
Comparison of hepatotoxicity and mechanisms induced by triclosan (TCS) and methyl-triclosan (MTCS) in human liver hepatocellular HepG2 cells†
Lu
Wang‡
 a,
Boyu
Mao‡
b,
Huixin
He
a,
Yu
Shang
a,
Yufang
Zhong
a,
Zhiqiang
Yu
c,
Yiting
Yang
*d,
Hui
Li
a and
Jing
An
*a
a,
Boyu
Mao‡
b,
Huixin
He
a,
Yu
Shang
a,
Yufang
Zhong
a,
Zhiqiang
Yu
c,
Yiting
Yang
*d,
Hui
Li
a and
Jing
An
*a
aSchool of Environmental and Chemical Engineering, Shanghai University, Shanghai 200444, China. E-mail: peace74839@shu.edu.cn; Tel: +86 21 66137736
bImplant Dentistry Department, Jiangbei Dental Hospital, Ningbo 315000, China
cState Key Laboratory of Organic Geochemistry, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou 510640, China
dDepartment of Neurology, Changhai Hospital, Second Military Medical University, Shanghai 200433, China. E-mail: yangytyd@163.com
First published on 12th October 2018
Abstract
Triclosan (TCS) is used as an antimicrobial agent and has been widely dispersed and detected in the environment and organisms including human samples. Methyl-triclosan (MTCS) is the predominant bacterial TCS metabolite. At present, the toxicological effects and mechanism of TCS and MTCS are still not fully understood. In this study, the cytotoxic effects of TCS and MTCS in HepG2 cells were investigated in terms of cell proliferation, comet assay, cell cycle, and apoptosis. In addition, the expressions of related proteins were detected with western blotting analysis. The results showed that TCS could significantly inhibit cell proliferation, while MTCS had no obvious effect on cell growth. Both TCS and MTCS caused oxidative injury associated with HO-1 induction and increased DNA strand breaks, which consequently initiated the damage repair process via up-regulation of DNA-PKcs. In addition, TCS blocked the HepG2 cells in S and G2/M phases of cell cycle through down-regulation of cyclin A2 and CDK; while MTCS induced cell cycle arrest at the S phase through up-regulation of cyclin A2 and CDK. Furthermore, TCS activated p53 mediated apoptosis in HepG2 cells in a caspase-independent manner, while MTCS induced apoptosis was dependent on caspase. Moreover, TCS exposure exhibited more severe toxicity in HepG2 cells as compared with MTCS exposure, indicating that the replacement of the ionizable proton in TCS by the methyl group in MTCS is correlated with the cellular toxicity and the molecular mechanism.
1. Introduction
Triclosan (5-chloro-2-[2,4-dichloro-phenoxy]-phenol, TCS), a broad-spectrum antimicrobial agent, has been widely used for more than 40 years in a variety of pharmaceutical and personal care products, such as soaps, deodorants, mouthwashes, toothpastes, and household cleaners. Since TCS can enter the environment through wastewater, the widespread use of TCS has led to elevated concentrations in environmental pollution causing health problems. Now TCS has been detected in almost all environmental and biological samples, including air, surface water, wastewater, water sediments, sludge, and fish,1–4 which may be potential sources of TCS exposure to humans. TCS has currently been detected in human plasma, urine, and breast milk samples, as well as in liver and brain tissues. Therefore, the potential risk of TCS to humans has attracted considerable attention worldwide.TCS was present in urine samples of children and adults in the USA in a concentration range of 2.3 to 3790 μg L−1, and 2.8 to 14 μg L−1 in anonymous lactating women.5,6 Azzouz et al. reported that TCS had been detected at a concentration of 0.31 to 2.1 μg L−1 in urine samples, 1.2 to 12 μg L−1 in blood, and 0.25 to 2.1 μg L−1 in breast milk in Spain.7 In Belgium, TCS was found in liver tissue with an average concentration of 3.14 ng g−1, followed by 0.61 ng g−1 in adipose tissue and 0.03 ng g−1 in brain tissue.8 In China, the TCS levels ranged from 1.8 to 7.0 μg L−1 in the urine of children and students, and from 0.1 to 89.36 μg L−1 in maternal urine samples.9,10
TCS can be degraded via microbial activity or abiotic mechanisms, and it forms the methylated transformation product of methyl-triclosan (MTCS) during wastewater treatment.11 Tohidi et al. reported that during anaerobic and aerobic sludge digestion processes for wastewater and sewage sludge, about 7.4% of TCS was transformed to MTCS through aerobic digestion.12 In typical conventional WWTPs, up to 50% of TCS can be degraded into MTCS as well as other unknown products.13,14 Compared with TCS, MTCS has a higher degree of environmental persistence, long-range transportation activity, and bioaccumulation potential in the sludge or living organisms.15 Researchers have reported that MTCS is detectable in environmental and biological samples. In marine sediments of Barker Inlet in South Australia, the concentration of MTCS was <11 μg kg−1.16 Balmer et al. also reported that the concentrations of MTCS in the white fish of Switzerland lakes ranged from 4 to 365 μg kg−1 on a lipid basis.17
Based on their high lipid solubility, both TCS and MTCS can be bio-accumulated in plants and animals, which would transfer into the human body through food chain and finally induce potential harmful effects.18,19 Cortez et al. reported that 12 ng L−1 of TCS could cause physiological stress in marine bivalves.20 The LC50 values of TCS under acute exposure (96 h) were 73.4 μg L−1 for amphipod, 74.3 μg L−1 for mysid,21 305 μg L−1 for adult grass shrimp, 154 μg L−1 for larvae shrimp, and 3.55 μg L−1 for Dunaliella tertiolecta.18 Oliveira et al. reported that the LC50 (96 h) of TCS was 420 μg L−1 for the zebrafish embryo and 340 μg L−1 for the adult zebrafish.22 Now TCS has been recognized as a member of the endocrine disrupting contaminants (EDCs) with estrogenic activity.23 Recently, research data showed that TCS could also initiate anti-inflammatory responses and suppress the function of natural killer cells in human cells. As a mitochondrial uncoupler, TCS could increase the oxygen consumption rate in rat and human mast cells to inhibit the mast cell function.24 Relatively, the study on the toxic effects and toxicological mechanism of MTCS is far from sufficient. Gaume et al. revealed the toxic effects of MTCS on immune (hemocytes) and/or respiratory cells (gill cells) of the abalone.25 MTCS of environmentally relevant concentrations induced a delay in the development of the sea urchin larvae.26 Lv et al. reported that MTCS could bind to human serum albumin (HSA) in vitro, which might be associated with the adverse impacts on the endocrine function.11 However, an exhaustive understanding of the potential bio-effects of TCS and MTCS and the underlying mechanisms is necessary to objectively evaluate the safety and potential hazard of these compounds.
