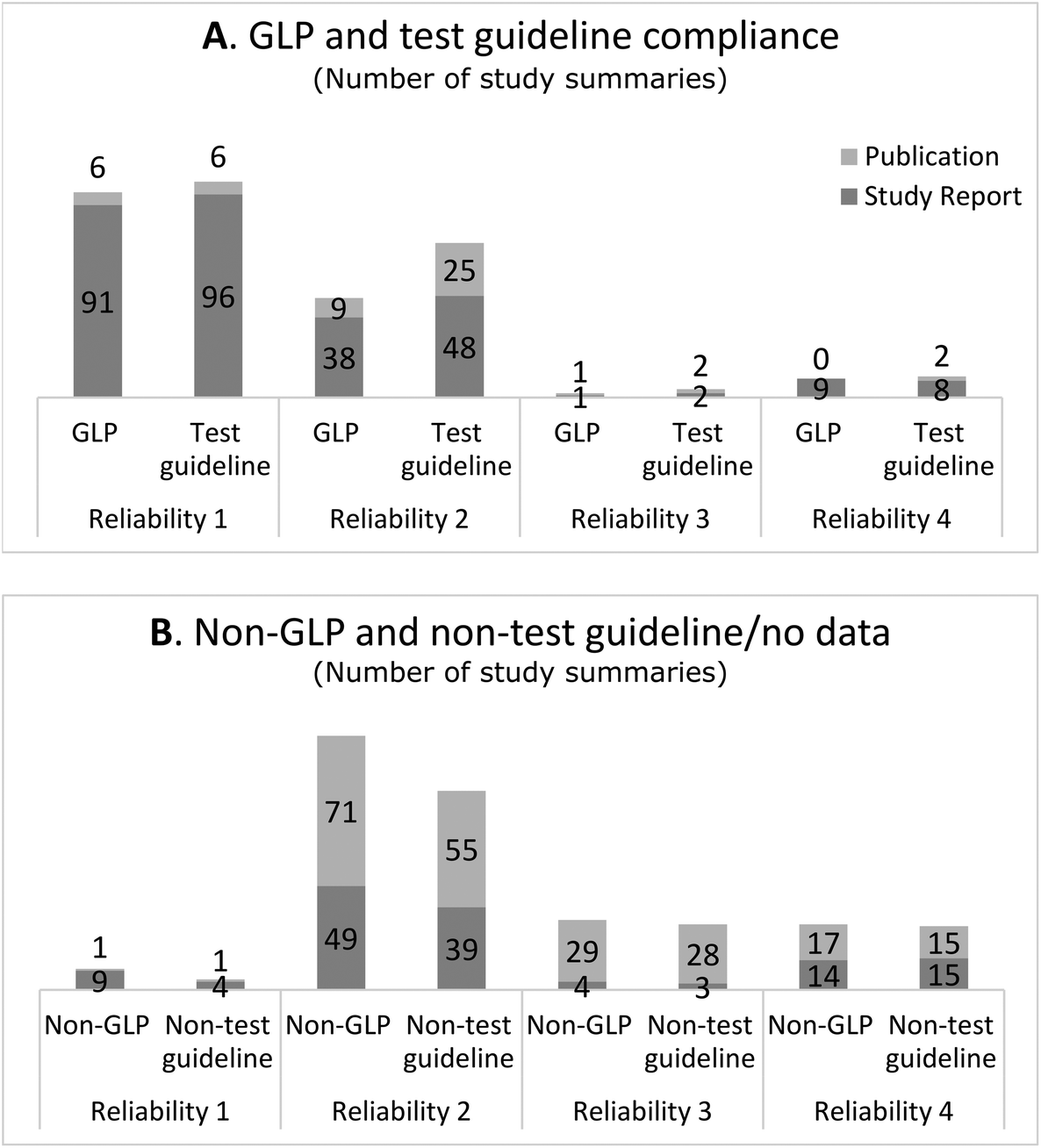 | ||
| Fig. 1 A. Number of study summaries based on the study report and publication reported to follow GLP and test guidelines. The categories include the pick-list options “according to” and “equivalent or similar to” test guidelines. B. Number of study summaries reported to not follow GLP and test guidelines or for which no information on GLP and test guideline compliance has been reported by the registrant. For GLP, this includes specifically the pick-list options “no GLP” or “no data” (i.e. GLP compliance not reported in the full study report) and for test guidelines “no guideline followed/required/available”. The distinctions within the GLP and test guideline categories are provided in ESI Table f.† | ||
In total, 7/107 or 7% of the study summaries assigned to reliability category 1 were based on a publication. Six of these were used as key studies and reported to conform to GLP and test guidelines (Fig. 1), which suggests that they were published industry reports.13,14 The bibliographic reference could only be identified for two of these study summaries which confirmed that they were based on an industry report (both referred to the same data source). Only one of the study summaries in reliability category 1 was identified as an academic research study, which was used as supporting evidence (Fig. 1). The academic research study did not mention compliance with GLP or test guidelines.
