
ZnS spheres wrapped by an ultrathin wrinkled carbon film as a multifunctional interlayer for long-life Li–S batteries†
Jin-Lin
Yang
ab,
Shi-Xi
Zhao
 *a,
Yi-Ming
Lu
ab,
Xiang-Tian
Zeng
ab,
Wei
Lv
a and
Guo-Zhong
Cao
*c
*a,
Yi-Ming
Lu
ab,
Xiang-Tian
Zeng
ab,
Wei
Lv
a and
Guo-Zhong
Cao
*c
aTsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055, China. E-mail: zhaosx@sz.tsinghua.edu.cn
bSchool of Materials Science and Engineering, Tsinghua University, Beijing, 100084, China
cDepartment of Materials Science and Engineering, University of Washington, Seattle, WA 98195, USA. E-mail: gzcao@uw.edu
First published on 9th November 2019
Abstract
The shuttling of soluble lithium polysulfides (LiPSs) in Li–S batteries during the cycling process is considered as a serious problem to handle. It often brings about sluggish redox kinetics, rapid capacity decay and limited sulfur utilization. In this study, we fabricate a novel functional interlayer by wrapping dispersed zinc sulfide (ZnS) nanospheres with a graphene-like ultrathin wrinkled carbon film (ZnS@WCF) through a facile hard-template method. The WCF acts as a unique 3D conductive network and ZnS nanospheres provide strong chemisorption of LiPSs. Owing to the synergistic effects between polar ZnS and the conductive WCF, the as-prepared interlayer can not only serve as a physical barrier but can also recycle the dissolved LiPSs with fast reaction kinetics. Li–S batteries based on such a ZnS@WCF modified separator exhibit remarkable capacity retention and outstanding rate performance. When cycling at 1C, a considerable specific capacity of 685 mA h g−1 can still be maintained after 600 cycles with an extremely low fading decay of 0.045% per cycle. This novel interlayer holds great potential in realizing the wide practical application of Li–S batteries.
1. Introduction
Since the demand for energy storage devices with high energy density and long life has sharply increased in recent years, conventional lithium-ion batteries have reached their energy density limit.1,2 As a promising next generation energy storage system, lithium–sulfur (Li–S) batteries have attracted extensive attention because of their high energy density (2600 W h kg−1), high specific capacity (1673 mA h g−1), low-cost and nontoxicity.3,4 However, the practical application of Li–S batteries is plagued by several drawbacks including (1) the rapid capacity fading caused by shuttling of intermediate LiPSs (shuttle effect); (2) the low electrical conductivity of sulfur and solid Li2S; and (3) the volume fluctuation of the cathode induced by density difference between sulfur and Li2S.5,6To deal with the issues mentioned above, many researchers are focusing on designing and synthesizing cathode materials with a novel sulfur host structure such as a carbon-based porous structure, hollow structure and 3D network sulfur hosts. These functional cathode materials not only exhibit outstanding electroconductivity but also can offer large pore volume to buffer the volume change and enhance the infiltration of electrolyte. Nevertheless, the nonpolar surface of carbon-based materials could do little to prevent LiPS species shuttling. In order to increase the polarity of host materials, a series of metal oxides, sulfides and nitrides have been applied due to their strong chemisorption to LiPSs through Li- or S- binding.7,8 Although they could trap LiPS species on their surface, such metallic compounds often have poor conductivity, affecting the electrochemical kinetics of the Li–S redox.9 So many studies pointed out that the conversion route from sulfur to soluble LiPSs and further to solid Li2S should be optimized by combining nanosized polar materials with a carbon matrix or inducing a heterostructure with a charge improved interface in order to achieve fast conversion kinetics during cycling.10–12 The “inside” design of sulfur cathodes often includes complex fabrication process or expensive producing cost of sulfur host materials. Thus, introducing an “outside” interlayer between the traditional sulfur–carbon cathode and separator is deemed to be a more sufficient and facile way to meet the above requirements.
Carbon materials such as carbon nanotubes, carbon paper and graphene are widely investigated as interlayers due to their high conductivity and good flexibility. Such a carbon-based interlayer not only serves as a physical barrier to block the diffusion of LiPSs but also acts as a second current collector to improve the conductivity of the whole cathode region.13 Meanwhile, as mentioned above, the nonpolar surface of the carbonaceous interlayer could only limit the shuttling of polar LiPS species by being a physical barrier, thus causing capacity loss. Similar to the strategies of sulfur host design, many polar inorganic compounds, such as SiO2,14 Al2O3,15 TiO2,16 SnO2,17 Ti4O7,18 MoO3,19 MoS2,20 TiN,21 Co9S8,22 InN,23 V2O5,24 and WO3,25 have been introduced into the interlayer to anchor LiPSs through strong chemical interaction. However, the separator modified by polar materials only would alleviate the shuttle effect at the expense of ion transfer and electrical conductivity. In this case, combining polar inorganics with a conductive carbon matrix is considered as a promising way to synergize the merits of absorptivity and conductivity. Typically, nanostructured polar materials are physically or chemically grown or coated on the surface of a carbon matrix such as carbon cloth,26 carbon fibres,27 and graphene sheets.28 With the rational design of a covalently bonded interface (TiO2–TiN,29 Co9S8−x–CNTs30) or cooperative interface (GN–LDH),31 such heterostructures are considered to accelerate the redox kinetics through the typical ‘adsorption–diffusion–conversion’ process and further sufficiently recycle the LiPS species from the cathode.
Herein, we present a 0D–3D combination hybrid material (ZnS@WCF) as an efficient LiPS-recycling mediator with fast reaction kinetics. Well dispersed ZnS nanospheres are wrapped by a graphene-like ultrathin wrinkled carbon film (WCF) via a hard template method. Within the resultant ZnS@WCF interlayer configuration, the micropore enriched WCF is considered as a flexible crosslinked 3D conductive framework and a robust physical barrier. Specifically, the curly and wrinkled structure of the porous WCF offers sufficient interspaces to hold the electrolyte and thus ensures the Li ion transfer. In addition to physical confinement by the WCF, ZnS nanospheres with the WCF coating serve as the anchoring sites for LiPS trapping. ZnS is a promising material for long life and fast kinetics Li–S batteries because it not only can capture LiPSs with a strong chemisorption but also can promote the decomposition of solid Li2S with a relatively low surface migration barrier of LiPSs.32–34 In this hybrid structure, the WCF separates the ZnS nanospheres away from each other and establishes abundant interfaces and interspaces to optimize the conversion route from soluble LiPSs to solid Li2S. The cells with the ZnS@WCF modified separator exhibit remarkable cycling stability and outstanding rate performance. Even the areal sulfur loading increases to 3.5 mg cm−2, and a reversible specific capacity of around 600 mA h g−1 can still be maintained after 100 cycles. Compared to the traditional graphene coating strategy, the WCF can deliver competitive electroconductivity and a uniform coating effect with a low cost and simpler fabrication process, and an additional pore-creating process can be omitted. We believe that such a novel hybrid structure could pave the way for wide application of long cycle life and high energy density Li–S batteries.
