A biodegradable functional water-responsive shape memory polymer for biomedical applications†
Received
19th September 2018
, Accepted 17th November 2018
First published on 20th November 2018
Abstract
Shape memory polymers (SMPs) have exhibited great potential in biomedical applications. However, the typical triggers of shape recovery such as heat, UV light, and electricity may be harmful to humans. Accordingly, water-responsive SMPs have become significant, especially for in vivo applications, due to the intrinsic biocompatibility and ready availability of water. However, the reported water-responsive SMPs are limited and relatively complicated. Here, we design a new water-responsive SMP, poly(butanetetrol fumarate) (PBF); the properties of PBF could be modulated by curing. The cured PBF scaffolds exhibited high shape recovery and fixity rates (>95%). PBF showed good biodegradability, and it could support the attachment, viability and alkaline phosphatase activity of osteoblasts. Furthermore, PBF could be readily functionalized via pendant hydroxyl groups, which was demonstrated by the immobilization and controlled release of bone morphogenetic protein 2. We expect that PBF will be useful for various biomedical applications including water-responsive scaffolds, sensors or actuators.
1. Introduction
Shape memory polymers (SMPs) are one type of the most active intelligent materials. They can be deformed into a desired temporary shape through the application of an external force when been touched stimulus. The temporary shape can be maintained for a long time after removing the stimulus and this external force. They can recover the original shape after re-touching stimulus.1 SMPs have attracted much attention and exhibited great potential in biomedical applications2 such as self-tightening sutures,3 controlled drug release,4 and minimally invasive surgical implants.5,6 The shape recovery of SMPs can be triggered by various stimuli such as heat,3 light,7 electricity,8 and magnetism.9 However, most of them are essentially temperature-responsive. There are some disadvantages associated with these existing SMPs for biomedical applications. First, it is difficult to accurately control the transition temperatures at around 37 °C (body temperature), which is necessary for in vivo applications of the temperature-responsive shape memory effect. Second, the triggers including heat, UV light, electricity, and magnetism are potentially harmful to humans. Third, additional devices or even penetration into the human body is usually required to generate the stimuli for SMPs, which is inconvenient for practical operations. Thus, it is highly desired to employ better triggers of SMPs for biomedical applications. Considering the fact that water is ubiquitous in the human body and intrinsically biocompatible,10 it is very promising to develop water-responsive SMPs, especially for in vivo applications.
However, reports on water-responsive SMPs are relatively limited. Some of them are based on natural biological materials, such as animal hair,11 peacock's tail covert feather12 and luffa sponges.13 They usually suffer from limited resources, difficulty of modification, batch variation, and potential disease transition. Some of them are based on composites such as thermoplastic polyurethane composites,10,14 PEG-based composites, polyvinyl alcohol/Al2O3 nanocomposites,15 chitosan/bioactive materials16–18 and glass nanoparticle scaffolds.5 However, the preparation of these shape memory composites is relatively complicated. The interfacial interaction between different components may be weak, leading to heterogeneous responses to water. Furthermore, only a few of these materials are both biocompatible and biodegradable, which are important properties for in vivo applications such as tissue engineering and drug delivery. Recently, water-responsive poly(lactide-co-glycolic acid) (PLGA) has been investigated as a potential smart nerve conduit.19 However, the resultant shape memory PLGA is hydrophobic and it has a very slow response to water (>1 month). Furthermore, PLGA is biologically inert and lacks modification sites for modulating its properties. For these reasons, it is highly desirable to develop new water-responsive SMPs for biomedical applications.