The liver exhibited the highest concentration of TCS followed by the gills, gonads, brain, and muscle tissues in P. vivipara. After depuration, TCS concentrations declined >80% in all tissues except the liver.3 In this study, the toxic effects of TCS and MTCS in human liver hepatocellular cells (HepG2) are firstly measured in terms of cell proliferation (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide, MTT assay), cell apoptosis (flow cytometry), cell cycle (flow cytometry), and DNA damage (comet assay). Moreover, the expression of related proteins was assayed with western blotting to explore the potential mechanism. The results of this study would promote the current understanding on the cytotoxicity of TCS and MTCS.
2. Materials and methods
2.1. Cell culture and treatments
Human hepatocellular carcinoma cells (HepG2) were obtained from ATCC (American Type Culture Collection). HepG2 cells were conventionally cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Paisley, UK). Based on preliminary experiments (concentrations ranging from 1 μM to 100 μM, data not shown), four concentrations (5, 10, 20, and 40 μM) of TCS (CAS: 3380-34-5) and MTCS (CAS: 4640-01-1) with mild to moderate mortality were selected in this study. HepG2 cells were treated with different concentrations of TCS or MTCS for 24 h, and then cell proliferation, apoptosis, cell cycle, DNA damage and related protein expressions were measured. The control group was incubated with 0.1% dimethyl sulfoxide (DMSO) (v/v) only. All experiments were carried out at least three times with more than three parallel samples.2.2. Cell growth assay
HepG2 cells were exposed to 5, 10, 20, or 40 μM of TCS/MTCS for 24 h. The growth of cells was detected with the MTT assay as described in the ESI.† The absorbance at OD 490 nm was recorded using a Multiscan Mk3 plate reader (Thermo Electron Corporation, USA).2.3. Cell apoptosis assay
After being treated with TCS or MTCS (5, 10, 20, and 40 μM) for 24 h, the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) detection kit (BD, MD, USA) was used to identify and quantify the apoptotic cells as described in the ESI.† The rate of apoptosis was analyzed by flow cytometry using the FL2 detector (Becton Dickinson FACScan) with an excitation wavelength of 488 nm. The data were analyzed with Summit 5.2 software. The apoptosis rates referred to the percentage of apoptotic cells in total cells (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per sample).
000 cells per sample).
2.4. Cell cycle assay
HepG2 cells were treated with TCS or MTCS (5, 10, 20, and 40 μM) for 24 h. Then cell cycle distribution of HepG2 was assessed using flow cytometry (Becton Dickinson FACScan) as described in the ESI.† All data were analyzed by Multicycle software. For each sample, a total of 10000 cells were recorded.
2.5. Comet assay
For DNA damage detection, HepG2 cells were treated with TCS or MTCS (5, 10, 20, and 40 μM) for 24 h. Then DNA damage was evaluated through the Olive tail moment (Olive™) using the CASP software version (University of Wroclaw, Poland) as described in the ESI.† For each sample, 300 randomly captured cells by an investigator were collected and measured. tert-Butyl hydroperoxide (tBHP, 10 μM) was used as a positive control.2.6. Western blotting