2. Experimental section
2.1 Synthesis of ZnS nanospheres
Well dispersed ZnS nanospheres were synthesized via a facile hydrothermal method. Typically, 0.015 mol of Zn(CH3COOH)2·2H2O and 0.018 mol of thiourea were dissolved in 100 ml of deionized water to obtain a transparent solution. Then, 1.5 g of gum arabic was added to the above solution and stirred for 2 hours until the powder was completely dissolved. After that, the transparent yellow solution was sealed in a 150 ml Teflon-lined stainless-steel autoclave and kept at 120 °C for 12 hours followed by centrifugation and filtration. Finally, the sample was dried in an oven at 80 °C for 12 hours.2.2 Preparation of the ZnS@wrinkled ultrathin carbon film (ZnS@WCF) composite
The as-prepared ZnS nanospheres were dispersed in a solution mixture containing 70 ml of ethyl alcohol, 10 ml of distilled water and 3 ml of NH3·H2O (25 wt%) by sonication. Afterward, 2 ml of tetrapropyl orthosilicate (TPOS) was dropped into the above solution and stirred for 15 min. Then, 50 mg of resorcinol and 70 μL of formaldehyde (37 wt%) were added to the solution successively and the system was kept stirring for 24 hours at room temperature. After that, the orange precipitate was separated by centrifugation and dried at 80 °C for 12 hours. Then, the products were kept in a tube furnace and carbonized at 750 °C for 4 hours under N2 flow, followed by removing the silica in 80 °C NaOH aqueous solution (1 M) for 12 hours. The pure WCF was prepared through the same procedure without the addition of ZnS.2.3 Fabrication of the ZnS@WCF hybrid interlayer modified separator
Typically, the ZnS@WCF modified separator was prepared by coating a slurry of ZnS@WCF and PVDF (with a mass ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) onto a polypropylene (PP) separator (Celgard 2400) by using a doctor blade. The average thickness was measured to be around 12 μm. The ZnS@WCF-PP was dried in a vacuum oven at 50 °C for 6 hours followed by punching it into disks with a diameter of 19 mm. In addition, ZnS-PP and WCF-PP were also prepared in the same way as the counter samples.
:1) onto a polypropylene (PP) separator (Celgard 2400) by using a doctor blade. The average thickness was measured to be around 12 μm. The ZnS@WCF-PP was dried in a vacuum oven at 50 °C for 6 hours followed by punching it into disks with a diameter of 19 mm. In addition, ZnS-PP and WCF-PP were also prepared in the same way as the counter samples.
2.4 Preparation of Li2S6 solution
A Li2S6 solution (0.2 M) was prepared for the adsorption test. Typically, sulfur powder and Li2S with a molar ratio of 1:5 were added to a solution mixture of 1,3-dioxolane (DOL) and dimethoxyethane (DME) (v/v, 1/1) and stirred at 50 °C for 12 hours with the protection of an argon atmosphere. Finally, a dark orange solution was obtained.
2.5 Materials characterization
Scanning electron microscopy (SEM, ZEISS SUORA®55) with an energy dispersive spectrometer (EDS) was operated to observe the sample morphology and surface element distribution, and high-resolution transmission electron microscopy (HRTEM, JEOL-201) was conducted to analyze the crystal and lattice structure. The phase constitution was investigated from XRD diffraction patterns, which were inspected using a diffractometer (XRD, Bruker D8 Advance, Cu Kα, λ = 0.15418, scan rate = 10° min−1). Raman spectra were collected using a laser Raman tester (HORIBA, LabRam HR800). X-ray photoelectron spectra (XPS, Thermo Fisher, ESCALAB 250X) were inspected to confirm the surface element valence states. The sulfur content was determined using thermal gravimetric analysis (TGA, NETZSCH STA 449F3 thermal analyzer) with a heating rate of 10 °C min−1.2.6 Assembly of symmetric cells
The electrodes in symmetric cells were prepared without active sulfur addition. Typically, ZnS@WCF, ZnS or WCF was dispersed in ethanol by sonication. Then the suspension was dropped onto carbon paper carefully with an areal mass loading of around 0.5 mg cm−2. After drying, the carbon paper disks were used as identical working and counter electrodes. 20 μL of electrolyte containing 0.2 M Li2S6 and 1 M LITFSI in DOL/DME (with a volume ratio of 1:1) solution was used as the catholyte for each cell. The cell was scanned within a voltage window between −0.8 and 0.8 V at a rate of 10 mV s−1.
2.7 Li2S nucleation test
The electrodes of each sample were fabricated in the same way as symmetric cells mentioned above. The catholyte for each cell was prepared by adding Li2S8 (0.2 M) to the electrolyte (1 M LITFSI in DOL/DME v/v = 1/1). Lithium foil was used as the anode in the cell. The cell was first discharged to 2.09 V galvanostatically under 0.134 mA and then the voltage would be held at 2.08 V until the current decreased to 0.01 mA. All the Li2S deposition capacities were calculated based on the mass of sulfur in the Li2S8 catholyte.2.8 Electrochemical measurements
A CR2032 coin cell was used to conduct the electrochemical measurements. The cathode materials were prepared by mixing carbon black and sulfur powder together with a mass ratio of 1:3 and heating at 155 °C for 24 h. The as-prepared CB/S composite was ground with Super P and PVDF binder (with a mass ratio of 8:1:1) in an agate mortar followed by addition of a certain amount of NMP. Then, the cathode slurry was coated on Al foil, and then dried at 60 °C for 12 h in a vacuum oven. The areal sulfur mass loading was measured as 1 mg cm−2 and 3.5 mg cm−2. Lithium metal was employed as the anode. Li–S cells using different types of separators (LS-ZnS, LS-WCF or LS-ZnS@WCF) were assembled in an Ar-filled glovebox. LITFSI (1 M in DOL/DME = 1/1 vol, 2 wt% LiNO3) was used as the electrolyte and the E/S ratio was measured as 15 μL mg−1. Galvanostatic charge and discharge tests were conducted using a LAND CT2001A in the potential range of 1.7–2.8 V. The specific capacity was calculated based on the weight of sulfur (1C = 1673 mA g−1). CV tests, symmetric cell tests, electrochemical impedance spectroscopy (EIS) tests and nucleation tests were operated on an electrochemical workstation (Gamry, Interface 1000, Germany) at room temperature.