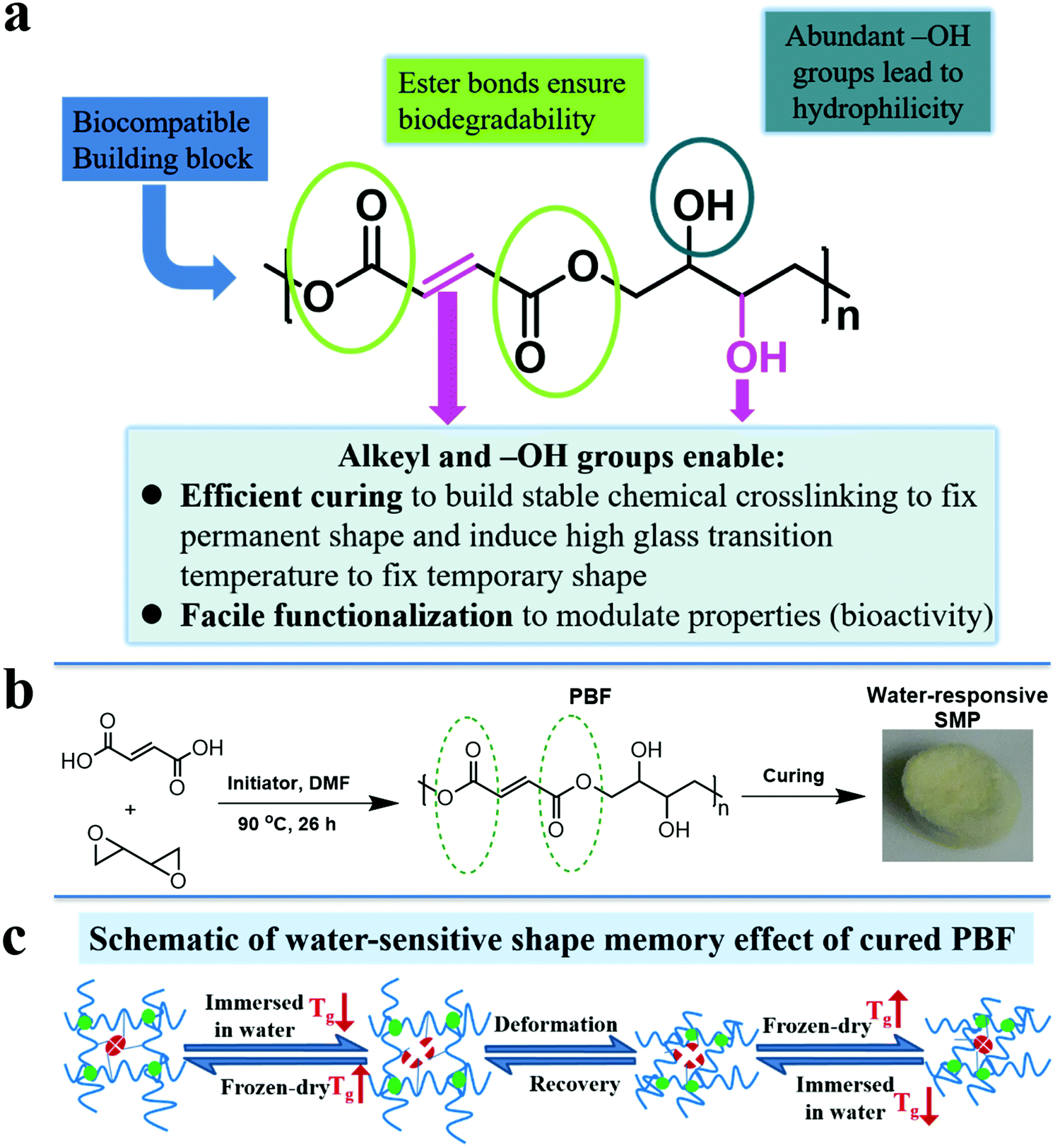
In view of above facts, we designed a functional polyester, poly(butanetetrol fumarate) (PBF) (Fig. 1a), for potential applications as a water-responsive SMP, according to following considerations: (1) multiple interactions for shape memory effect. PBF contains both pendent hydroxyl groups and terminal carboxylic groups, which enable facile chemical crosslinking to build three dimensional covalent networks for determining the permanent shape (Fig. 1b). The H-bonds between the extensive hydroxyl and ester groups and the π–π interaction between the rigid fumarates can lead to a high glass transition temperature (Tg) to fix the temporary shape at ambient and body temperatures (Fig. 1c). The PBF scaffold can be plasticized by water and becomes elastic for reversible deformation into a temporary shape, which is fixed after drying. After being immersed in water, the PBF scaffold becomes elastic again for restoration to its permanent shape. (2) Good hydrophilicity for water responsiveness. The moieties of PBF including fumaric acid and butanetetrol are all water soluble. The resultant PBF contains extensive hydroxyl groups. Thus, we expect that PBF can have good hydrophilicity and water can efficiently plasticize cured PBF through weakening of the interchain physical interactions to trigger the shape memory process. (3) Facile modification for diverse functionalization. The hydroxyl, alkenyl, and carboxyl groups in PBF enable multiple chemical transformations such as bioconjugation to efficiently modulate the physical, mechanical and biological properties. (4) Concise synthesis for large scale applications. PBF can be readily synthesized from commercially available fumaric acid and 2,2′-bioxirane via acid-induced epoxide ring-opening polymerization in one step.20 (5) Potentially good biodegradability and biocompatibility. The hydrolyzable ester bonds in the main chain ensure biodegradability. The edible building blocks including fumaric acid and butanetetrol would likely lead to good biocompatibility. Furthermore, the abundant hydroxyl and carboxyl groups may promote cell adhesion and proliferation.21 Herein, we report the synthesis, characterization, and cytocompatibility of PBF and a detailed investigation of PBF-based water-responsive SMPs.
 |
| | Fig. 1 Design, synthesis, and working mechanism of PBF-based shape memory polymer (a) design principles. (b) Synthesis of shape memory cured PBF. (c) Schematic of water-responsive shape memory mechanism of PBF. | |
2. Experimental section
2.1. Chemical reagents
1,3-Butadiene diepoxide (Alfa Aesar, 95%) was distilled under vacuum, and fumaric acid (J&K Chemical, 99%) was recrystallized from deionized water. Tetrabutylammonium bromide (TCI, 98%) and anhydrous N,N-dimethylformamide (DMF, J&K Chemical, 98.8%) were used as received. Tetrahydrofuran (THF, 99%) and acetone were purchased from Sinopharm Chemical Reagent Co., Ltd, China.
2.2. Synthesis of PBF
An equimolar amount of distilled 1,3-butadiene diepoxide and recrystallized fumaric acid and 0.6 mol% tetrabutylammonium bromide were mixed and dissolved in anhydrous DMF in a reaction bottle and sealed in a glove box under nitrogen. The vial was transferred out of the glove box. The mixture in the reaction bottle was reacted at 85 °C under a nitrogen atmosphere for 26 h. The resultant PBF solution was purified via precipitation in acetone and dried under vacuum at ambient temperature for one day.