Study summaries assigned to reliability 2 were based on study report and publication to roughly the same extent, 52% and 48%, respectively (Table 1). Regardless of the type of data source, study summaries assigned to reliability category 2 were generally used as supporting information and not as key studies (Table 1).
Most of the study summaries assigned to reliability category 3 were based on publication (30/35 or 86%). Study summaries assigned to reliability category 4 were based on study reports and publications to about the same extent: 58% and 42%, respectively (Table 1).
3.4. Registrants’ rationales for reliability
The following section describes the registrants’ justifications for assigning a study to a certain reliability category, which is provided in the field rationale for reliability incl. deficiencies (ESI Tables a–d†). Some general observations regarding the rationales are given followed by an analysis of the justifications for each reliability category.The justifications were observed to differ in some aspects. First, the amount of text in this field varied from two to three words to full sentences and paragraphs. For three study summaries, no justification or text was provided in the field.
Second, the rationales for reliability ranged from generic statements, such as “acceptable for assessment”, “insufficient documentation for assessment”, “meets scientific principles” and “basic data given”, to more specific statements related to limitations in reporting or methodology for that particular study, such as “no purity” and “insufficient number of animals” (Table 2 and ESI Tables a–d†).
| Reliability category | No. of rationales | Description of rationales |
|---|---|---|
| 1 (Reliable without restriction) | 107 | Rationale for reliability explicitly stated in 19 rationales (18%) |
| Restrictions/deficiencies mentioned in 9 rationales, and specified in 1 rationale | ||
| Typical statements: | ||
| [According to/closely adhere to] GLP and/or guideline study (105 rationales) | ||
| The only information in 70 rationales | ||
| Other common statements: | ||
| “Well-documented” | ||
| “Scientifically acceptable/sound” | ||
| “Acceptable scientific principles” | ||
| “Fully adequate for assessment” | ||
| 2 (Reliable with restriction) | 167 | Rationale for reliability explicitly stated in 18 rationales (11%) |
| Restrictions/deficiencies mentioned in 80 rationales, and specified in 47 rationales | ||
| Typical statements: | ||
| [According to, equivalent, comparable similar, near] GLP and/or guideline study (68 rationales) | ||
| [Prior to or non] GLP and/or guideline study (20 rationales) | ||
| GLP and/or guideline study the only information in 16 rationales | ||
| Other common statements: | ||
| “Well-documented” | ||
| “Acceptable for assessment” | ||
| “Meets generally accepted scientific standards” | ||
| “Meets basic scientific principles” | ||
| 3 (Not reliable) | 35 | Rationale for reliability explicitly stated in 3 rationales (9%) but in general the reasons were implicitly understood |
| Restrictions/deficiencies mentioned in 33 rationales, and specified in 20 rationales | ||
| Typical statements: | ||
| “Methodological deficiencies” (10 rationales) | ||
| “Insufficient documentation/data” (16 rationales) | ||
| 4 (Not assignable) | 40 | Rationale for reliability explicitly stated in 3 rationales (8%), but in general the reasons were implicitly understood |
| Typical statements: | ||
| “Insufficient documentation/data” | ||
| “Only abstract available” | ||
| “Documentation not available” (referring to EU RAR and TSCATS) | ||
Third, in some cases the rationales contained information concerning relevance, completeness or general informative aspects of the study. This included registrants stating that the study focused on a particular endpoint, did not cover the full scope of a guideline study, was published in peer reviewed literature and was a probe study (i.e. studies performed to find appropriate dose ranges in subsequent toxicity studies). When the rationale contained several statements and information not considered related to reliability, it was difficult to judge what the reliability assessment and the assigned reliability category was based on. The reason for assigning a study a certain reliability category was only explicitly stated in 43/349 study summaries (12%) by stating “this study is classified as reliability category × because…” or similar in the rationale (ESI Tables a–d†). The reason for the reliability category was less clear for studies assigned to reliability category 2 than for the other categories since they comprised more diverse statements.