2.9 Theoretical calculations
All the calculations were conducted in the framework of density functional theory with the projector augmented plane-wave method, as implemented in the Vienna ab initio simulation package.35 The generalized gradient approximation proposed by Perdew, Burke, and Ernzerhof was selected for the exchange-correlation potential.36 The cut-off energy for the plane wave was set to 337 eV. The energy criterion was set to 10−6 eV in the iterative solution of the Kohn–Sham equation. The slab model contains atomic layers of 14 Å and a vacuum layer of 15 Å. The top two layers were allowed to relax during the adsorption of Li2S4, Li2S6, and Li2S8 molecules on ZnS (102). All the structures were relaxed until the residual forces on the atoms declined to less than 0.03 eV Å−1.3. Results and discussion
The schematic illustration of ZnS@WCF fabrication is shown in Fig. 1. Well dispersed ZnS nanospheres were first synthesized, and then the ZnS nanospheres were successively coated with SiO2 and RF through the hydrolysis of tetrapropyl orthosilicate (TPOS) and the polymerization of resorcinol–formaldehyde (RF) oligomers.37,38 After carbonization at 750 °C in nitrogen, the RF layer transformed into an ultrathin carbon layer and formed a ZnS@SiO2@C core shell structure. Finally, the middle SiO2 layer was etched using NaOH and the outer carbon layer collapsed into a wrinkled graphene-like film and partially attached to the ZnS nanospheres.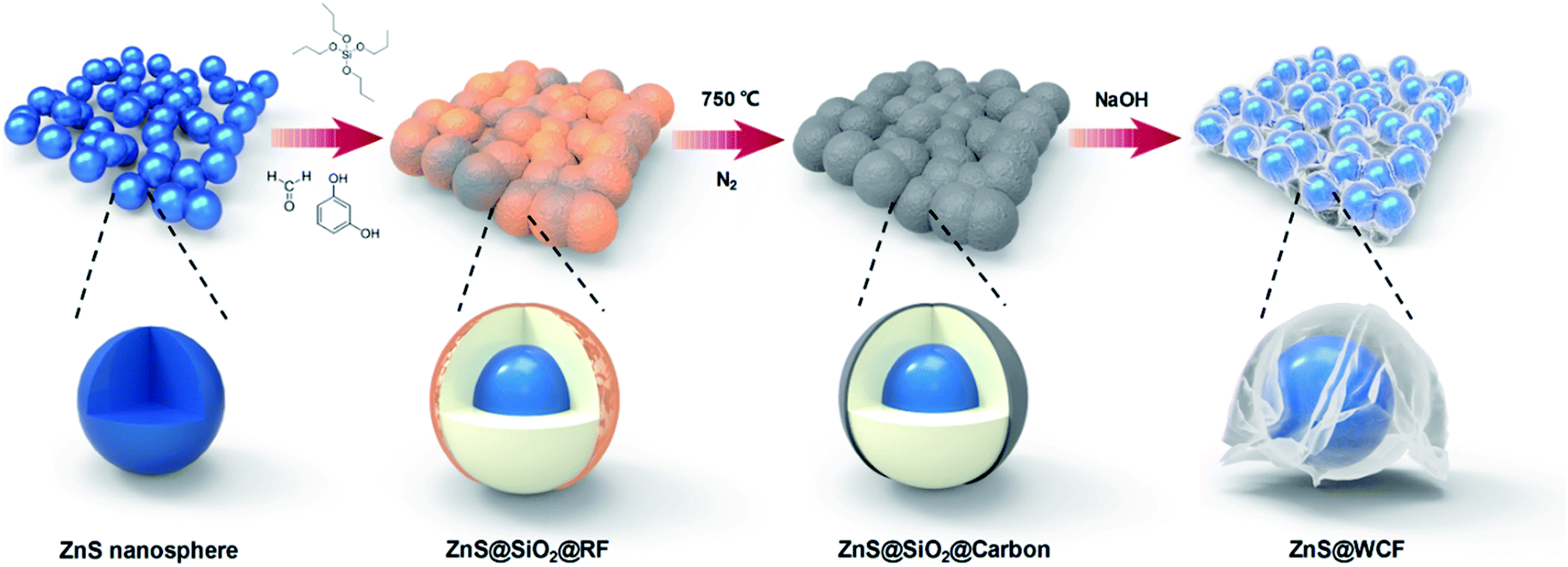 | ||
| Fig. 1 Schematic representation of the preparation process of the ZnS@WCF heterarchical structure. | ||
Fig. 2 shows the morphology of the intermediate products at different stages of the process. As shown in Fig. 2a and d, the ZnS nanospheres are uniform with an average diameter of about 250 nm. Furthermore, the high-resolution transmission electron microscopy (HRTEM) image in Fig. 2g shows an interplanar spacing of 0.311 nm, which matches well with the (102) plane of ZnS. The SEM and TEM images of ZnS@SiO2@C demonstrate that such core spheres tend to have an aggregate morphology (Fig. 2b). From the TEM image of a broken sphere (Fig. 2e and h) it can be observed that the ZnS nanospheres are wrapped inside the SiO2 and carbon layer, and the thickness of the carbon layer is only about 8 nm. After etching with NaOH, SiO2 was removed and all the ZnS nanospheres were wrapped entirely by the carbon film. As shown in Fig. 2c, f and S1 (ESI),† the ultrathin carbon film exhibits a fluctuant and wrinkled structure just like graphene sheets. A pure wrinkled carbon film without ZnS addition was also prepared as a counter sample (Fig. S2, ESI†). The amount of NH3·H2O, TPOS and RF precursors is the key technical parameter in this procedure. The details of the synthesis of ZnS@WCF are listed in Table S1 and Fig. S3 (ESI).† The 3D wrinkled ultrathin carbon film (WCF) constructs a conductive network and wraps up 0D polar ZnS nanospheres to form a heterarchical architecture which can simultaneously provide outstanding electroconductivity and strong chemisorption of LiPSs.