2.3. Polymer characterization
The molecular weight was determined via gel permeation chromatography (GPC) on a Malvern GPC system consisting of GPC Max with a Dual 270 triple detector array (differential refractive index and right-angle light scattering, low-angle static light scattering, and four-capillary differential viscometer detectors), and DMF (HPLC grade) was used as the eluent at a flow rate of 0.7 mL min−1 at 40 °C. The 1H NMR spectrum was recorded on Bruker 400 NMR. The attenuated total reflectance-Fourier transformed infrared (ATR-FTIR) spectra were recorded on a Thermo Nicolet 6700 spectrometer. Thermo-gravimetric analysis (TGA) of PBF was performed on Discovery TGA (TA, American) from 40 °C to 500 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere. Different scanning calorimetry (DSC) was performed on TA DSC Q200 at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The glass transition temperature (Tg) was referred to the midpoint of the glass transition. The static air–water contact angles of the polymer films were tested on an AST Products Inc. VCA2000 instrument. PBF solution was dropped on a glass slide and dried at room temperature in the air. After the solvent evaporated, a PBF film was obtained. The average values and errors were calculated from at least three independent data for each specimen in this paper.
2.4. Fabrication and characterization of PBF scaffolds
The porous PBF scaffolds were readily prepared by the salt leaching method.22 Briefly, THF solution of PBF (10% w/v) was added to the salt template made of salt particles with a size range from 75 to 150 μm and then, THF was allowed to evaporate completely in a fume hood for 1 h. The mold was transferred to a vacuum oven and cured at 150 °C and 1 Torr vacuum for a predetermined amount of time (24 h and 36 h, marked as PBF-24 and PBF-36, respectively). The resultant PBF-impregnated salt template was immersed in deionized water for 24 h and then freeze-dried to obtain a porous PBF scaffold.
Thermal properties were investigated by TGA and DSC. TGA of the PBF scaffold was performed on Discovery TGA (TA, American) from 40 °C to 500 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere. DSC was performed on TA DSC Q200 from 0 °C to 170 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere. Tg was referred to the midpoint of the glass transition.
The morphology was investigated by scanning electron microscopy (SEM). The porosity of the PBF scaffold was determined by the following equation:
| | 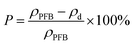 | (1) |
Here,
ρd refers to the apparent density, and
ρPBF refers to the real density determined
via a Vacuum density meter (UL TRAPYCNOMETER100, Quanta chrome).
The mechanical properties of the PBF scaffolds were evaluated by compression tests at room temperature using an Instron 5969 universal testing machine equipped with a 5 N sensor. The specimen (10.8 mm diameter, 5 mm height) was compressed at a speed of 1.00 mm min−1 to 40% strain with an initial load of 0.01 N. In the cyclic compression test, the sample was compressed to a strain of 40% and recovered to 5% before immediately compressing to 40% five times.
| | 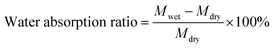 | (2a) |
Here,
Mdry is the original weight of the dry sample and
Mwet is the weight of the scaffold absorbing de-ionized water.
| | 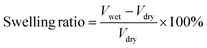 | (2b) |
Here,
Vdry is the original volume of the dry sample and
Vwet is the volume of the scaffold absorbing de-ionized water.
2.5. Shape memory behavior of PBF scaffolds
The following experiment was carried out at room temperature to characterize the shape memory behavior of the PBF scaffold. The PBF scaffold was immersed in de-ionized water for 3 min and then compressed to 30% strain of its original height. After freeze-drying, the sample maintained its compressed shape. Then, it was re-immersed in de-ionized water and recovered to its original shape. Shape fixity (Rf) and shape recovery (Rr) were used to evaluate the shape memory effect. Rf and Rr were calculated from the following equation:| | 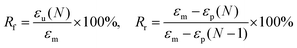 | (3) |
Here, εm is the maximum strain in the compression cycle, εu(N) is the residual strain after unloading in the dry state, and εp(N) and εp(N − 1) are the final strains for two continuous cycles after shape recovery in water.
2.6. Functionalization of PBF scaffolds
PBF scaffolds were soaked in 2-(N-morpholino) ethanesulfonic acid hydrate buffer solution (MES, 0.05 mol L−1, pH = 5.5), which contained N-hydroxysuccinimide (NHS, 0.06 M) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 0.6 M). PBF scaffolds were rinsed three times with Dulbecco's phosphate buffered saline (DPBS) after soaking for 30 min. Bone morphogenetic protein 2 (BMP2, 100 ng) solution in DPBS (1% bovine serum albumin, 40 μL) was dropped in the PBF scaffold and placed in an incubator (37 °C, 5% CO2) to fix. The fixed amount of BMP2 in the PFB scaffolds and its subsequent release from the PBF scaffolds can be determined by enzyme-linked immunosorbent assay (ELISA).
2.7. Cell adhesion
Osteoblasts were isolated by sequential trypsin–collagenase digestion of calvaria obtained from neonatal (2–3 days old) Sprague–Dawley rats.23 Generally, passage 4 osteoblasts were used for all cell function tests.
Osteoblasts were seeded on 24-well TCPS plates coated with PBF and PLGA, which were prepared as previously described for 2, 4, and 24 h, respectively.22 At each time point, adhered cells on the plate were enzymatically detached by 0.25% trypsin–0.1% EDTA-Gibco and counted using a hemocytometer. Cell adhesion rate was calculated by the percentage of original seeded cells (2 × 104 cells well−1).