Western blotting was performed according to our previous study.27 After treatment of TCS or MTCS (5, 10, 20, and 40 μM) for 24 h, the total protein was collected and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Then the protein was transferred to the polyvinylidene difluoride (PVDF) membrane, and incubated with the primary and secondary antibodies as described in the ESI.† The blots were visualized using chemiluminescence, and the optical densities of individual bands were quantitated using a Chemi-Imager digital imaging system (Alpha Innotech, San Leandro, CA, USA).2.7. Data analysis
Data were presented as the mean ± standard error of mean (SEM) of at least three independent experiments. Differences between groups were determined by a one-way ANOVA test with Bonferroni's correction. A p-value <0.05 was considered statistically significant.3. Results
3.1. Cytotoxicity in HepG2 cells induced by TCS and MTCS
In this study, cell viability after TCS and MTCS exposure was examined using the MTT assay. As shown in Fig. 1A, under lower concentration exposure (<20 μM), TCS had no effect on HepG2 cell proliferation. After being treated with 20 and 40 μM of TCS for 24 h, the cell viability rate reduced to 90.5 ± 3.1% and 66.5 ± 5.9%, respectively, as compared with the control groups. However, MTCS had no obvious effect on cell growth under all of the concentrations (5–40 μM). The morphological changes were observed using an inverted phase contrast microscope (Fig. 1B), showing deformed cell shapes and disrupted cell junctions after 24 h treatment of 40 μM of TCS, while the cell morphology maintained roughly the original state after MTCS treatment.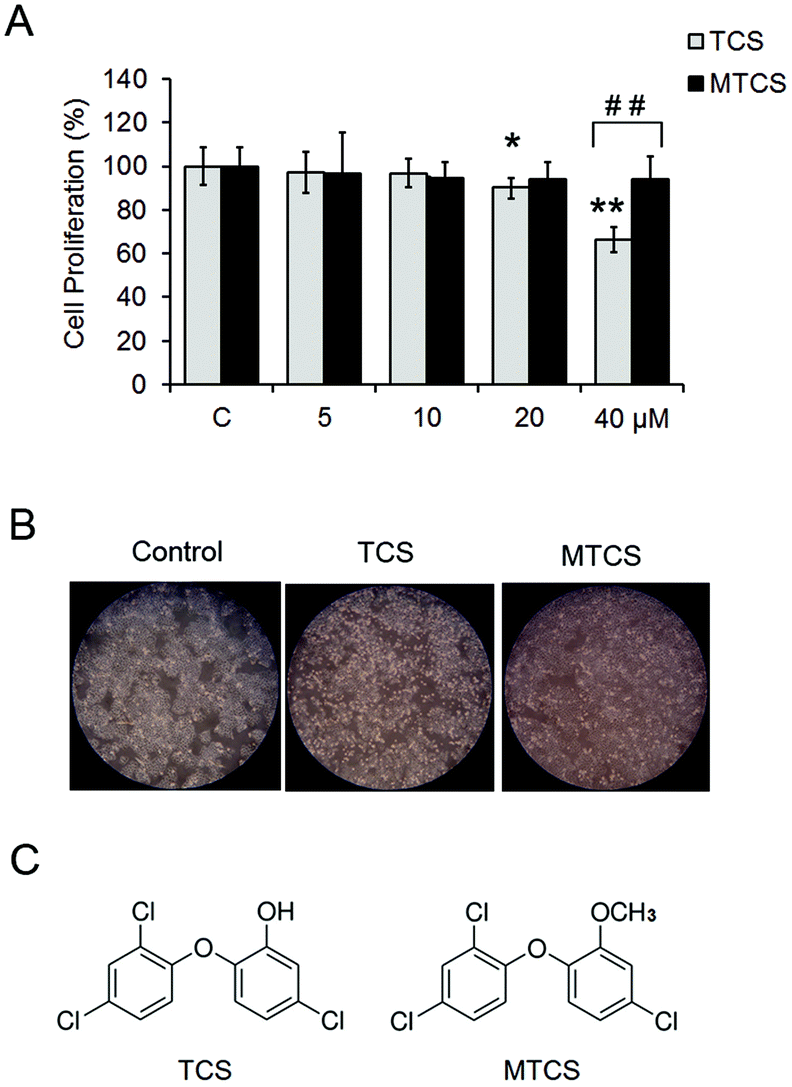 | ||
| Fig. 1 Cell proliferation in HepG2 cells treated with TCS and MTCS. (A) HepG2 cells were treated with TCS or MTCS (5, 10, 20, and 40 μM) for 24 h. The cell proliferation was determined by the MTT assay. (B) HepG2 cells were treated with 40 μM of TCS or MTCS for 24 h. The morphological changes were observed using an inverted phase contrast microscope. (C) The chemical structure of TCS and MTCS. C: Control groups. HepG2 cells treated with DMSO only. * p < 0.05, ** p < 0.01 compared with the control groups (C). ## p < 0.01 compared with the TCS groups. | ||
3.2. Oxidative stress and DNA damage in HepG2 cells induced by TCS and MTCS
The expression of heme oxygenase-1 (HO-1), an oxidative stress responsive enzyme, was measured with western blotting. As shown in Fig. 2A and B, compared with the control groups, the expression of HO-1 was up-regulated to 1.4, 2.3 and 2.6-fold and 1.6, 1.9 and 2.3-fold, respectively (p < 0.05), after treatment of 10, 20 and 40 μM of TCS or MTCS. The results of the comet assay displayed that TCS and MTCS (20 and 40 μM) treatments could cause an increase of the tail moment in HepG2 cells from 5.58% to 14.30%, 16.69% (p < 0.05), and to 11.20%, 12.82% (p < 0.05), respectively, indicating the intensification of DNA strand breaks (Fig. 2C).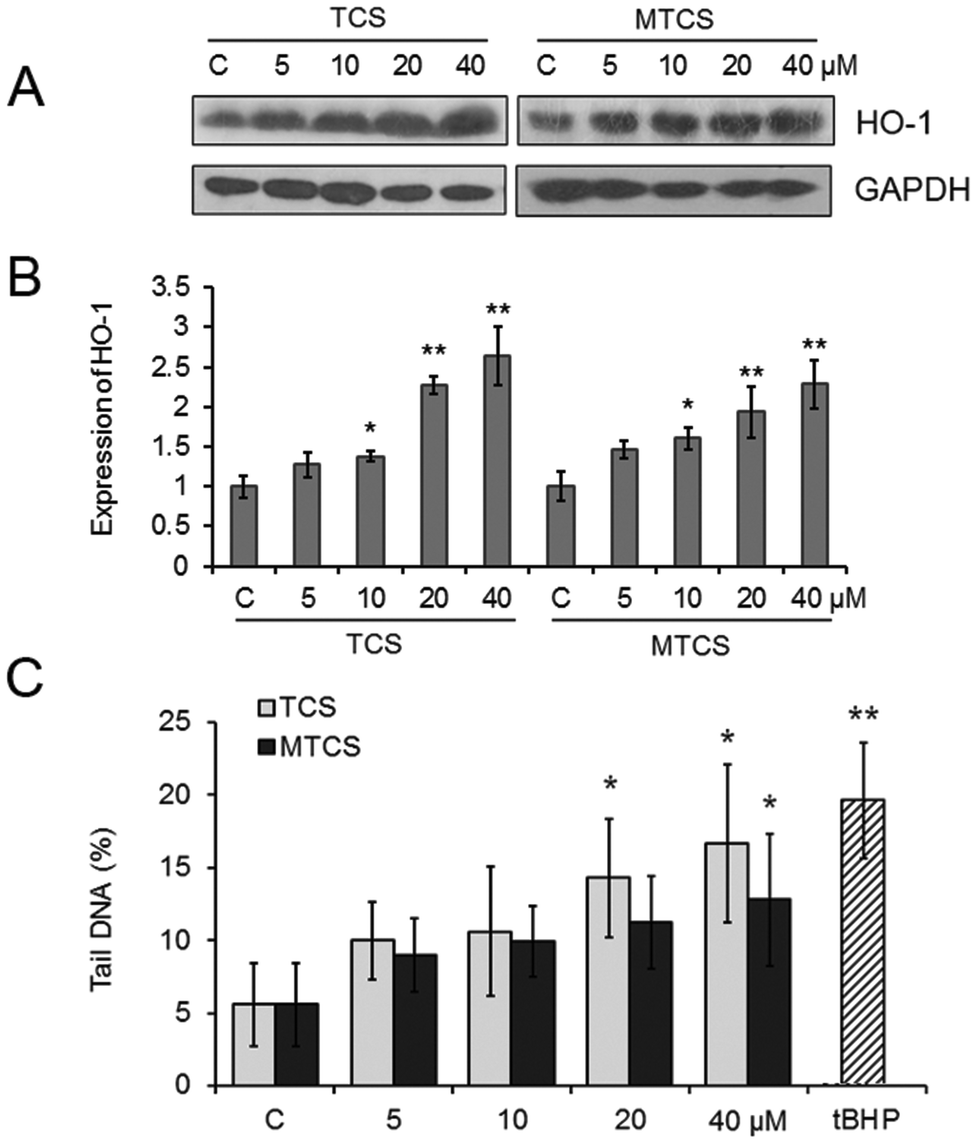 | ||
| Fig. 2 Oxidative stress and genetic toxicity in HepG2 cells induced by TCS and MTCS exposure. HepG2 cells were treated with TCS or MTCS (5, 10, 20, and 40 μM) for 24 h. (A) The expression of HO-1 was examined by western blotting. GAPDH protein expression was used as an internal reference. (B) The quantization of HO-1 expression levels. (C) DNA damage was evaluated by the comet assay. C: Control groups. HepG2 cells treated with DMSO only. * p < 0.05, ** p < 0.01 compared with the control (C) groups. | ||
3.3. Cell cycle arrest in HepG2 cells induced by TCS and MTCS