 | ||
| Fig. 2 Morphological characterization. (a and d) SEM images of ZnS nanospheres. (g) HRTEM image of ZnS nanospheres. (b and e) SEM images of the ZnS@SiO2@C core–shell structure. (h) TEM image of ZnS@SiO2@C and the inset is the measurement of the carbon layer thickness. (c and f) SEM images of ZnS@WCF. (i) TEM image of ZnS@WCF. | ||
The crystal structure of ZnS@WCF was investigated by XRD and the pattern is shown in Fig. 3a. All the peaks are indexed to hexagonal ZnS (JCPDS no. 89-2739), which also indicates the amorphous structure of the WCF (Fig. S4, ESI†). Raman spectra of ZnS@WCF in Fig. S5 (ESI†) show two characteristic peaks of carbon (D and G bands), which correspond to the sp3-type disordered and sp2-type graphitic carbon, respectively. The band intensity ratio (ID/IG) was calculated to be 0.992, which demonstrates that the WCF has a certain amount of defects or functional groups on its surface.39,40 XPS spectra of C 1s in ZnS@WCF also illustrate the existence of some defects such as –COOH and –OH on the WCF surface (Fig. S6, ESI†). The carbon and ZnS contents in this hybrid structure were evaluated by thermogravimetric analysis (TGA) from room temperature to 1000 °C in air (Fig. 3b). Two exothermic peaks located at 590 °C and 602 °C can be easily observed in the DSC curve. The 3.2 wt% mass loss below 250 °C can be assigned to the evaporation of absorbed water, the weight loss of about 16 wt% from 250 to 590 °C corresponds to the combustion of carbon and the 11 wt% weight loss between 590 and 700 °C can be ascribed to the oxidation of ZnS. Considering the 17% weight change from ZnS to ZnO,41 the ZnS content was calculated as 68 wt%/(100–17%) = 81 wt%, and the content of WCF was 16 wt%. The N2 isotherms of ZnS nanospheres, WCF and ZnS@WCF are illustrated in Fig. S7a, b (ESI†) and Fig. 3c, respectively. ZnS nanospheres possess the lowest BET specific surface area of 12.532 m2 g−1 compared to the WCF (768.05 m2 g−1) and ZnS@WCF (446.95 m2 g−1). Meanwhile, the BJH pore size distribution of each sample was calculated by from the adsorption branches (Table S2, ESI†). As shown in the inset of Fig. 3c, ZnS@WCF reveals a pore size of 1.64 nm. When applied in Li–S batteries, as shown in Fig. 3d, the micropore enriched WCF serves as a flexible physical barrier to block LiPS shuttling, and the polar ZnS nanospheres wrapped by the WCF provide sufficient anchor sites to capture LiPSs. Based on the synergistic effect between the ZnS and WCF, such a composite with a high pore volume (0.7 cm3 g−1) can also act as an electrolyte container and establish excellent ion pathways to promote the redox of LiPS species. The cross-sectional SEM images of the ZnS@WCF modified separator are shown in Fig. 3e and f. The thickness of the dense coating layer was about ∼12.6 μm and the EDS maps illustrated in the inset of Fig. 3f show the uniform distribution of each element. The digital photographs of the modified separator (inset of Fig. 3e) indicate that the ZnS@WCF slurry uniformly deposited on the pristine PP separator without any exfoliation, exhibiting strong adhesion ability and remarkable flexibility which are suitable for practical utilization.
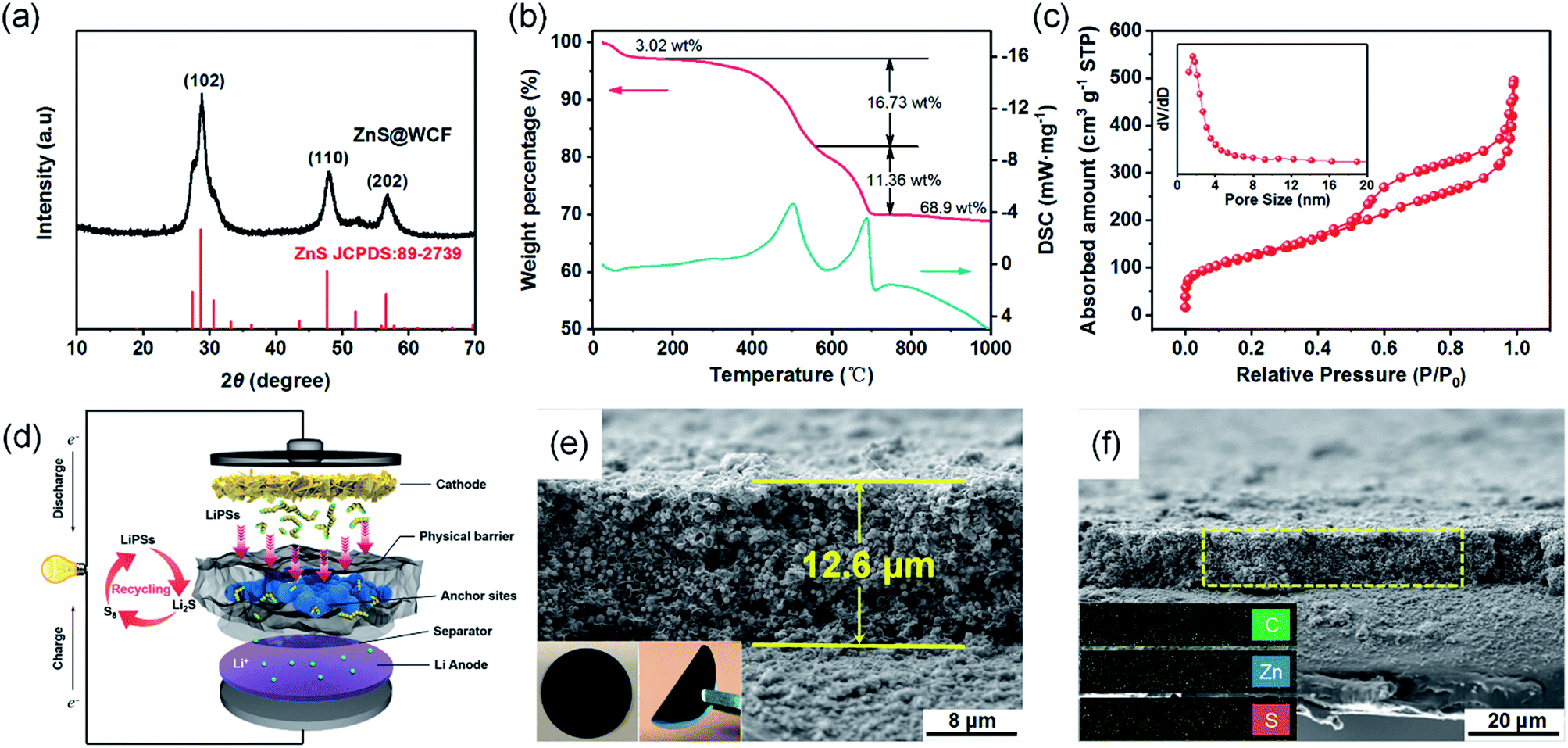 | ||
| Fig. 3 (a) XRD pattern of the ZnS@WCF hybrid. (b) TG-DSC curves of ZnS@WCF in air. (c) Nitrogen adsorption–desorption isotherm of ZnS@WCF. The inset is the pore size distribution. (d) Schematic illustration of a Li–S battery employing a ZnS@WCF modified separator. (e) Cross-sectional SEM image of the ZnS@WCF coated separator. The inset is the digital photograph. (f) Cross-sectional SEM image of the ZnS@WCF coated separator and the corresponding EDS elemental mapping. | ||