2.8. Cell morphology
Osteoblasts were cultured on TCPS covered with PBF and PLGA in a humidified incubator. Phase contrast images were obtained using an inverted microscope Eclipse Ti (Nikon, Melville, NY) equipped with a RETI-GA-SRV digital camera (Q-Imaging, BC, Canada) after cell seeding for 1 h. The culture media were removed after 24 h cell seeding on PBF and PLGA. Cells were fixed for 10 min with 4% paraformaldehyde and permeated with 0.1% Triton-X 100 for 5 min. Bovine serum albumin (BSA) (2%) was added to plates to protect protein as confining liquid. Then, cells were cultivated in rhodamine phalloidin (Cytoskeleton, #PHDR1, 14l M stock solution was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 in 0.5% BSA) for 30 min at room temperature (in darkness) and the nuclei were stained with DAPI for 20 min at room temperature in darkness. F-Actin fluorescence images were captured by Fluorescent micrographs (Nikon Eclipse Ti microscope) at 594 nm to present the structure of cytoskeleton. Besides, cells were cleaned with DPBS (3 × 1 mL) three times before each step.
:200 in 0.5% BSA) for 30 min at room temperature (in darkness) and the nuclei were stained with DAPI for 20 min at room temperature in darkness. F-Actin fluorescence images were captured by Fluorescent micrographs (Nikon Eclipse Ti microscope) at 594 nm to present the structure of cytoskeleton. Besides, cells were cleaned with DPBS (3 × 1 mL) three times before each step.
2.9. Cell viability (live/dead assay)
Cell viability was determined using a calcein and ethidium homodimer live/dead viability assay according to the manufacturer's instructions. Stained cells were visualized using a fluorescence microscope (Nikon Eclipse Ti microscope) and imaged with Nikon Digital Sight DS-SMc for further analysis. Live cells were stained positive with green fluorescence, whereas dead cells were visualized by red-stained nuclei. The red and green images were superimposed to yield the final live/dead results.
2.10. Alkaline phosphatase activity assay
Osteoblasts were seeded on 24-well plates (2 × 104 well−1) coated with PBF or PLGA for 7, 14, and 21 days. At each time point, cell lysis (200 μL) and Protease Inhibito were added to the clean plates to quiescence in ice for 15 min and then, the cells were scraped. The lysis was collected and centrifuged (13362 × g) at 4 °C for 15 min. The supernatant (10 μL) was mixed with p-nitrophenylphosphate (pNPP) (100 μL), as a phosphatase substrate, for 30 min at room temperature in darkness to generate a yellow product, which was spectrophotometrically quantified at 405 nm to present the concentration of protein under a SynergyMX plate reader (Biotek, Winooski, VT).
2.11.
In vitro degradation experiment
The in vitro enzymatic degradation test was performed in DPBS containing lipase enzyme from Thermomyces lanuginosus (100000 U g−1, Sigma) at an activity of 2000 U mL−1. A cylindrical PBF scaffold (5 mm diameter, 5 mm height) was weighed and immersed in 6 mL DPBS with lipase enzyme and then incubated at 37 °C. The sample was retrieved, washed with distilled water, and dried every 12 h. The degree of degradation was determined by the dry-weight change.
2.12. Statistical analysis
Statistical analysis was performed using one-way ANOVA; p < 0.05 was the considered level for statistical significance. All values were reported as mean ± standard deviation.
3. Results and discussion
3.1. Synthesis and characterization of PBF
PBF (Mn = 59.7 kD, PDI = 2.5) was readily synthesized based on acid-induced epoxide ring-opening polymerization in one step according to our previous report.20,24 The structure of PBF was characterized by 1H NMR (Fig. 2a) and ART-FTIR (Fig. 2b). In the 1H NMR spectrum, the sharp peak at 7.00 ppm corresponded to the alkene protons of the fumarate moiety in the polymer backbone. The multiple peaks at 4.5–3.5 ppm corresponded to the signals of CH2 or CH protons of the butane-1,2,3,4-tetraol moiety in the polymer backbone. The FTIR spectra also confirmed the presence of different functional groups. The broad peak at 3390 cm−1 corresponded to the stretch vibration of O–H bond. The peak at 3090 cm−1 corresponded to the stretch vibration of C–H bond in alkenyl groups. The sharp peaks at 1710 cm−1 and 1640 cm−1 corresponded to the stretch vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds.
C and CO bonds.
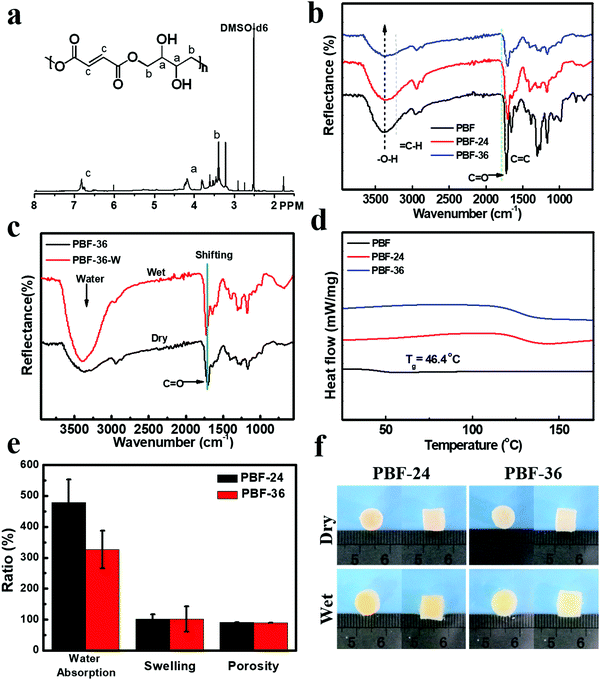 |
| | Fig. 2 Structural and morphology characterization of PBF polymer and scaffolds. (a) 1H NMR spectrum of PBF. (b) FTIR spectra of PBF and scaffolds. (c) Comparison of the FTIR spectra of dry and wet PBF-36 scaffolds. (d) DSC curves of PBF and scaffolds. (e) Water absorption, volume swelling ratio and porosity of PBF scaffolds. (f) Images of dry and wet PBF scaffolds. | |
The thermal decomposition temperature of PBF was 237 °C, as determined by TGA, revealing good thermostability of PBF at body temperature. The Tg value of PBF was 46.5 °C (Fig. 2d), indicating that it was a glassy material at body temperature. The water contact angle of PBF film was 15.6 ± 1.7°, confirming its good hydrophilicity likely due to abundant hydroxyl groups. All these properties revealed the potential of PBF for water-responsive shape memory materials.