As shown in Table 1, both TCS and MTCS could induce a significant cell cycle arrest at the S phase in a dose dependent manner. After treatment of 20 and 40 μM TCS or MTCS for 24 h, the percentage of S phase cells increased 44.3%, 34.0%, and 26.7%, 26.3%, respectively (p < 0.05). At 48 h after treatment, the population of S phase cells was still higher than the control group, with the increased percentage of 34.2%, 50.8%, and 22.1%, 31.2%, respectively (p < 0.05). In addition, the G2/M phase cells also increased 34.1% after treatment with 40 μM of TCS for 48 h (p < 0.05).| Group | 24 h | 48 h | |||||
|---|---|---|---|---|---|---|---|
| G0/G1 | S | G2/M | G0/G1 | S | G2/M | ||
| HepG2 cells were treated with TCS or MTCS (5, 10, 20, and 40 μM) for 24 h or 48 h, and the distribution of cell cycle was evaluated with flow cytometry. C: Control groups. HepG2 cells treated with DMSO only. * p < 0.05, ** p < 0.01 compared with the control (C) groups. | |||||||
| Control | 68.59 ± 0.66 | 20.15 ± 1.22 | 11.26 ± 0.79 | 67.53 ± 1.03 | 22.08 ± 1.72 | 10.39 ± 0.69 | |
| TCS | 5 μM | 69.23 ± 0.77 | 22.36 ± 1.10 | 8.41 ± 0.7 | 69.95 ± 0.44 | 21.84 ± 1.44 | 8.22 ± 0.70 |
| 10 μM | 68.80 ± 0.78 | 23.95 ± 0.55 | 7.25 ± 0.68 | 68.85 ± 0.43 | 22.93 ± 0.56 | 8.22 ± 0.14 | |
| 20 μM | 65.19 ± 2.10 | 29.08 ± 1.02** | 5.73 ± 0.66 | 66.00 ± 1.31 | 29.63 ± 0.99* | 4.37 ± 0.32 | |
| 40 μM | 60.78 ± 1.20 | 27.01 ± 0.66* | 12.21 ± 0.42 | 52.73 ± 0.44 | 33.30 ± 0.57** | 13.97 ± 0.12 | |
| MTCS | 5 μM | 72.14 ± 1.20 | 19.16 ± 0.46 | 8.76 ± 1.44 | 65.06 ± 0.43 | 24.27 ± 2.18 | 10.68 ± 1.75 |
| 10 μM | 68.36 ± 0.49 | 20.30 ± 0.64 | 11.34 ± 1.22 | 65.68 ± 0.25 | 25.72 ± 0.20 | 8.61 ± 0.05 | |
| 20 μM | 67.98 ± 2.42 | 25.53 ± 0.62* | 6.49 ± 0.12 | 65.87 ± 1.15 | 26.95 ± 0.46 | 7.18 ± 0.88 | |
| 40 μM | 68.05 ± 0.68 | 25.45 ± 0.02* | 6.50 ± 0.21 | 65.23 ± 0.34 | 28.97 ± 1.01* | 5.80 ± 0.78 | |
TCS could significantly inhibit the expressions of cyclin A2 and cyclin-dependent kinases 2 (CDK2) in a concentration-dependent manner (Fig. 3). The expressions of cyclin A2 and CDK2 decreased 9.0%, 30.1%, 48.4% and 49.7%, 57.8%, 60.1%, respectively, after 10, 20, and 40 μM of TCS treatment. However, completely opposite alterations on the expression of cyclin A2 and CDK2 were found after MTCS exposure. The expressions of cyclin A2 and CDK2 increased 93.6%, 109.1%, 122.6%, 205.7% and 26.6%, 33.5%, 36.9%, 60.4%, respectively, after 5, 10, 20, and 40 μM of MTCS treatment. In addition, the expression of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) increased in a concentration-dependent manner after treatment of TCS and MTCS.
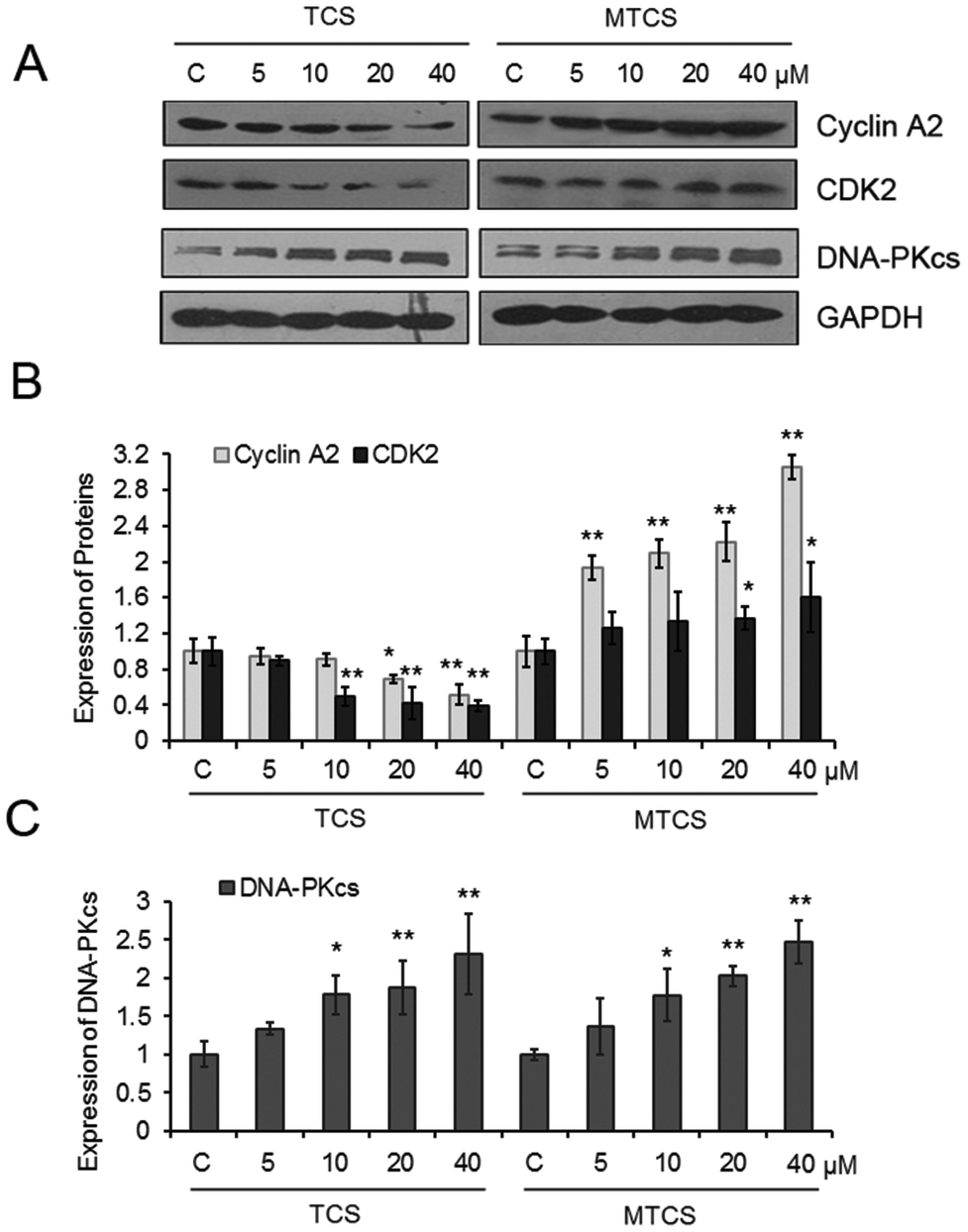 | ||
| Fig. 3 Effect of TCS and MTCS on related protein expressions in HepG2 cells. HepG2 cells were treated with TCS or MTCS (5, 10, 20, and 40 μM) for 24 h. (A) The expressions of related protein were examined by western blotting. GAPDH protein expression was used as the internal reference. (B) and (C) The quantization of protein expression levels. C: Control groups. HepG2 cells treated with DMSO only. * p < 0.05, ** p < 0.01 compared with the control (C) groups. | ||
3.4. The apoptosis in HepG2 cells induced by TCS and MTCS