To probe the strong chemisorption of LiPS species by the ZnS, the WCF and ZnS nanospheres were soaked in Li2S6 solution (3 mM) (Fig. 4a). After 2 hours, the Li2S6 solution with ZnS nanospheres became clear and transparent; however, the WCF showed inferior adsorption of Li2S6. X-ray photoelectron spectroscopy (XPS) was further carried out to demonstrate the surface chemistry of ZnS nanospheres before and after adsorption of Li2S6. As shown in the Zn 2p spectrum (Fig. 4b), two peaks located at 1045.25 eV and 1022.18 eV are indexed to the 2p 1/2 and 2p 3/2 of Zn2+ in ZnS nanospheres, respectively.32 After soaking in Li2S6 solution, the peaks of both Zn2+ 2p 1/2 and 2p 3/2 shift overall towards lower binding energies (1044.29 eV and 1021.29 eV, respectively), indicating increased electron density at the metal center, which can be assigned to the polarization of electrons away from the terminal sulfur atoms (ST−1) in Li2S6 to the electropositive Zn at the surface.42,43 Meanwhile, in the S 2p spectrum of pristine ZnS (Fig. 4c), two peaks at 161.47 eV and 162.74 eV correspond to the S2− in ZnS. When it comes to ZnS–Li2S6, the peaks of bridge sulfur atoms (SB0, 163.82 eV) and ST−1 (162.59 eV) in Li2S6 can be easily detected, and at the same time, the peaks belonging to the S2− in ZnS can also be found (160.69 eV and 161.79 eV). According to the reported literature, pristine Li2S6 has two S 2p peaks at 161.7 eV and 163.1 eV.44 In that case, the peaks of Li2S6 in the ZnS–Li2S6 sample show positive shifts compared to the pristine one, and the peaks belonging to the S in ZnS show negative shifts. This confirms the strong chemical interaction between ZnS and Li2S6 through the combination of S*–Zn and Li–S binding (S*, Li atoms belong to Li2S6). To further elucidate the chemical interaction between ZnS and long-chain LiPSs, density functional theory (DFT) calculations were conducted to elucidate the absorption energy. As shown in Fig. 4d, the schematic diagrams confirm the coexistence of S*–Zn and Li–S binding between the ZnS surface and LiPSs, which is consistent with the XPS results. The binding energies (EB) between the (102) plane of ZnS and Li2S8, Li2S6 and Li2S4 clusters were calculated to be −4.6 eV, −7.23 eV and −7.11 eV, respectively, indicating the outstanding LiPS confinement by ZnS nanospheres. Apart from chemical affinity, the enhanced conversion kinetics of long-chain LiPS species in ZnS@WCF were verified by cyclic voltammetry (CV) tests of symmetric cells. Each cell was assembled by sandwiching two identical electrodes (ZnS, WCF or ZnS@WCF) in an electrolyte with 0.2 M Li2S6 addition. Obviously, in Fig. 4e, the ZnS@WCF electrode shows larger current density than the bare ZnS and WCF electrodes, indicating utilization of more active materials. Meanwhile, as marked in CV curves, the ZnS@WCF electrode shows two reversible distinct oxidation peaks at 0.133 V and 0.333 V, and two well-defined reduction peaks at −0.133 V and 0.333 V. In contrast, the electrodes with ZnS or the WCF show wider peak separation than ZnS@WCF, exhibiting that ZnS@WCF can combine the polarity of ZnS with the conductivity of the WCF and further promote the kinetics of LiPS conversion. Fig. 4f shows the electrochemical impedance spectroscopy (EIS) results of Li2S6 symmetric cells based on different electrodes. The semicircle of ZnS@WCF (Rct) is smaller than that of the WCF and ZnS nanospheres, confirming the enhanced charge transfer at the interface between ZnS@WCF and LiPSs. The slope of the linear fitting plot of Z′–ω−1/2 is illustrated in Fig. 4g, which defines the Warburg factors of different electrodes.45 Compared to the ZnS–Li2S6 (2.85) and WCF–Li2S6 (5.61) electrodes, the slope of the ZnS@WCF–Li2S6 electrode is much smaller (2.58), indicating its better ion diffusion capacity.
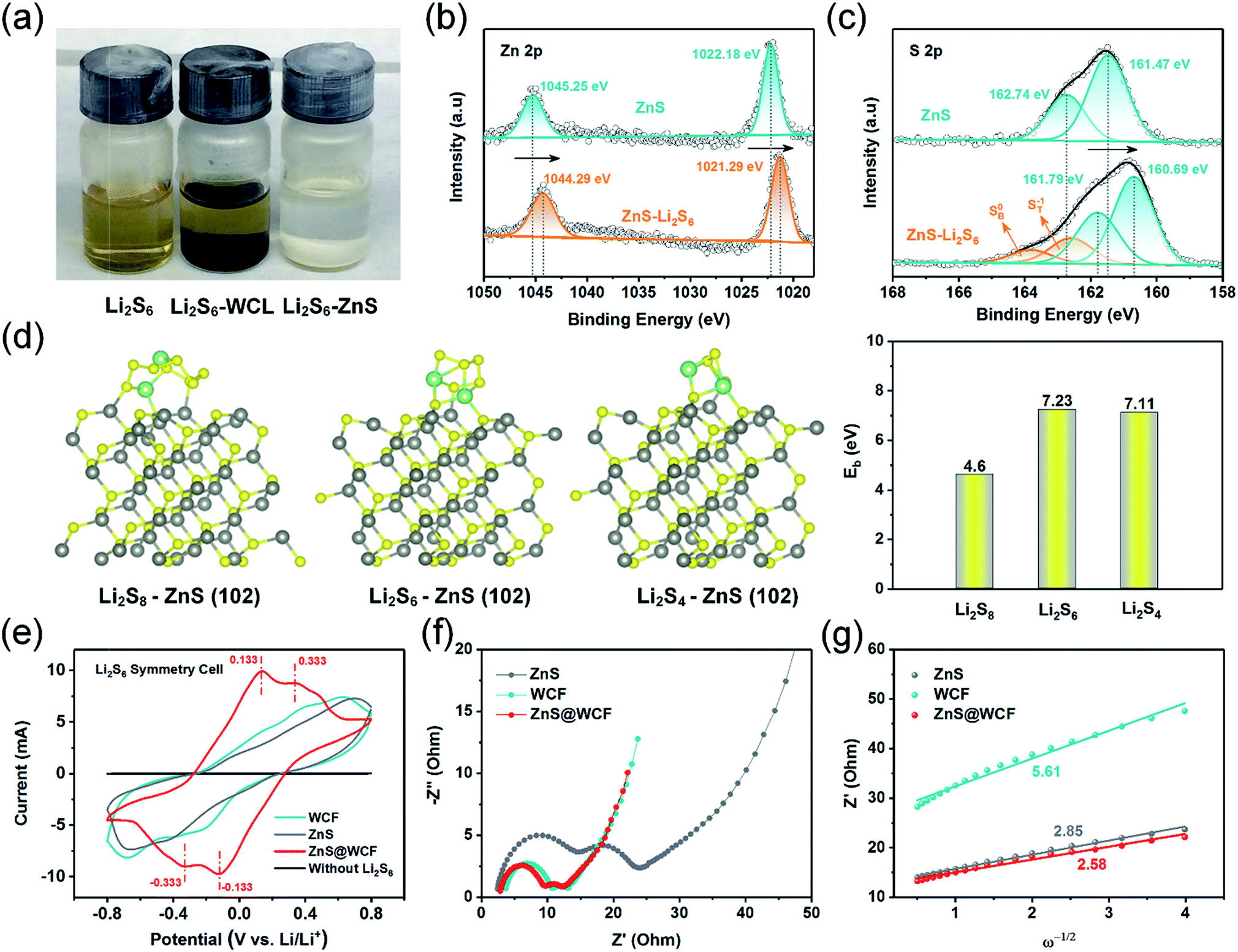 | ||
| Fig. 4 (a) Digital photograph of pure Li2S6 in DOL/DME solution, WCF–Li2S6 and ZnS–Li2S6. XPS spectra of (b) Zn 2p in ZnS before and after Li2S6 adsorption, and (c) S 2p in ZnS before and after Li2S6 adsorption. (d) Optimized geometries and related binding energies of Li2S8, Li2S6 and Li2S4 species with the ZnS (102) surface. (e) CV curves of symmetric cells using ZnS, WCF and ZnS@WCF as electrodes. (f) Electrochemical impedance spectroscopy (EIS) curves of Li2S6 symmetric cells based on different electrodes and (g) the corresponding Z′–ω−1/2 relationships (symbols, real data; lines, fitting results). | ||