3.2. The fabrication and structural characterization of PBF scaffolds
PBF porous scaffolds were fabricated by thermal curing and salt leaching method, as described previously. To investigate the effect of curing time, we prepared two types of scaffolds PBF-24 and PBF-36 cured at 150 °C for 24 h and 36 h, respectively. The condensation reaction between pendent hydroxyl groups and terminal carboxyl groups resulted in covalent networks. As shown in the ATR-FTIR curves (Fig. 2b), the intensity of the characteristic peaks of O–H stretching decreased significantly with the increase in curing time, indicating the progress of the condensation reaction. The resultant PBF scaffolds could not be dissolved in common solvents such as acetone, THF, and DMF, confirming the stable covalent crosslinks.
ATR-FTIR was used to investigate the interaction between water molecules and PBF scaffolds (Fig. 2c). There were significant differences between the spectra of PBF scaffolds in the dry and wet states. The characteristic CO stretching peak for the PBF-36 scaffold shifted to a higher wavenumber from 1708 cm−1 (dry sample) to 1719 cm−1 (wet sample), indicating the formation of intermolecular hydrogen bonds between the PBF scaffold and water molecules. The broad and strong peak around 3400 cm−1 in the wet sample corresponded to absorbed water.
3.3. Thermal properties of PBF scaffolds
Both PBF-24 and PBF-36 scaffolds were stable up to 220 °C. Thus, they were thermally stable for typical biomedical applications at room and body temperatures. PBF-24 and PBF-36 scaffolds exhibited Tg values of 127.3 °C and 128.4 °C, respectively, which were much higher than that of the PBF polymer because the covalent crosslinks in the scaffolds greatly constrained the mobility of the molecular chains. Thus, the PBF scaffolds were in a glassy state, which could provide solid interactions to fix their temporary shapes.
3.4. The morphology of PBF scaffolds
The main macropores (75–150 μm) regulated via salt particles were distributed throughout the scaffold and were connected to each other. The PBF scaffolds had a porous structure with average porosity values of 91.08 ± 0.01% and 88.40 ± 0.04% after curing for 24 h and 36 h, respectively (Fig. 2e). Both PBF-24 and PBF-36 scaffolds could absorb significant amount of water at the weight ratios of 478.9 ± 74.5% and 326.7 ± 60.6%, respectively. These results confirmed the good hydrophilicity of the PBF scaffolds. The decrease in the water absorption ratio with increasing curing time was ascribed to the consumption of hydrophilic hydroxyl and carboxylic groups in the curing reaction. At the same time, the volume swelling ratios of PBF-24 and PBF-36 in water were only 102.3 ± 14.4% and 101.7 ± 41.3%, respectively, indicating their good shape stability in a water environment (Fig. 2f), which was possibly due to their stable covalent networks.
3.5. Mechanical properties of PBF scaffolds
The typical stress vs. strain curves of the PBF scaffolds in simple compression tests are shown in Fig. 3a. Both PBF-24 and PBF-36 scaffolds could resist a strain of up to 40%. Their elastic moduli could be well regulated in a wide range by curing duration. Dry PBF-24 scaffolds exhibited a modulus of 26.52 ± 2.3 kPa. After extending the curing time to 36 h, the modulus of the PBF-36 scaffolds significantly increased to 182.27 ± 22.35 kPa, which was around 7 times that of the PBF-24 scaffolds.