Reliability category 1 should be assigned to studies that are considered reliable without restriction. Nevertheless, for nine study summaries assigned to reliability category 1, the registrant stated possible or minor restriction, deficiency or deviation of the study in the rationale. However, only for one of the study summaries this was further specified in the rationale, which concerned a deviation from the test guidelines. In seven of the rationales indicating a restriction, the registrant stated either “possibly” or “no or minor” deviations or deficiencies although not “affect[ing] the quality of the results”. These possible or minor deviations or deficiencies were not further specified other than it could concern incomplete reporting, methodological deficiencies or deviations from standard guidelines. Two rationales included statements regarding the scope of the study and it is unclear whether this was considered as a restriction by the registrant (“but the study scope does not cover a full OECD study” and “guideline adapted to the needs of a shorter study duration (subchronic to subacute)”).
Some rationales included information that was difficult to judge whether the registrant considered it to influence the reliability assessment or if it was merely included as supporting information. This included statements such as “preferred study for this SIDS endpoint” (Screening Information Dataset), “taken from EU RAR” (European Union Risk Assessment Report), “range-finding study/probe study”, “available as unpublished report”, “study conducted to investigate specific endpoints”, “publication with summarized results” and “published in peer reviewed literature”.
The reason for assigning the study to reliability category 2 was explicitly stated for 18/167 study summaries (ESI Table b†). In most cases, the reliability was justified by stating “GLP-compliant but not conforming to test guidelines” or vice versa. Some justifications are of interest to highlight further. For example, one rationale stated that the study was carried out according to test guidelines and following GLP but that the NOAEL (No observed adverse effect level) was questionable due to limitations in the study, although not further specified. This pinpoints that the result of a study is not necessarily considered reliable even though it has been performed according to GLP and test guidelines.
Another study was assigned to reliability category 2 because the study was performed prior to the implementation of GLP and did not follow OECD guidelines. However, the registrant also stated in the rationale that an audit did not identify any problems with the study and that studies conducted under the U.S. National Toxicology Program generally follow current guidelines. This rationale indicates how stating GLP and test guideline compliance seems to be more important for the resulting reliability category than the study's inherent scientific quality. The registrant also stated that the study was assigned to reliability category 2 because the complete study report was publicly available. It is, however, unclear how the registrant considered this to affect the reliability of the study.
Read-across was stated to be one of the reasons for assigning the study to reliability category 2 for four study summaries. One of the rationales stated that the ECHA guidance recommends assigning read-across to reliability category 2. This is misleading since read-across is a relevance aspect and is not related to the reliability of the study. In read-across, the properties of the registered substance is predicted based on data from a similar substance.
One rationale stated “reliability from SIDS summary”. SIDS is a data set that was required under the High Production Volume Chemicals Programme on existing chemicals.24 This rationale was interpreted to mean that the registrant used the reliability evaluation from the SIDS summary. If a registrant agrees with an evaluation of the study performed in a previous assessment, it could still be argued that the justification should be provided in the dossier for transparency reasons.
GLP and/or guidelines or national standard methods were mentioned in 88 out of 167 rationales of the studies assigned to reliability category 2 (Table 2 and ESI Table b†). In 77% of these rationales (68/88), studies were stated to be similar or according to the guideline study and/or GLP, or not GLP but guideline. This was more commonly stated for study summaries based on study reports than publication.
In 20 out of 88 rationales, the study was stated to be conducted prior to GLP and test guidelines (mostly based on study reports) or non-GLP and/or non-guideline study” (mostly based on publication) (Table 2 and ESI Table b†). In 16 rationales, compliance with GLP and/or test guideline was the only information provided as justification. Apart from GLP and test guideline compliance, other common statements in the justification field included “well documented”, “acceptable for assessment”, “meets generally accepted scientific standards”, “meets basic scientific principles” or similar (Table 2 and ESI Table b†).
For 80/167 (48%) study summaries assigned reliability category 2, some types of restrictions, deficiencies or deviations from guidelines were either explicitly or implicitly stated in the rationale. The restriction was further specified in 46/80 rationales or 58%. Restrictions or deficiencies included not following GLP or test guidelines due to excluding test parameters, such as urinalysis or full histopathology, using few exposure concentrations or a lower top dose than recommended and reporting no purity. Rationales stating a restriction without specification were expressed as “with [acceptable] restrictions”, “limitations in design and/or reporting”, “limited experimental detail” or similar.