In addition to the promotion of liquid–liquid transformation from soluble long-chain LiPSs to short-chain LiPSs, facilitating the liquid–solid conversion and optimizing the precipitation of solid Li2S also play pivotal roles in the whole electrochemical reaction. Considering the insulating nature of the solid product Li2S, it's extremely significant to make the Li2S deposition more controllable. To deeply investigate the positive effect of the ZnS@WCF on the Li2S nucleation, a Li2S nucleation test was conducted by using Li2S8 as the active material in the catholyte.46 The current–time transient profiles of different samples (ZnS, WCF or ZnS@WCF) obtained at 2.09 V are shown in Fig. 5a–c, and the capacities of Li2S precipitation on ZnS nanospheres, WCF and ZnS@WCF are calculated to be 171.35, 92.99 and 217.01 mA h g−1, respectively. It demonstrates that ZnS@WCF is more favorable to reduce the nucleation energy and facilitate the liquid–solid phase transformation. To reveal the kinetics properties of Li2S deposition on different samples, we used a dimensionless current–time transient based on four classical models (equation S1–4, ESI†) of electrochemical deposition to fit the current–time response profiles (Fig. 5d–f).47 The results indicate that the deposition of Li2S on the bare ZnS nanosphere electrode follows closely the 3D progressive nucleation model (3DP). During this process, Li2S nucleated with an uneven size distribution because of the impeded charge transfer between the polar ZnS nanospheres, and the subsequent growth on the surface is controlled by bulk diffusion of LiPS precursors. Thus, the final distribution of Li2S is inhomogeneous and results in the increase of dead sulfur (Fig. 5g). Meanwhile, the Li2S growth on the bare WCF or ZnS@WCF electrode is governed by surface diffusion of adatoms (2D model). Both of them illustrate a typical 2D instantaneous nucleation model (2DI) at the beginning (Fig. 5e and f), and their nucleation rate is extremely higher than that of ZnS nanospheres with the 3D growth model. We further compared the growth rate of Li2S on the WCF and ZnS@WCF based on the nucleus lateral growth-rate constant (kg). As shown in Table 1, ZnS@WCF exhibits a higher lateral growth rate (2.966 × 10−10 mol cm−6 s−2) than the bare WCF (1.780 × 10−10 mol cm−6 s−2). Meanwhile, the WCF shows a higher tendency to obey the 3DP nucleation model than ZnS@WCF at the end of the growth process. For the bare WCF electrode, its outstanding electroconductivity allows the relatively fast lateral growth of Li2S but may also lead to fast passivation of the surface due to the early termination of the precipitation process (Fig. 5h). In that case, the subsequent growth of Li2S species will be along 3D directions together with a serious shuttle effect. In contrast, ZnS@WCF with a heterarchical structure not only can accelerate the nucleation rate owing to the strong interaction between ZnS and LIPSs but also can optimize the distribution of solid Li2S on the interfaces between ZnS and the conductive WCF (Fig. 5i). It has been reported that both Li2S4 species and Li ions have an extraordinarily low migration barrier on the ZnS surface,32 and thus the whole process can be divided into three steps: first, long-chain soluble intermediates were trapped by polar ZnS; second, the adsorbed LiPSs smoothly diffused to the interfaces between ZnS and the WCF; third, the LiPS species transformed into Li2S and nucleated. Based on the above results, the LiPS surface diffusion on ZnS and the Li2S nucleation on the nearby WCF led to a combination of 2DI and 3DP growth, which ensured the unsaturated state of LiPSs and enhanced redox kinetics. In a word, the unique morphology of ZnS@WCF plays an important role in optimizing the Li2S distribution through a typical adsorption–diffusion–conversion of LiPSs.29,48
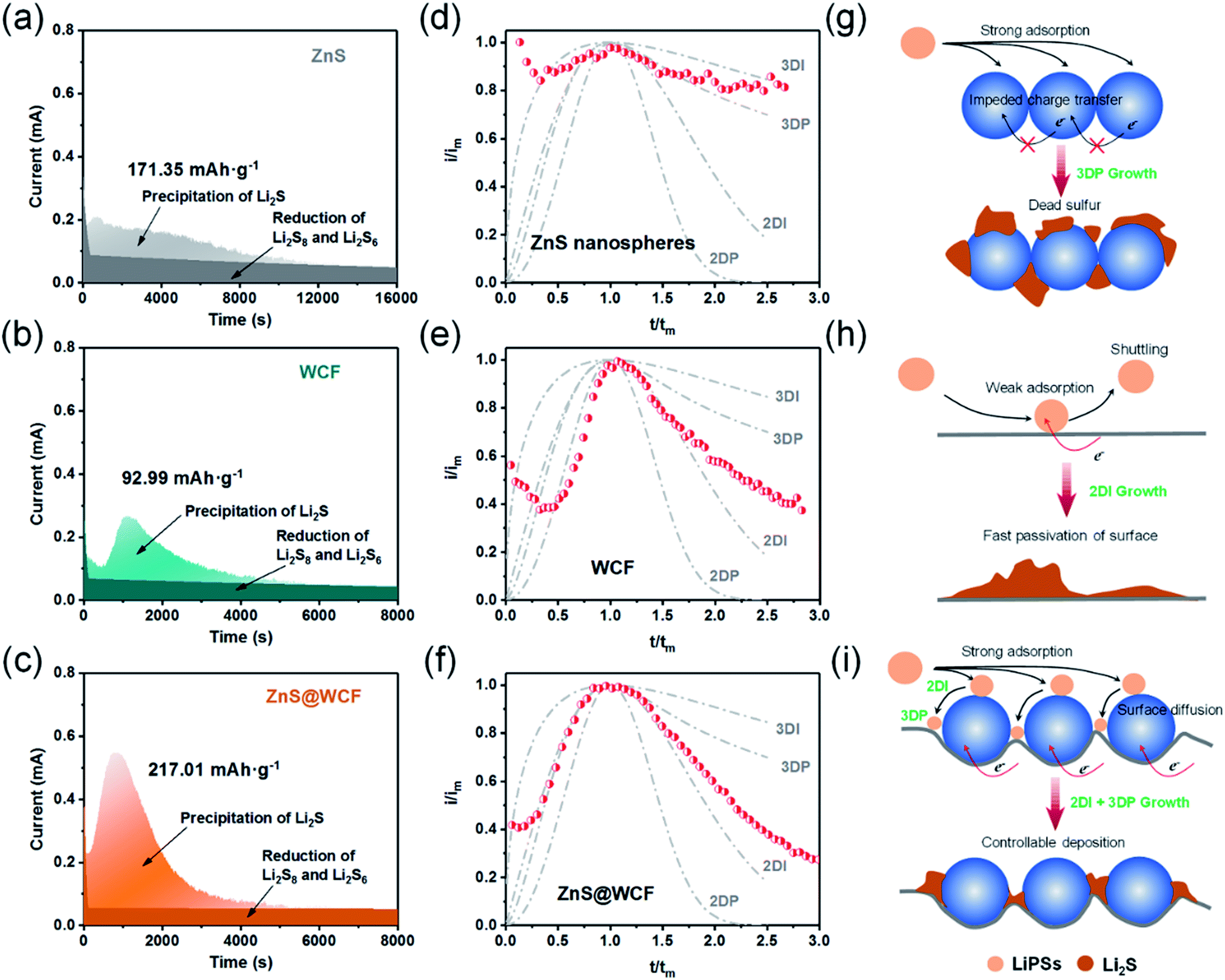 | ||
| Fig. 5 Li2S nucleation test and analysis. (a–c) Current–time transients of ZnS, WCF and ZnS@WCF obtained at 2.08 VLi. (d–f) The corresponding dimensionless transients of ZnS, WCF and ZnS@WCF in comparison with theoretical 2D and 3D growth models. (g–i) Schematic illustration of the Li2S deposition and growth process on different sample surfaces. | ||
| Model: 2DI | t m [s] | N 0 k g 2 [mol cm−6 s−2] | |
|---|---|---|---|
| a M Li2S = 46 g mol−1, ρLi2S = 1.66 g cm−3. | |||
| WCF |
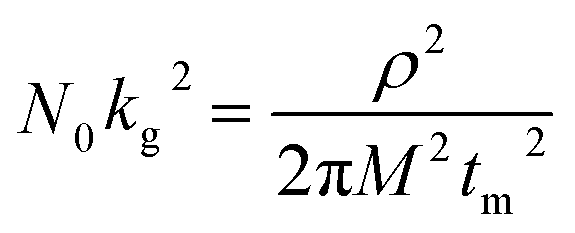
|
1078 | 1.780 × 10−10 |