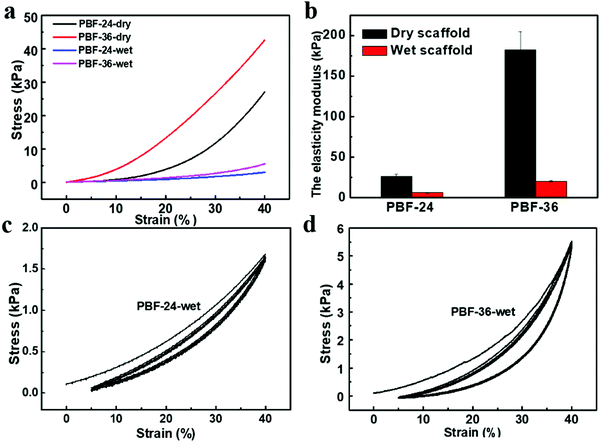 |
| | Fig. 3 Mechanical properties of PBF scaffolds. (a) Typical simple compression stress vs. strain curves of the PBF scaffolds. (b) Comparison of the elastic moduli of dry and wet scaffolds revealed the water-responsive mechanical behavior and the water-triggered stiff-soft transition. (c and d) Cyclic compression tests from 5% to 40% for 5 cycles of wet PBF-24 and PBF-36 scaffolds, indicating their good elasticity. | |
To evaluate the potential of PBF scaffolds as shape memory devices, we investigated their water-responsive mechanical behavior. The moduli of wet PBF-24 and PBF-36 scaffolds greatly reduced to 6.34 ± 0.27 kPa and 20.36 ± 0.97 kPa, respectively (Fig. 3b). This confirmed their water-responsive sensitivity due to their high hydrophilicity. Water molecules could efficiently diffuse into the crosslinked network to plastify it, triggering stiff-soft transition of the PBF scaffolds, which was the key for water-responsive shape memory effect. The modulus of the PBF-36 scaffolds was around three times that of the PBF-24 scaffolds in the wet state. This further revealed the efficiency of modulation of the elastic moduli of PBF scaffolds via adjusting the crosslinking degree. In addition, the cyclic compression (Fig. 3c and d) tests showed that both wet PBF scaffolds recovered well from dynamic deformation with limited hysteresis. This revealed that the wet PBF scaffolds have good elasticity, which is favorable for implantation in mechanically dynamic environments.
3.6. Water-responsive shape-memory effect
We investigated the water-induced shape-memory effect of the PBF scaffolds in water (Fig. 4). Fig. 4a and b illustrate the macroscopic and microscopic water-responsive shape-recovery progress, respectively. Original the PBF scaffold displayed extensive micropores. The cylindrical sample was immersed in water for 10 minutes, compressed, and then freeze-dried to fix its temporary shape. The sample maintained its shape fixity until it was submerged in water, where it recovered its original shape and microporous morphology within a few minutes. Both the macrostructure and the microstructure evolution demonstrated the high efficiency of shape fixity and recovery of PBF-24 scaffold. We used the shape fixity ratio Rf and shape recovery ratio Rr to evaluate the ability of PBF scaffolds to memorize their temporary shape and permanent shape, respectively (Fig. 4c–e). Fig. 4c shows the relationship between recovery rate and recovery time. Rr reached around 90% within 1 min in water. Fig. 4d and e present Rf and Rr after 2 min corresponding to the testing cycles. Both PBF-24 and PBF-36 exhibited high Rf and Rr over 95% for at least five cycles.
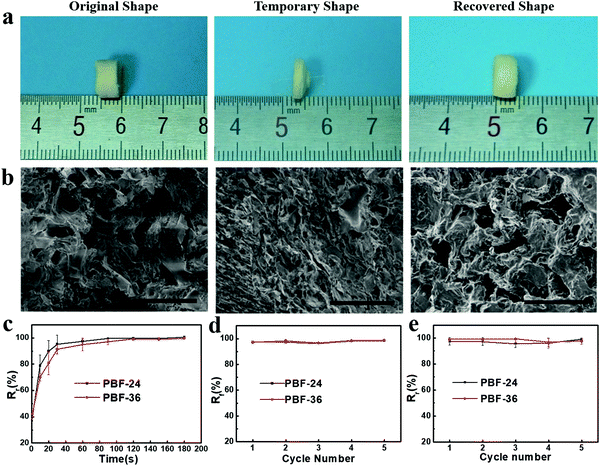 |
| | Fig. 4 (a) Photo images and (b) SEM images (magnification 100×, scale bar = 200 μm) of water-responsive shape-memory process of the PBF-24 scaffold. (c–e) Quantification of water-responsive shape-memory effect of PBF scaffolds. (c) Rrvs. recovery time curves of PBF scaffolds. Shape fixity (d) and recovery (e) ratios of PBF scaffolds for five cycles. | |
Next, we used the PBF-24 scaffolds as an example to further investigate different shape memory processes (Fig. 5). The PBF scaffolds were deformed to different shapes in a roll, fold and helix in water and fixed to the temporary state via freeze drying. All the PBF scaffolds readily recovered their original shapes after around 3 min in water, showing excellent water-responsive shape-memory property.
 |
| | Fig. 5 Water-responsive shape-memory effect of PBF scaffolds from various temporary shapes. | |
3.7. Functionalization of PBF scaffolds
As designed, the extensive free hydroxyl groups in PBF enable facile modifications of PBF including biofunctionalization, which are highly desirable for biomedical applications. Here, immobilization of BMP2 was used as an example to prove the potential of the functionalization of PBF. BMP2 was readily incorporated to PBF scaffolds via a typical NHS/EDC-mediated esterification reaction between the hydroxyl groups of PBF and the carboxylic acid groups of BMP2. BMP2 was sustainably released from the scaffold with an amount of 93% over 21 days due to the gradual hydrolysis of ester bonds in the DPBS buffer (Fig. 6).