The apoptosis rate of HepG2 cells treated with TCS and MTCS was investigated using flow cytometry with the Annexin V-FITC/PI staining assay. As shown in Fig. 4A, both TCS and MTCS could significantly induce cell apoptosis in a concentration-response manner. After being treated with 20 and 40 μM of TCS and MTCS for 24 h, the rates of apoptosis increased 2.8, 3.9 fold, and 1.7, 2.1 fold, respectively, as compared with the control groups (p < 0.05). The results of western blotting showed that TCS exposure could significantly inhibit the expressions of cysteine–aspartic acid–specific proteases (caspase) -3 and -9, while MTCS could significantly stimulate them with the change indexes close to 2 after the treatment of 40 μM of TCS and MTCS (Fig. 4B and C). Furthermore, TCS exposure could significantly inhibit the expression of B cell lymphoma 2 (Bcl-2) associated X protein (Bax) by 24.9% and 47.6% after 20 and 40 μM exposures, respectively. However, MTCS stimulated Bax expression by 30.7%, 74.2%, 112.1% and 180.6%, respectively (Fig. 4B and D). The expression of p53 was obviously up-regulated by different concentrations of TCS and MTCS treatment.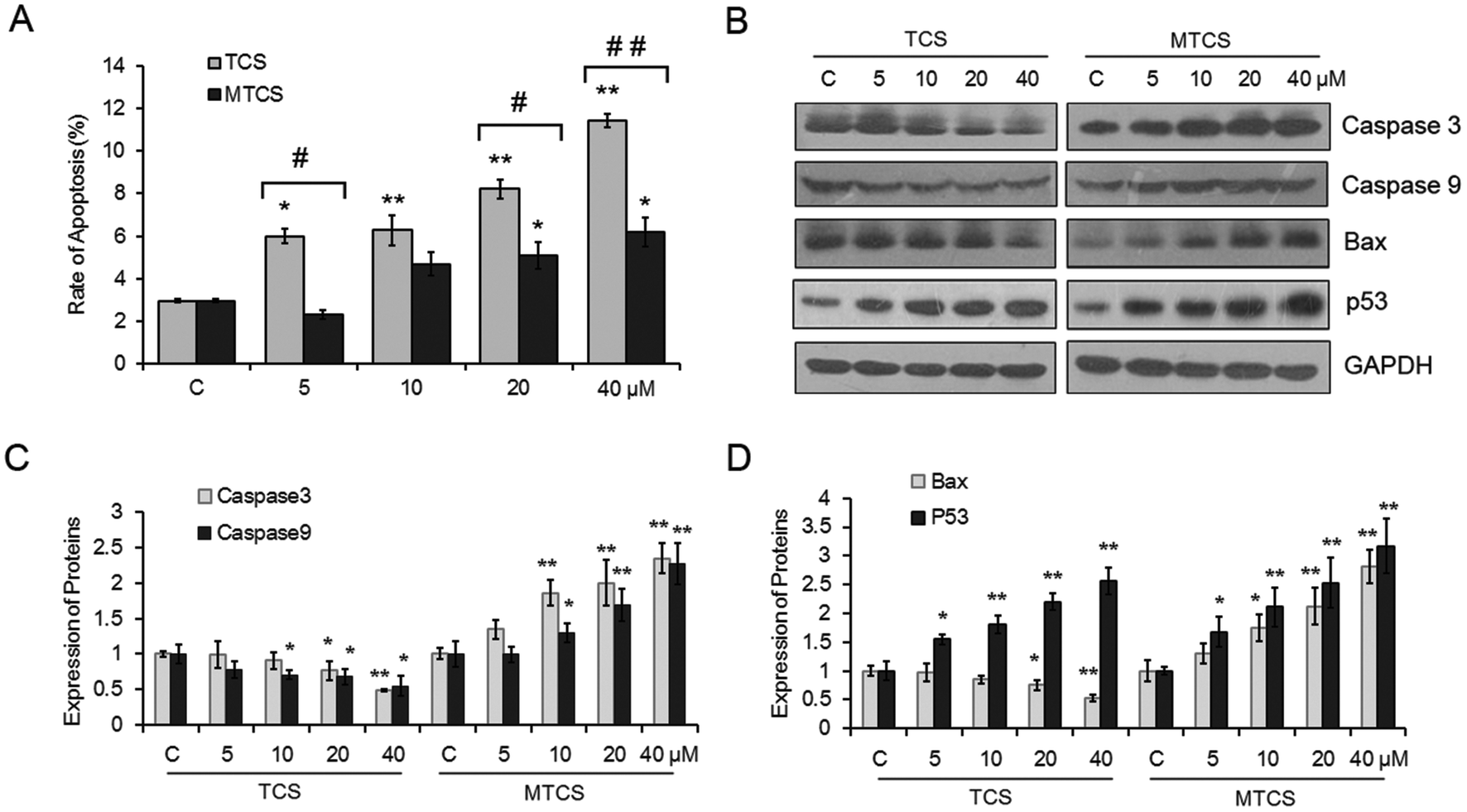 | ||
| Fig. 4 Apoptosis and expressions of related proteins in HepG2 cells treated with TCS and MTCS. HepG2 cells were treated with TCS or MTCS (5, 10, 20, and 40 μM) for 24 h. (A) The apoptosis was measured and evaluated using flow cytometry with the Annexin V-FITC/PI apoptosis assay kit. (B) Protein expressions of caspase-3, caspase-9, Bax, and p53 were measured with western blotting assay. GAPDH protein expression was used as the internal reference. (C) and (D) The quantization of protein expression levels. C: Control groups. HepG2 cells treated with DMSO only. * p < 0.05, ** p < 0.01 compared with the control (C) groups. # p < 0.05, ## p < 0.01 compared with the TCS groups. | ||
4. Discussion
Recently, Yueh et al. found that long term exposure of TCS could enhance the liver fibrogenesis and tumorigenesis in mice.28 MTCS is the predominant bacterial TCS metabolite through methylation of the hydroxyl group (Fig. 1C), which might influence its biological function. Unlike TCS, MTCS can disrupt the thyroid hormone (TH) action by regulating the TH responsive transcripts in mammalian cells.29 Our results revealed a more severe cytotoxicity of TCS than MTCS in HepG2 cells, which suggested that the chemical structure difference between TCS and MTCS might impact the biological response in mammalian cells.As a mitochondrial uncoupler, TCS could inhibit ATP production and increase the oxygen consumption rate in rat basophilic leukemia mast cells (RBL-2H3).24 Wang et al. reported that TCS induced lipid oxidative damage, DNA damage, and genetic toxicity in the liver of goldfish, accompanied by the increased content of antioxidant reduced glutathione (GSH) to eliminate the excess reactive oxidative species (ROS).30 TCS could also stimulate the ROS production in mouse neocortical neurons.31 Zhang et al. reported that TCS exposure during pregnancy could induce potential genetic damage to the Caenorhabditis elegans offspring.32 In the present study, the up-regulation of HO-1 indicated the activation of oxidative stress response in HepG2 cells exposed to TCS. And the following DNA damage mediated by oxidative stress may be an important mechanism of TCS and MTCS-induced cytotoxicity in HepG2 cells. As far as we know, the genetic toxicity of TCS in mammalian cells has been presented for the first time. Until now, there was no study conducted on the oxidative stress and genotoxic effects of MTCS. Our results showed that MTCS also induced oxidative stress response and caused DNA damage in HepG2 cells. Consistent with cell growth results, TCS had more severe cytotoxicity than MTCS in terms of inducing DNA damage, although there was no significant difference between these two treatment groups.