GLP and guidelines were mentioned in 4/35 rationales. One study based on a study report was judged to be “comparable to” the guideline study and “mainly GLP” but the test substance did not correspond to the registered substance, which is related to relevance rather than reliability. Another study was reported to deviate from a specified guideline and conducted prior to GLP was made mandatory in addition to other restrictions specified in the rationale (based on study report). Two studies based on publication were stated to be “not according to GLP nor to specific testing guideline” in addition to the statements “insufficient data for assessment” and “not reliable”.
GLP and guideline/standard study was mentioned in three rationales. Two of the rationales stated that the study was a “non-standard study” or “conducted prior to GLP and guideline” (in addition to providing few data on the test material). In the third rationale, the study was stated to be a GLP and guideline study but with limited documentation on histopathology.
4. Discussion
Evaluation of reliability and relevance of data is a critical step in chemical risk assessment that can influence the final conclusion on hazards and risks. Since chemical risk management decisions are based on REACH registration data, these evaluations need to be systematic and transparent. However, the system for evaluating data under REACH as well as the format for reporting these evaluations in IUCLID is inadequate for supporting systematic and transparent evaluations of (eco)toxicity data. Four major issues were identified:(1) The current framework focuses mainly on reporting and evaluating reliability, thereby overlooking the relevance aspect. In addition, the ECHA guidance confuses relevance aspects with reliability for read-across studies.
(2) The reliability evaluations follow the Klimisch method, which does not promote a systematic and transparent evaluation of data and is likely to favour studies conducted in compliance with GLP and standardised test guideline.
(3) The rationales for reliability provided by registrants were not always clear.
(4) Poor reporting of a study was sometimes confused with poor quality when evaluating studies as not reliable.
4.1. Procedures for evaluating data under REACH
Registrants must evaluate reliability as well as relevance when assessing the adequacy of studies. However, IUCLID only has a designated field for reporting the reliability evaluation in the study summary and no corresponding field for relevance.4 As a possible consequence, relevance aspects were sometimes reported in the reliability field. This included aspects such as the test substance was similar to the registered substance, focus on a particular endpoint, and whether the route of exposure in a study compared with human exposure. Furthermore, there is no field for reporting how the registrant has weighed and combined reliability and relevance to conclude on the adequacy of the study even though this is crucial for understanding how data have been selected and used in the hazard and risk assessment. Adequacy is merely reported through a pick-list field with the options key, supporting, weight of evidence or disregarded study.Furthermore, there seems to be no adequacy option for indicating studies that have been excluded from the risk assessment, i.e. studies that have been considered neither reliable nor relevant for hazard and risk assessment. For clarity, it can be important to know whether data have been considered in the risk assessment process and for what reasons the studies have been omitted. According to ECHA guidance, the adequacy option “disregarded study” should be used for studies that are flawed but show critical results, i.e. low reliability but relevant.2,21,22 The lack of a suitable label for such studies may explain why no adequacy was reported for 18 study summaries.
In four cases, the registrants assigned studies to reliability category 2 due to read-across, which involves using data from a similar substance to predict the properties of the registered substance. In one rationale, this was supported by referring to guidance provided by the ECHA. Indeed, ECHA guidance states that studies used for read-across purposes can at most be assigned reliability category 2 to reflect the uncertainty in assuming that the substances have similar properties.26,27 However, this is confusing since similarity of substances is related to relevance and not to reliability. Reliability and relevance are two separate aspects that should be distinguished for the sake of transparency, although we recognise that making this distinction is not always clear-cut.15
The terminology in the field of evaluating data for risk assessment is not standardised, which can lead to confusion. For example, ECHA guidance states that evaluating data quality under REACH involves assessing adequacy, reliability and relevance.4,22 However, using the term “data quality” while referring to the process of evaluating reliability as well as relevance can be confusing. Reliability is generally defined as the “inherent quality” of a study and therefore quality is sometimes used interchangeably with reliability.