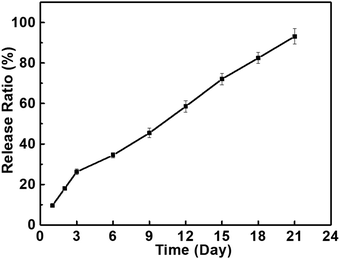 |
| | Fig. 6 The release profiles of BMP2 on the PBF-24 scaffolds in DPBS buffer (containing 1% BSA, 37 °C, 5% CO2). | |
3.8. Cell morphology and adhesion
Cell morphology was investigated through phase contrast and fluorescence (F-actin staining) images (Fig. 7a–d). Osteoblasts showed typical polygonal morphology on both PBF and PLGA. The cell adhesion rate on both polymers significantly increased from 2 h to 4 h. Also, 93.3 ± 16.3% cells adhered to PBF within 24 h (Fig. 7e). At all time points, the cell adhesion rates on PBF were as much as that on PLGA. These results demonstrated that PBF is a good substance for osteoblast attachment compared to the standard biocompatible polymer PLGA.
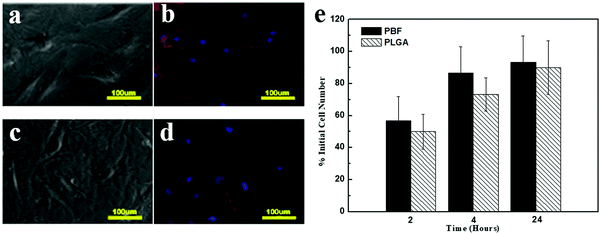 |
| | Fig. 7 Cell attachment. Phase contrast images (a and c) and fluorescence images of stained F-actin (b and d) of osteoblasts 24 h after seeding onto PBF (a and b) and PLGA (c and d) (magnification 200×, scale bar = 100 μm). (e) Osteoblast adhesion rates on PBF and PLGA. | |
3.9. Cell viability
The cytotoxicity of PBF was examined via Live/Dead assay at 24 and 48 h (Fig. 8a–e). Live and dead cells were stained as green and red fluorescences, respectively. High cell viability of the osteoblasts was viewed on both polymers, and few dead cells (red) were observed at all detections. Also, 90.5 ± 0.07% cells were alive on PBF after 48 h culture.
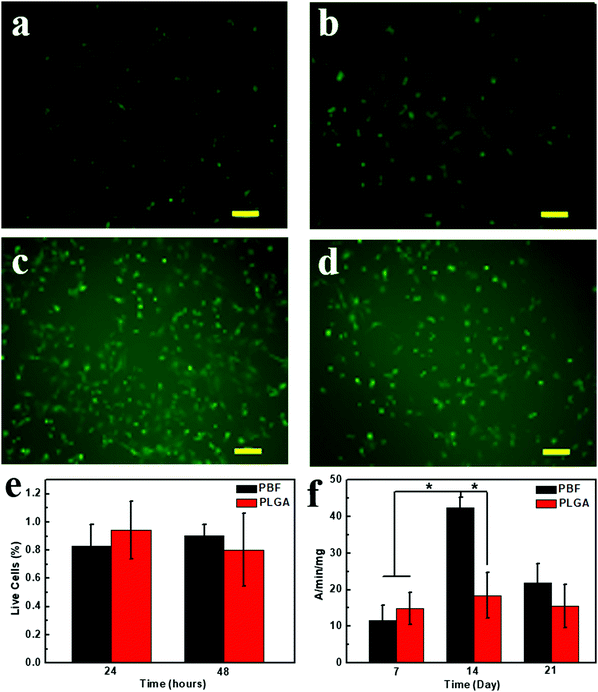 |
| | Fig. 8 (a–e) Live/Dead assay and ALP activity of osteoblasts cultured on PBF and PLGA. Fluorescence images of osteoblasts cultured on PBF (a and c) and PLGA (b and d) at 24 h (a and b) and 48 h (c and d). (e) The percentage of viable cells on both polymers at 24 h and 48 h. (f) Alkaline phosphatase activity assay. ALP activity of osteoblasts cultured on PBF and PLGA (magnification 40×, scale bar 100 μm). | |
3.10. Alkaline phosphatase activity assay
An alkaline phosphatase (ALP) activity assay was used to investigate the activity and function of osteoblasts on PBF (Fig. 8f). Osteoblasts on PLGA and PBF showed similar ALP activities at day 7. ALP activity of osteoblasts on PBF dramatically increased from day 7 to day 14 and was significantly higher than that on PLGA. Since ALP is the early marker of osteogenesis, ALP activity of the osteoblasts on PBF decreased from day 14 to day 21. The higher maturation of osteoblasts on PBF than on PLGA was likely due to the abundant hydroxyl groups of PBF.23
3.11. Degradation of PBF scaffolds
The PBF scaffolds exhibited good biodegradability in DPBS of lipase enzyme at activity of 2000 U mL−1 at 37 °C. A total of 62.7 ± 5.7% mass of PBF-24 scaffolds was lost within 7 days (Fig. 9). Compared to PBF-24 scaffolds, the PBF-36 scaffolds exhibited slower degradation with mass loss of 22.3 ± 7.1% within 7 days, which was possibly due to the higher crosslink density. With the increase in lipase enzyme activity to 8000 U mL−1, the degradations of PBF scaffolds significantly accelerated, indicating that the ester bonds were their primary degradation sites (Fig. S1, ESI†).