| ZnS@WCF | 805 | 2.966 × 10−10 | |
To investigate the superiority of the ZnS@WCF modified separator in LiPS blocking, recycling and improved redox kinetics, CR 2023 type Li–S batteries based on CB/S cathodes with separators modified with different interlayer materials were tested. The sulfur content in the CB/S hybrid was measured to be 75 wt% (Fig. S8, ESI†). Fig. 6a shows the CV results for Li–S batteries with different interlayer materials within the voltage window of 1.7–2.8 V at a scan rate of 0.1 mV s−1. Two well-defined reduction peaks located at 2.25 V and 2.05 V are ascribed to the conversion from S8 to LiPSs and LiPSs to Li2S2/Li2S, respectively.49 One distinct oxidation peak is assigned to the transformation from Li2S2/Li2S to S8. Obviously, the Li–S battery with the ZnS@WCF modified separator (LS-ZnS@WCF) delivers an identifiable positive shift in the reduction process, which indicates lower electrochemical polarization and enhanced LiPS redox kinetics compared to the separator coated by bare ZnS nanospheres or the WCF. Furthermore, the two cathodic peaks and one anodic peak remain almost constant in the subsequent four cycles (Fig. S9, ESI†), indicating the excellent reversibility. A CV test with various scan rates was also conducted (Fig. S10†), and the Li+ storage mechanism of the ZnS@WCF coated separator in Li–S batteries is discussed in the ESI.† At the same time, in the charge–discharge voltage profiles at 0.2C (Fig. 6b), we calculated the polarization potential (η) based on the gap between the second discharge plateau and the charge plateau. LS-ZnS@WCF exhibits the smallest polarization potential (η1 = 153.8 mV), compared with LS-ZnS (η3 = 223.2 mV) and LS-WCF (η2 = 183.8 mV), which is consistent with the CV results. What's more, the initial activation energy of solid Li2S on various interlayer materials was also evaluated to verify the enhanced oxidation process kinetics. As shown in Fig. 6c, the small voltage jump before the charge voltage plateaus is related to the activation of solid Li2S.50,51 It is clear that LS-ZnS@WCF displays the smallest polarization potential compared with the cells based on the bare ZnS or WCF modified separator. According to our dimensionless current–time transient results mentioned before, this can be attributed to the formation of dead sulfur or passivation of the ZnS or WCF surface (Fig. 5g and h). The calculations of different cell discharge plateau capacities (QH and QL) are shown in Fig. 6d, which represent the conversion from S to LiPSs (S8 → Li2S6 → Li2S4) and further reduction of LiPSs to solid Li2S (Li2S4 → Li2S2 → Li2S), respectively. As expected, LS-ZnS@WCF shows higher QH and QL values than LS-ZnS and LS-WCF, which is a good indicator of high sulfur utilization and enhanced conversion kinetics. Rate performance at different currents ranging from 0.2C to 2C was also investigated (Fig. 6e). LS-ZnS@WCF exhibits outstanding rate capability among all the samples, and delivers an extremely high initial capacity of 1370.3 mA h g−1 at 0.2C. When cycled at 0.5C, 1C and 2C, it also acquires remarkable capacities of 1070.4, 957.4, and 806.5 mA h g−1, respectively, much higher than that of the bare ZnS or WCF modified separator. When the current density returned to 0.5C after 40 cycles, a high capacity of 951 mA h g−1 is still recovered in the cell based on the ZnS@WCF modified separator, demonstrating high stability and electrochemical reversibility. Fig. 6f illustrates the galvanostatic charge–discharge voltage profile of LS-ZnS@WCF at different current densities. When the current increased to 2C (Fig. S11, ESI†), two typical discharge plateaus can still be detected in LS-ZnS@WCF and both of them are significantly longer than that of LS-ZnS and LS-WCF (372 mA h g−1 and 628 mA h g−1 respectively), suggesting the lower polarization, higher sulfur utilization and improved redox reaction kinetics. It's worth noting that ZnS@WCF can only contribute negligible capacity to the whole battery during cycling performance. As shown in Fig. S12,† the ZnS@WCF hybrid only exhibits a capacity of 50 mA h g−1, which can be ignored compared to the high capacity of sulfur when coupled with CB/S electrodes.