4.2. Reliability evaluation method
The choice of method used for evaluating data reliability may significantly affect how and what type of data are considered adequate for the risk assessment.8,9 As seen in this investigation, the majority of the studies assigned to reliability category 1 were used as key studies compared to studies assigned to reliability category 2, which were mainly used as supporting evidence. Thus, the resulting reliability evaluation appears to matter for how data were used in the risk assessment, which makes it important to consider what criteria are used for evaluating reliability and assigning studies to the different reliability categories.The recommendation to use the Klimisch method likely contributed to registrants assigning GLP and test guideline studies to a higher reliability category. Standardised test guidelines were developed to produce reliable and reproducible results, but merely stating that a study complies with such guidelines does not ensure that the study is reliable. The same applies to GLP, which is a system for documentation and following certain procedures. Such studies may still be flawed in how the study is designed, conducted or in how the results are interpreted.28–30 Standardised test guidelines also differ in how much they allow for flexibility in the study design.31–33 Thus, studies must be systematically evaluated based on their inherent scientific quality. This applies to GLP and/or guideline studies as well as non-GLP and non-standardised studies. Since neither the Klimisch method nor ECHA guidance provides clearly defined criteria for evaluating reliability, methodological as well as reporting flaws can easily be overlooked.9 Lack of criteria and guidance for evaluating studies also requires expert judgment to a higher degree, which can result in inconsistent evaluations.15
Studies assigned to reliability category 1 were furthermore almost exclusively based on study reports, i.e. industry reports, which is not surprising since industry financed studies must generally comply with GLP and standardised test guidelines to fulfill regulatory requirements.13,14 According to ECHA guidance, studies can be assigned to reliability category 1 if they conform to “generally accepted scientific standards” and are “described in sufficient detail”.4,21 However, only one study summary assigned to reliability category 1 was not conducted according to GLP and test guidelines and based on a publication. The majority of the study summaries referring to a publication were assigned to reliability category 2. Thus, reliability criteria based on GLP and test guideline compliance may result in GLP and test guideline studies being considered reliable by default while giving less weight to peer-reviewed studies. This contradicts the requirement under REACH to include all available and relevant information in the risk assessment, Art. 12, REACH.
It should be acknowledged that study summaries based on publication could be academic research studies as well as published industry studies and publications could be assigned to reliability category 2 for other reasons. The category may accurately reflect restrictions in the study due to, for example, outdated test designs. Insufficient reporting of information required for evaluating the study may also be perceived to affect the quality of the study. The documentation requirements of GLP could contribute to GLP and test guideline studies to be considered reliable to a greater extent than published studies. Underreporting of peer-reviewed papers has been extensively discussed in the scientific community and has resulted in reporting guidelines for academic (eco)toxicological studies to improve their reproducibility and use in regulatory processes.28,34,35 Despite these efforts, improvement in the reporting of peer-reviewed studies has been proved to be slow.36 Further actions have been taken to raise academic researchers’ awareness of regulatory processes and their requirements on data37 as well as to encourage scientific journals to introduce reporting requirements in the peer-review process.38
4.3. Registrants’ rationale for reliability
The rationale for reliability provided by the registrant should presumably relate to the reliability assessment and ideally provide an unambiguous reason for assigning the study to a certain reliability category. However, the reliability justifications were frequently observed to be vague, confusing and lack information necessary information for understanding the assessment. For example, the rationales typically contained statements, such as “well documented” and “scientifically acceptable”, that are vague and influenced by the evaluator's expertise and experience. The reporting system contributes to rationales being broad and unspecific since the rationale for the reliability field consists of a pick-list with general statements as well as a free-text field.21 Consequently, any broad statement selected by the registrant from the pick-list needs to be further specified in the free-text field for clarity.For some rationales, it was difficult to determine to what extent the information in the rationale was part of the reliability assessment of the study. This was particularly the case for rationales with a diverse content, as for studies assigned to reliability category 2, and where the information was seemingly not related to reliability, but rather to relevance, compliance with REACH requirements or other miscellaneous information. Only in some rationales were the reasons for the reliability categorisation explicitly stated.
Rationales were also found to lack important information required for understanding the registrant's reasoning and to scrutinise the assessment. For example, restrictions in reliability were only clearly specified for one fourth and two thirds of the studies that were assigned to categories 2 and 3, respectively. Deficiencies or restrictions in reliability need to be explicitly stated since experts may judge how this influences reliability differently, which affects how the study is used in the risk assessment. Interestingly, restrictions in reliability were also mentioned for some studies assigned to reliability category 1 that are supposed to be reliable without restrictions. Although these restrictions were stated not to affect the quality of the results, these should also be specified to enable a third party to review the assessment.
Rationales for studies assigned to reliability category 4 were also lacking more detailed information. The assigned category was generally justified by not having access to the full study reports (“documentation not available”) or not providing sufficient experimental information on the study. However, what information is required for evaluating the reliability of the study needs to be further specified. Some information will inevitably be considered more critical than other for evaluating reliability.