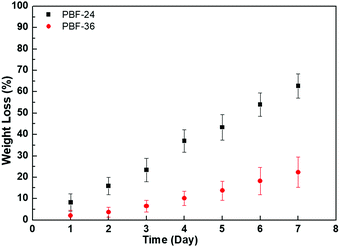 |
| | Fig. 9
In vitro enzymatic degradation of PBF scaffolds in lipase DPBS solutions at 37 °C. | |
4. Conclusions
We designed and synthesized a new kind of SMP, i.e., PBF. PBF porous scaffolds showed excellent water-responsive shape-memory effect with high Rr (>95%) and Rf (>95%) for multiple cycles. The mechanical properties of the PBF scaffolds can be adjusted by changing the curing conditions over a wide range. PBF exhibited good biocompatibility and biodegradability. More importantly, PBF can be readily functionalized via abundant hydroxyl and alkenyl groups, exemplified by the immobilization of BMP2, which is highly desired for diverse applications. This research provides a new class of shape-memory materials, which combine diverse functionalizability and favorable water-responsiveness for in vivo utilization. We expect that these materials will be very useful for biomedical applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21574019, 81320108010, 81470662, 31570984), the Natural Science Foundation of Shanghai (18ZR1401900), International Joint Laboratory for Advanced fiber and Low-dimension Materials (18520750400), and the Science and Technology Commission of Shanghai (17DZ2260100).
References
- J. S. Leng, X. Lan, Y. J. Liu and S. Y. Du, Prog. Mater. Sci., 2011, 56, 1077–1135 CrossRef CAS.
- L. Sun, W. M. Huang, Z. Ding, Y. Zhao, C. C. Wang, H. Purnawali and C. Tang, Mater. Des., 2012, 33, 577–640 CrossRef CAS.
- A. Lendlein and R. Langer, Science, 2002, 296, 1673–1676 CrossRef PubMed.
- A. T. Neffe, B. D. Hanh, S. Steuer and A. Lendlein, Adv. Mater., 2009, 21, 3394–3398 CrossRef CAS.
- C. O. Correia, A. J. Leite and J. F. Mano, Carbohydr. Polym., 2015, 123, 39–45 CrossRef CAS.
- T. M. Filion, J. W. Xu, M. L. Prasad and J. Song, Biomaterials, 2011, 32, 985–991 CrossRef CAS.
- A. Lendlein, H. Y. Jiang, O. Junger and R. Langer, Nature, 2005, 434, 879–882 CrossRef CAS.
- C. W. Li, L. Qiu, B. Q. Zhang, D. Li and C. Y. Liu, Adv. Mater., 2016, 28, 1510–1516 CrossRef CAS PubMed.
- Z. W. He, N. Satarkar, T. Xie, Y. T. Cheng and J. Z. Hilt, Adv. Mater., 2011, 23, 3192–3196 CrossRef CAS PubMed.
- Y. Zhu, J. L. Hu, H. S. Luo, R. J. Young, L. B. Deng, S. Zhang, Y. Fan and G. D. Ye, Soft Matter, 2012, 8, 2509–2517 RSC.
- X. L. Xiao and J. L. Hu, Sci. Rep., 2016, 6 DOI:10.1038/srep26393.
- Z. Q. Liu, D. Jiao and Z. F. Zhang, Biomaterials, 2015, 65, 13–21 CrossRef CAS.
- J. H. Shen, Y. M. Xie, S. W. Zhou, X. D. Huang and D. Ruan, J. Mech. Behav. Biomed., 2014, 34, 283–293 CrossRef CAS.
- T. F. Wu, K. O'Kelly and B. Q. Chen, J. Mech. Behav. Biomed. Mater., 2014, 52, 55–62 CAS.
- Q. M. Bai, G. Z. Zhang, B. Xu, X. Q. Feng, H. Y. Jiang and H. J. Li, RSC Adv., 2015, 5, 91213–91217 RSC.
- Y. L. Cui, M. Tan, A. D. Zhu and M. Y. Guo, J. Mater. Chem. B, 2014, 2, 5478 RSC.
- A. V. Salvekar, W. M. Huang, R. Xiao, Y. S. Wong, S. S. Venkatrarnan, K. H. Tay and Z. X. Shen, Acc. Chem. Res., 2017, 50, 141–150 CrossRef CAS.
- Y. S. Wong, A. V. Salvekar, K. Da Zhuang, H. Liu, W. R. Birch, K. H. Tay, W. M. Huang and S. S. Venkatraman, Biomaterials, 2016, 102, 98–106 CrossRef CAS PubMed.
- C. Chen, J. Hu, H. Huang, Y. Zhu and T. Qin, Adv. Mater. Technol., 2016, 1, 1600015 CrossRef.
- Z. W. You, H. P. Cao, J. Gao, P. H. Shin, B. W. Day and Y. D. Wang, Biomaterials, 2010, 31, 3129–3138 CrossRef CAS.
- R. Vasita, K. Shanmugam and D. S. Katti, Curr. Top. Med. Chem., 2008, 8, 341–353 CrossRef CAS.
- P. Huang, X. P. Bi, J. Gao, L. J. Sun, S. F. Wang, S. Chen, X. Q. Fan, Z. W. You and Y. D. Wang, J. Mater. Chem. B, 2016, 4, 2090–2101 RSC.
- X. P. Bi, Z. W. You, J. Gao, X. Q. Fan and Y. D. Wang, Acta Biomater., 2014, 10, 2814–2823 CrossRef CAS.
- Z. W. You, X. Bi and Y. D. Wang, Macromol. Biosci., 2012, 12, 822–829 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02462f |
| ‡ Co-first author. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Ziying
Lv‡
a,
Yiru
Huo
a,
Lijie
Sun
a,
Ziying
Lv‡
a,
Yiru
Huo
a,
Lijie
Sun