The cell growth and division is tightly controlled during the progress of cell cycle phases (including G1, S, G2/M phases). Our results showed that both TCS and MTCS could induce a significant cell cycle arrest at the S phase in a dose dependent manner (Table 1). The higher cell cycle arrest rate in TCS suggested a slower cell proliferation efficacy, which was in agreement with the MTT results that TCS was more efficient in inhibiting cell growth. The molecular mechanism to regulate the passage of cells through cell cycle controlling is complicated and diverse. Key components mainly include a series of cyclin proteins and CDK partners. The S phase is a critical DNA replication period; the transition from the G1 into S phase and the preparation of cells to enter the M phase are regulated by cyclin A and CDK2 complexes. Down-regulation of cyclin A2 and CDK2 is closely related to the S phase cell cycle arrest.33,34 Our results indicated that TCS could obstruct the transition from the S to M phase via down-regulation of cyclin A2 and CDK2, while MTCS induced cell cycle arrest at the S phase through completely opposite alterations on the expression of cyclin A2 and CDK2. Ming et al. reported that an arylbenzofuran could induce G1/S phase arrest through increased cyclin A2 and CDK2 proteins in cervical cancer cells which was consistent with our finding.35
Moreover, DNA repair and cell cycle checkpoint are simultaneously initiated and interlinked to guarantee genomic stability. Generally, the G1/S and intra-S checkpoints prevent the cells from unfaithful replication, and the G2/M checkpoint is in charge of DNA damage repair prior to mitosis. In this study, the expression of DNA-PKcs which is required in the non-homologous end joining (NHEJ) pathway of DNA double-strand break repair was measured with western blotting.36 The up-regulation of DNA-PKcs after TCS and MTCS treatment suggested the initiation of the DNA damage repair pathway.
Apoptosis is widely used as one of the reliable markers of cellular injury. Research has shown that apoptosis appears to be prevailing as a toxicity effect induced by TCS.37,38 Our results also showed that both TCS and MTCS could significantly induce cell apoptosis in HepG2 cells. Consistent with the cell growth and DNA damage results, TCS had higher capability to induce apoptosis than MTCS (p < 0.05). Caspases are a family of protease enzymes playing essential roles in a relatively early stage of apoptosis. Among the various caspase family members, caspase-9 is an apoptosis initiator and caspase-3 is an executioner to induce mitochondria mediated apoptosis.39 Our data suggested that the process of apoptosis induced by TCS was caspase-independent, while MTCS induced apoptosis was dependent on caspase in HepG2 cells. Bax, a pro-apoptotic effector, can directly bind to Bcl-2 to regulate mitochondria mediated apoptosis.40 The opposite expression alterations of Bax further confirmed the different apoptotic mechanisms induced by TCS and MTCS. MTCS might cause cell death mainly through the mitochondrial apoptosis pathways in HepG2 cells, whereas TCS does not. Our results were inconsistent with previous studies that TCS could evaluate caspase-3 activity in the lung tissue homogenate of Sprague Dawley rats or in mouse neocortical neurons.31,37 These findings suggested that species differences or cell types might affect the apoptosis mechanism induced by TCS. Meanwhile, the structure difference between TCS and MTCS leading to various response mechanisms was further confirmed.
In addition, the activation of the p53 gene by the DNA damage signal plays a major role in regulating cell apoptosis. He et al. reported that ROS mediated DNA damage could activate the p53–p21 pathway to cause cell cycle arrest and apoptosis,41 which were consistent with our results that both TCS and MTCS stimulated oxidative stress response, mediated DNA strand break, and activated p53 to induce apoptosis in HepG2 cells.