4.4. Poor reporting vs. poor quality
In some rationales, we could also see that registrant considered insufficient documentation to influence the reliability of the study, thereby assigning the study to reliability category 3 “not reliable”. However, poor documentation or reporting is not equivalent to poor study design, but rather affects the possibility for the evaluator to judge the reliability of the study. There could be a possibility of retrieving more information necessary for evaluating its reliability by for example contacting the authors of the study. Therefore, it would be more appropriate to assign studies that are not sufficiently documented to reliability category 4 “not assignable”, unless the study would be considered unreliable for methodological reasons.16,284.5. Limitations of the study
This analysis was based on 349 study summaries registered for the lead dossier and the endpoint repeated dose toxicity for a total of 60 substances. This only represents a small number of the ∼21000 substances registered under REACH (August 2018).3 To what extent the results for the selected dossiers and endpoint are relevant to other dossiers and endpoints is not known and needs to be confirmed. The ratio of study summaries based on publications or study reports could differ for another endpoint or another chemical. For example, more data were registered for the substance DEHP than for any of the other substances, which also had a high proportion of study summaries based on publication.
Another limitation of the study is that the analyses were based on each study summary. Several study summaries can refer to the same study, which could have resulted in information being counted and included more than once. The data source for study reports is not disseminated due to personal data protection and individual studies cannot therefore be identified.
Finally, analysing the information in the field rationale for reliability incl. deficiencies is a matter of interpretation, since it is a free text field and the explicitness of the information may vary. Any interpretation of the text, for example as a restriction or deficiency, is reported in the supporting information for transparency.
5. Conclusions
The lack of systematic and transparent procedures for evaluating and reporting data evaluations under REACH may result in registration data not being satisfactorily evaluated and limits the possibilities for third parties to understand and scrutinise the assessments. This is disconcerting considering that REACH registration data are used by regulatory authorities to identify hazardous chemicals and take appropriate risk management measures. Therefore, the ECHA is strongly recommended to revise their guidance and procedures for registrants to evaluate and report data evaluations under REACH to ensure that data are systematically evaluated and improve transparency.Abbreviations
| BfR | German federal institute for risk assessment (Bundesinstitut für Risikobewertung) |
| DEHP | Di-ethyl hexyl phthalate |
| ECHA | European CHemicals Agency |
| EU RAR | European Union Risk Assessment Report |
| GLP | Good laboratory practice |
| IUCLID | International Uniform Chemical Information Database |
| OECD | Organisation for Economic Co-operation and Development |
| RDT | Repeated dose toxicity |
| REACH | Registration, Evaluation, Authorisation and restriction of CHemicals |
| ESI | Electronic supplementary information |
| SIDS | Screening information dataset |
| TSCATS | Toxic Substances Control Act Test Submissions database |
Conflicts of interest
Christina Rudén was a member of ECHA's management board between 2012 and 2017. However, ECHA has not been involved in funding nor taking part in this research project. The other authors declare no conflict of interest.Acknowledgements
This project was funded by Stockholm University faculty grants.References
- EC, Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC.
- ECHA, IUCLID 5. End-user Manual, 2013. https://iuclid6.echa.europa.eu/archive-iuclid-5 Search PubMed.
- ECHA, REACH Registration database, https://echa.europa.eu/sv/information-on-chemicals/registered-substances, accessed August 1, 2018 Search PubMed.
- ECHA, Evaluation of available information. Version 1.1, in Guidance on information requirements and chemical safety assessment, 2011, ch. R.4. https://echa.europa.eu/guidance-documents/guidance-on-reach Search PubMed.
- N. Roth and P. Ciffroy, A critical review of frameworks used for evaluating reliability and relevance of (eco)toxicity data: Perspectives for an integrated eco-human decision-making framework, Environ. Int., 2016, 95, 16–29 CrossRef CAS PubMed.
- G. O. Samuel, S. Hoffmann, R. A. Wright, M. M. Lalu, G. Patlewicz, R. A. Becker, G. L. DeGeorge, D. Fergusson, T. Hartung, R. J. Lewis and M. L. Stephens, Guidance on assessing the methodological and reporting quality of toxicologically relevant studies: A scoping review, Environ. Int., 2016, 92–93, 630–646 CrossRef CAS PubMed.
- H. J. Klimisch, M. Andreae and U. Tillmann, A systematic approach for evaluating the quality of experimental toxicological and ecotoxicological data, Regul. Toxicol. Pharmacol., 1997, 25, 1–5 CrossRef CAS PubMed.