5. Conclusion
In general, we concluded that both TCS and MTCS could induce oxidative DNA damage and initiate the DNA damage repair process. TCS could block the cell cycle process to inhibit HepG2 cell proliferation, which was mediated by the down-regulation of cyclin A2 and CDK2, while the expression of cyclin A2 and CDK2 in MTCS treated HepG2 was up-regulated. Furthermore, TCS activated p53 mediated apoptosis in a caspase-independent manner in HepG2 cells, while MTCS induced apoptosis was regulated in a caspase-dependent manner (the potential regulation mechanism shown in Fig. 5). In addition, TCS presented more severe cytotoxicity in HepG2 cells as compared with MTCS, which may be due to the difference in chemical structures.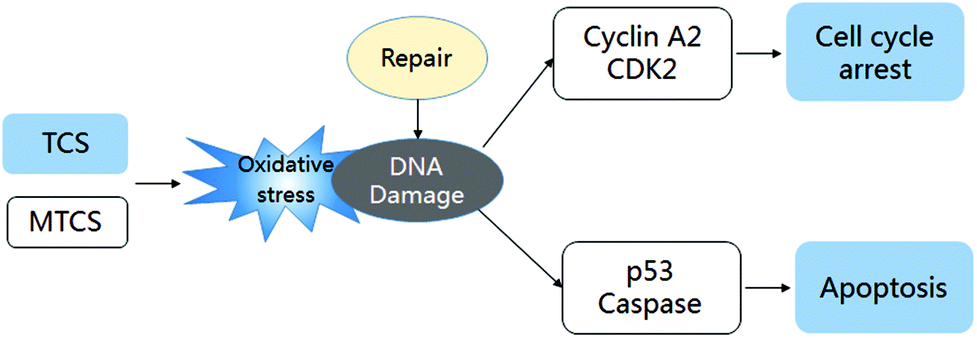 | ||
| Fig. 5 The regulation mechanism of TCS and MTCS on cytotoxicity. | ||
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The study was supported by the National Natural Science Foundation of China (No. 91543123); the Joint NSFC-ISF Research Program, jointly funded by the National Natural Science Foundation of China and the Israel Science Foundation (No. 41561144007); and Innovative Research Team in University (No. IRT13078).References
- K. Bester, Fate of triclosan and triclosan-methyl in sewage treatment plantsand surface waters, Arch. Environ. Contam. Toxicol., 2005, 49, 9–17 CrossRef CAS PubMed.
- J. M. Buth, P. O. Steen, C. H. Sueper, P. J. Vikesland and W. A. Arnold, Dioxinphotoproducts of triclosan and its chlorinated derivatives in sediment cores, Environ. Sci. Technol., 2010, 44, 4545–4551 CrossRef CAS PubMed.
- A. L. Escarrone, S. S. Caldas, E. G. Primel, S. E. Martins and L. E. Nery, Uptake, tissue distribution and depuration of triclosan in the guppy Poecilia vivipara acclimated to freshwater, Sci. Total Environ., 2016, 560–561, 218–224 CrossRef CAS PubMed.
- D. E. Haggard, P. D. Noyes, K. M. Waters and R. L. Tanguay, Phenotypically anchored transcriptome profiling of developmental exposure to the antimicrobial agent, triclosan, reveals hepatotoxicity in embryonic zebrafish, Toxicol. Appl. Pharmacol., 2016, 308, 32–45 CrossRef CAS PubMed.
- A. M. Calafat, X. Ye, L. Y. Wong, J. A. Reidy and L. L. Needham, Urinary concentrations of triclosan in the U.S. population: 2003–2004, Environ. Health Perspect., 2008, 116(3), 303–307 CrossRef CAS PubMed.
- X. Ye, A. M. Bishop, L. L. Needham and A. M. Calafat, Automated on-linecolumn-switching HPLC–MS/MS method with peak focusing for measuringparabens, triclosan, and other environmental phenols in human milk, Anal. Chim. Acta, 2008, 622, 150–156 CrossRef CAS PubMed.
- A. Azzouz, A. J. Rascón and E. Ballesteros, Simultaneous determination of parabens, alkylphenols, phenylphenols, bisphenol A and triclosan in human urine, blood and breast milk by continuous solid-phase extraction and gas chromatography-mass spectrometry, J. Pharm. Biomed. Anal., 2016, 119, 16–26 CrossRef CAS PubMed.
- T. Geens, H. Neels and A. Covaci, Distribution of bisphenol-A, triclosan and n-nonylphenol in human adipose tissue, liver and brain, Chemosphere, 2012, 87(7), 796–802 CrossRef CAS PubMed.
- X. Li, G. G. Ying, J. L. Zhao, Z. F. Chen, H. J. Lai and H. C. Su, 4-Nonylphenol, bisphenol-A and triclosan levels in human urine of children and students in China, and the effects of drinking these bottled materials on the levels, Environ. Int., 2013, 52, 81–86 CrossRef CAS PubMed.
- X. Wang, F. Ouyang, L. Feng, X. Wang, Z. Liu and J. Zhang, Maternal Urinary Triclosan Concentration in Relation to Maternal and Neonatal Thyroid Hormone Levels: A Prospective Study, Environ. Health Perspect., 2017, 125(6), 067017 CrossRef PubMed.
- W. Lv, Y. Chen, D. Li, X. Chen and J. Leszczynski, Methyl-triclosan binding to human serum albumin: multi-spectroscopic study and visualized molecular simulation, Chemosphere, 2013, 93(6), 1125–1130 CrossRef CAS PubMed.
- F. Tohidi and Z. Cai, Fate and mass balance of triclosan and its degradation products: Comparison of three different types of wastewater treatments and aerobic/anaerobic sludge digestion, J. Hazard. Mater., 2017, 323(Pt A), 329–340 CrossRef CAS PubMed.
- J. Heidler and R. U. Halden, Mass balance assessment of triclosan removal during conventional sewage treatment, Chemosphere, 2007, 66(2), 362–369 CrossRef CAS PubMed.
- N. Lozano, C. P. Rice, M. Ramirez and A. Torrents, Fate of triclocarban, triclosan and methyltriclosan during wastewater and biosolids treatment processes, Water Res., 2013, 47(13), 4519–4527 CrossRef CAS PubMed.
- M. A. Coogan, R. E. Edziyie, T. W. La Point and B. J. Venables, Algal bioaccumulation of triclocarban, triclosan, and methyl-triclosan in a North Texas wastewater treatment plant receiving stream, Chemosphere, 2007, 67, 1911–1918 CrossRef CAS PubMed.
- M. Fernandes, A. Shareef, R. Kookana, S. Gaylard, S. Hoare and T. Kildea, The distribution of triclosan and methyl-triclosan in marine sediments of Barker Inlet, South Australia, J. Environ. Monit., 2011, 13(4), 801–806 RSC.
- M. E. Balmer, T. Poiger, C. Droz, K. Romanin, P. A. Bergqvist, M. D. Müller and H. R. Buser, Occurrence of methyl triclosan, a transformation product of the bactericide triclosan, in fish from various lakes in Switzerland, Environ. Sci. Technol., 2004, 38(2), 390–395 CrossRef CAS PubMed.
- M. E. Delorenzo, J. M. Keller, C. D. Arthur, M. C. Finnegan, H. E. Harper, V. L. Winder and D. L. Zdankiewicz, Toxicity of the antimicrobial compound triclosan and formation of the metabolite methyltriclosan in estuarine systems, Environ. Toxicol., 2008, 23, 224–232 CrossRef CAS PubMed.
- F. M. Zarate, S. E. Schulwitz Jr., K. J. Stevens and B. J. Venables, Bioconcentration of triclosan, methyl-triclosan, and triclocarban in the plants and sediments of a constructed wetland, Chemosphere, 2012, 88(3), 323–329 CrossRef CAS PubMed.