- M. Ågerstrand, M. Breitholtz and C. Rudén, Comparison of four different methods for reliability evaluation of ecotoxicity data: a case study of non-standard test data used in environmental risk assessments of pharmaceutical substances, Environ. Sci. Eur., 2011, 23, 17 CrossRef.
- R. Kase, M. Korkaric, I. Werner and M. Agerstrand, Criteria for Reporting and Evaluating ecotoxicity Data (CRED): comparison and perception of the Klimisch and CRED methods for evaluating reliability and relevance of ecotoxicity studies, Environ. Sci. Eur., 2016, 28, 7 CrossRef PubMed.
- C. Rudén, Doctoral thesis, From data to decision. A case study of controversies in cancer risk assessment, Karolinska Institute, 2002 Search PubMed.
- B. Wandall, Values in science and risk assessment, Toxicol. Lett., 2004, 152, 265–272 CrossRef CAS PubMed.
- L. Molander, M. Ågerstrand, A. Beronius, A. Hanberg and C. Rudén, Science in Risk Assessment and Policy (SciRAP) – An Online Resource for Evaluating and Reporting In Vivo (Eco) Toxicity Studies, Hum. Ecol. Risk Assess., 2015, 21, 753–763 CrossRef CAS.
- OECD, More about OECD Test Guidelines, http://www.oecd.org/env/ehs/testing/more-about-oecd-test-guidelines.htm, accessed April 6, 2018 Search PubMed.
- ECHA, Guidance on Registration. Guidance for the implementation of REACH, Version 2.0, 2012. https://echa.europa.eu/guidance-documents/guidance-on-reach Search PubMed.
- C. Rudén, J. Adams, M. Ågerstrand, T. C. Brock, V. Poulsen, C. E. Schlekat, J. R. Wheeler and T. R. Henry, Assessing the relevance of ecotoxicological studies for regulatory decision making, Integr. Environ. Assess. Manage., 2017, 13, 652–663 CrossRef PubMed.
- C. Moermond, A. Beasley, R. Breton, M. Junghans, R. Laskowski, K. Solomon and H. Zahner, Assessing the reliability of ecotoxicological studies: An overview of current needs and approaches, Integr. Environ. Assess. Manage., 2017, 13, 640–651 CrossRef PubMed.
- J.-P. Bourguignon, R. Slama, Å. Bergman, B. Demeneix, R. Ivell, A. Kortenkamp, G. Panzica, L. Trasande and R. T. Zoeller, Science-based regulation of endocrine disrupting chemicals in Europe: which approach?, Lancet Diabetes Endocrinol., 2016, 4, 643–646 CrossRef PubMed.
- N. B. Hartmann, M. Ågerstrand, H.-C. H. Lützhøft and A. Baun, NanoCRED: A transparent framework to assess the regulatory adequacy of ecotoxicity data for nanomaterials – Relevance and reliability revisited, NanoImpact, 2017, 6, 81–89 CrossRef.
- R. E. Alcock, B. H. MacGillivray and J. S. Busby, Understanding the mismatch between the demands of risk assessment and practice of scientists - The case of Deca-BDE, Environ. Int., 2011, 37, 216–225 CrossRef CAS PubMed.
- BfR, REACH Compliance: Data Availability of REACH Registrations. Part 1: Screening of chemicals >1000 tpa, 2015 Search PubMed.
- OECD, OECD Template #67: Repeated dose toxicity: oral (Version [5.17]-[April 2016]), 2016. http://www.oecd.org/ehs/templates/harmonised-templates-health-effects.htm Search PubMed.
- ECHA, Practical Guide 3: How to report robust study summaries. Version 2.0, 2012. https://echa.europa.eu/practical-guides Search PubMed.
- ECHA, Dissemination and confidentiality under REACH regulation, 2016. https://echa.europa.eu/manuals Search PubMed.
- OECD, OECD Cooperative Chemicals Assessment Programme (CoCAP), http://www.oecd.org/chemicalsafety/risk-assessment/oecdcooperativechemicalsassessmentprogramme.htm, accessed August 14, 2017 Search PubMed.
- USEPA, Toxic substances control act test submissions (TSCATS), https://cfpub.epa.gov/si/si_public_record_Report.cfm?dirEntryId=2855, accessed April 14, 2018 Search PubMed.
- ECHA, Practical guide 6 - How to report read-across and categories, 2012. https://echa.europa.eu/practical-guides Search PubMed.