- F. S. Cortez, C. D. Seabra Pereira, A. R. Santos, A. Cesar, R. B. Choueri, G. A. Martini and M. B. Bohrer-Morel, Biological effects of environmentally relevant concentrations of the pharmaceutical Triclosan in the marine mussel Perna perna (Linnaeus, 1758), Environ. Pollut., 2012, 168, 145–150 CrossRef CAS PubMed.
- M. M. Perron, K. T. Ho, M. G. Cantwell, R. M. Burgess and M. C. Pelletier, Effects of triclosan on marine benthic and epibenthic organisms, Environ. Toxicol. Chem., 2012, 31(8), 1861–1866 CrossRef CAS PubMed.
- R. Oliveira, I. Domingues, C. K. Grisolia and A. M. V. M. Soares, Effects of triclosan on zebrafish early-life stages and adults, Environ. Sci. Pollut. Res., 2009, 16, 679–688 CrossRef CAS PubMed.
- M. Farré, D. Asperger, L. Kantiani, S. González, M. Petrovic and D. Barceló, Assessment of the acute toxicity of triclosan and methyl triclosan in wastewater based on the bioluminescence inhibition of Vibrio fischeri, Anal. Bioanal. Chem., 2008, 390(8), 1999–2007 CrossRef PubMed.
- L. M. Weatherly, J. Shim, H. N. Hashmi, R. H. Kennedy, S. T. Hess and J. A. Gosse, Antimicrobial agent triclosan is a proton ionophore uncoupler of mitochondria in living rat and human mast cells and in primary human keratinocytes, J. Appl. Toxicol., 2016, 36(6), 777–789 CrossRef CAS PubMed.
- B. Gaume, N. Bourgougnon, S. Auzoux-Bordenave, B. Roig, B. Le Bot and G. Bedoux, In vitro effects of triclosan and methyl-triclosan on the marine gastropod Haliotis tuberculata, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2012, 156(2), 87–94 CAS.
- S. Macedo, T. Torres and M. M. Santos, Methyl-triclosan and triclosan impact embryonic development of Danio rerio and Paracentrotus lividus, Ecotoxicology, 2017, 26(4), 482–489 CrossRef CAS PubMed.
- J. An, P. P. Guo, Y. Shang, Y. F. Zhong, X. Y. Zhang and Z. Q. Yu, The “adaptive responses” of low concentrations of HBCD in L02 cells and the underlying molecular mechanisms, Chemosphere, 2016, 145, 68–76 CrossRef CAS PubMed.
- M. F. Yueh, K. Taniguchi, S. Chen, R. M. Evans, B. D. Hammock and M. Karin, Tukey RH. The commonly used antimicrobial additive triclosan is a liver tumor promoter, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(48), 17200–17205 CrossRef CAS PubMed.
- A. Hinther, C. M. Bromba, J. E. Wulff and C. C. Helbing, Effects of triclocarban, triclosan, and methyl triclosan on thyroid hormone action and stress in frog and mammalian culture systems, Environ. Sci. Technol., 2015, 45(12), 5395–5402 CrossRef PubMed.
- F. Wang, R. Xu, F. Zheng and H. Liu, Effects of triclosan on acute toxicity, genetic toxicity and oxidative stress in goldfish (Carassius auratus), Exp. Anim., 2018, 67(2), 219–227 CrossRef PubMed.
- K. A. Szychowski, A. Wnuk, M. Kajta and A. K. Wójtowicz, Triclosan activates aryl hydrocarbon receptor (AhR)-dependent apoptosis and affects Cyp1a1 and Cyp1b1 expression in mouse neocortical neurons, Environ. Res., 2016, 151, 106–114 CrossRef CAS PubMed.
- A. Zhang, X. Gu, X. Wang, L. Wang, L. Zeng, X. Fan, C. Jiang, Z. Fu, X. Cui, C. Ji, H. Qu and X. Guo, Potential genetic damage to nematode offspring following exposure to triclosan during pregnancy, Mol. Med. Rep., 2017, 16(2), 1321–1327 CrossRef CAS PubMed.
- G. Chang, W. Xiao, Z. Xu, D. Yu, B. Li, Y. Zhang, X. Sun, Y. Xie, S. Chang, L. Gao, G. Chen, L. Hu, B. Xie, B. Dai, W. Zhu and J. Shi, Pterostilbene induces cell apoptosis and cell cycle arrest in T-cell leukemia/lymphoma by suppressing the ERK1/2 pathway, BioMed Res. Int, 2017, 2017, 9872073 Search PubMed.
- Y. Wang, C. Compton, G. O. Rankin, S. J. Cutler, Y. Rojanasakul, Y. Tu and Y. C. Chen, 3-Hydroxyterphenyllin, a natural fungal metabolite, induces apoptosis and S phase arrest in human ovarian carcinoma cells, Int. J. Oncol., 2017, 50(4), 1392–1402 CrossRef CAS PubMed.
- P. Ming, T. Cai, J. Li, Y. Ning, S. Xie, T. Tao and F. Tang, A novel arylbenzofuran induces cervical cancer cell apoptosis and G1/S arrest through ERK-mediated Cdk2/cyclin-A signaling pathway, Oncotarget, 2016, 7(27), 41843–41856 CrossRef PubMed.
- N. Jette and S. P. Lees-Miller, The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis, Prog. Biophys. Mol. Biol., 2015, 117(2–3), 194–205 CrossRef CAS PubMed.
- A. T. Mohammed, A. A. Mohamed and H. Ali, Pulmonary apoptotic and oxidative damaging effects of Triclosan alone or in combination with Fluoride in Sprague Dawley rats, Acta Histochem., 2017, 119(4), 357–363 CrossRef CAS PubMed.
- E. Movahed, G. M. Tan, K. Munusamy, T. C. Yeow, S. T. Tay, W. F. Wong and C. Y. Looi, Triclosan demonstrates synergic effect with amphotericin B and fluconazole and induces apoptosis-like cell death in cryptococcus neoformans, Front. Microbiol., 2016, 7, 360 Search PubMed.
- L. Galluzzi, S. Kumar and G. Kroemer, Caspases connect cell-death signaling to organismal homeostasis, Immunity, 2016, 44(2), 221–231 CrossRef CAS PubMed.
- R. Brown, The Bcl-2-family of proteins, Br. Med. Bull., 1997, 53, 466–477 CrossRef CAS PubMed.
- G. He, Z. H. Siddik, Z. Huang, R. Wang, J. Koomen, R. Kobayashi, A. R. Khokhar and J. Kuang, Induction of p21 by p53 following DNA damage inhibits both cdk4 and cdk2 activities, Oncogene, 2005, 24(18), 2929–2943 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tx00199e |
| ‡ Co-first author. |
| This journal is © The Royal Society of Chemistry 2019 |