- ECHA, Practical Guide: How to use alternatives to animal testing to fulfil your information requirements for REACH registration. Version 2.0, 2016. https://echa.europa.eu/practical-guides Search PubMed.
- C. T. A. Moermond, R. Kase, M. Korkaric and M. Ågerstrand, CRED: Criteria for reporting and evaluating ecotoxicity data, Environ. Toxicol. Chem., 2016, 35, 1297–1309 CrossRef CAS PubMed.
- J. P. Myers, F. S. vom Saal, B. T. Akingbemi, K. Arizono, S. Belcher, T. Colborn, I. Chahoud, D. A. Crain, F. Farabollini, L. J. Guilleffe, T. Hassold, S. M. Ho, P. A. Hunt, T. Iguchi, S. Jobling, J. Kanno, H. Laufer, M. Marcus, J. A. McLachlan, A. Nadal, J. Oehlmann, N. Olea, P. Palanza, S. Parmigiani, B. S. Rubin, G. Schoenfelder, C. Sonnenschein, A. M. Soto, C. E. Taisness, J. A. Taylor, L. N. Vandenberg, J. G. Vandenbergh, S. Vogel, C. S. Watson, W. V. Welshons and R. T. Zoeller, Why public health agencies cannot depend on good laboratory practices as a criterion for selecting data: The case of bisphenol A, Environ. Health Perspect., 2009, 117, 309–315 CrossRef CAS PubMed.
- G. Stieger, M. Scheringer, C. A. Ng and K. Hungerbühler, Assessing the persistence, bioaccumulation potential and toxicity of brominated flame retardants: Data availability and quality for 36 alternative brominated flame retardants, Chemosphere, 2014, 116, 118–123 CrossRef CAS PubMed.
- A. Beronius, A. Hanberg, R. Heimeier and H. Håkansson, IMM-rapport 1/2013. Risk assessment of developmental neurotoxicity: Evaluation of the OECD TG 426 test guideline and guidance documents. Institutet för miljömedicin - IMM. Karolinska Institutet, Institutet för miljömedicin - IMM. Karolinska Institutet, 2013 Search PubMed.
- OECD, OECD Test Guideline No. 203. Fish, Acute Toxicity Test, 1992. http://www.oecd.org/chemicalsafety/testing/oecdguidelinesforthetestingofchemicals.htm Search PubMed.
- OECD, OECD Test Guideline No. 301. Ready Biodegradability, 1992. http://www.oecd.org/chemicalsafety/testing/oecdguidelinesforthetestingofchemicals.htm Search PubMed.
- A. Beronius, L. Molander, C. Rudén and A. Hanberg, Facilitating the use of non-standard in vivo studies in health risk assessment of chemicals: a proposal to improve evaluation criteria and reporting, J. Appl. Toxicol., 2014, 34, 607–617 CrossRef CAS PubMed.
- C. Kilkenny, W. J. Browne, I. C. Cuthill, M. Emerson and D. G. Altman, Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research, Osteoarthritis Cartilage, 2012, 20, 256–260 CrossRef CAS PubMed.
- M. Enserink, Sloppy reporting on animal studies proves hard to change Scientists appear to ignore guidelines adopted 7 years ago, Science, 2017, 357, 1336–1338 CrossRef PubMed.
- M. Agerstrand, A. Sobek, K. Lilja, M. Linderoth, L. Wendt-Rasch, A.-S. Wernersson and C. Ruden, An academic researcher's guide to increased impact on regulatory assessment of chemicals, Environ. Sci.: Processes Impacts, 2017, 19, 644–655 RSC.
- M. Agerstrand, S. Christiansen, A. Hanberg, C. Ruden, L. Andersson, S. Andersen, H. Appelgren, C. Bjorge, I. H. Clausen, D. M. Eide, N. B. Hartmann, T. Husoy, H. P. Halldorsson, M. van der Hagen, E. Ingre-Khans, A. D. Lillicrap, V. M. Beltoft, A. K. Mork, M. Murtomaa-Hautala, E. Nielsen, K. Olafsdottir, J. Palomaki, H. Papponen, E. M. Reiler, H. Stockmann-Juvala, T. Suutari, H. Tyle and A. Beronius, A call for action: Improve reporting of research studies to increase the scientific basis for regulatory decision-making, J. Appl. Toxicol., 2018, 38(5), 783–785 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tx00216a |
| This journal is © The Royal Society of Chemistry 2019 |