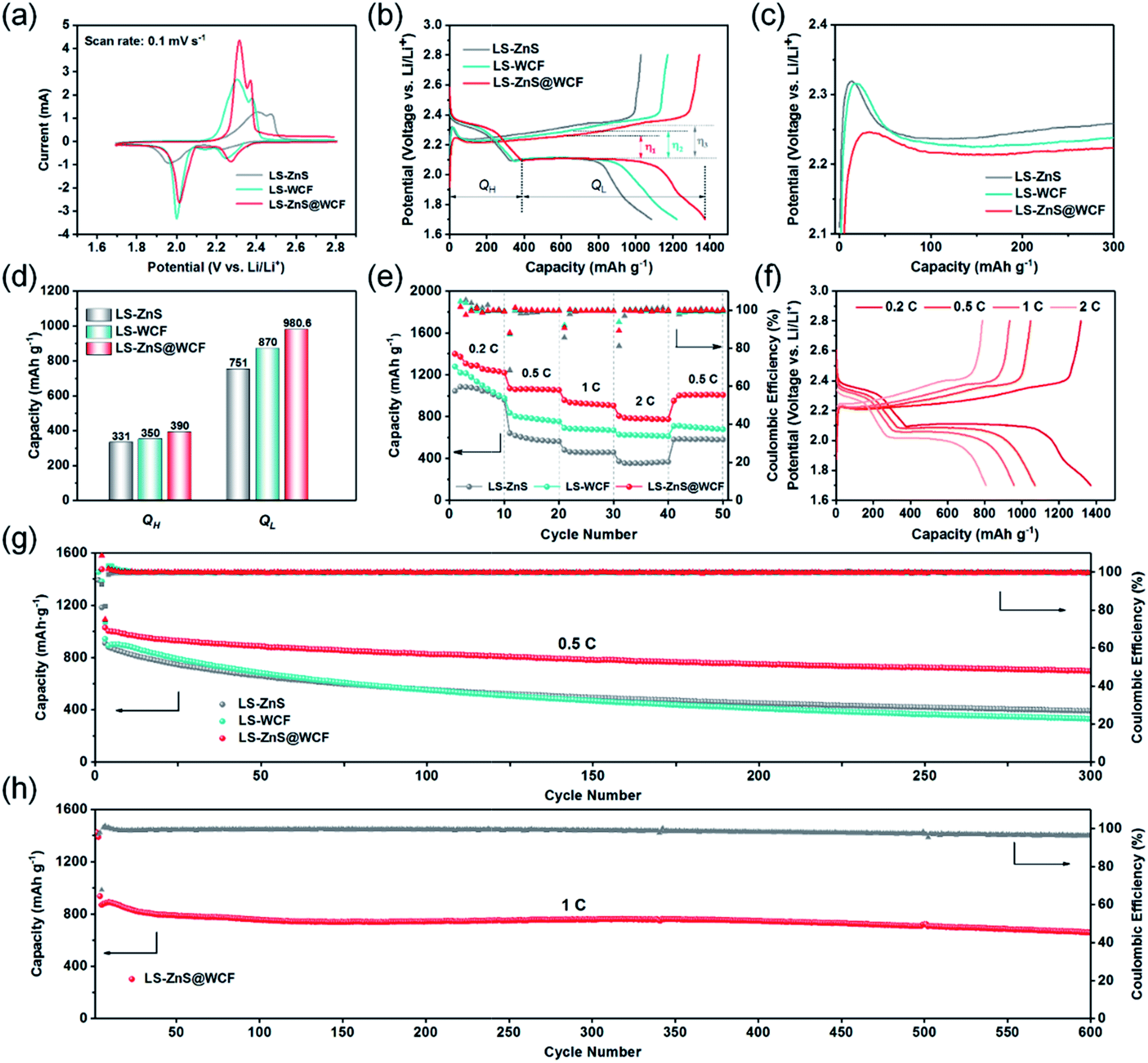 | ||
| Fig. 6 Electrochemical performances of Li–S batteries with different sample coated separators. (a) CV curves recorded from 1.7 to 2.8 V. (b) Representative charge–discharge voltage profiles of cells based on different separators at 0.2C and (c) the corresponding partial magnification of charge profiles. (d) The calculation of two discharge plateau capacities, QH and QL. (e) Rate performance from 0.2C to 2C and (f) the corresponding charge–discharge voltage profiles of LS-ZnS@WCF. (g) Cycling performance of LS-ZnS, LS-WCF and LS-ZnS@WCF at 0.5C. (h) Long-term cycling stability of LS-ZnS@WCF at 1C for 600 cycles. | ||
The cycling performance of Li–S batteries with separators coated with different interlayers was also evaluated. As shown in Fig. 6g, it is quite evident that LS-ZnS@WCF delivers higher cycling performance at 0.5C: an initial discharge specific capacity of 1029 mA h g−1 which decreased to 700 mA h g−1 after 300 cycles, with a fading decay of 0.1% per cycle and a high coulombic efficiency of over 99.5%. Meanwhile, the capacity retention of LS-WCF and LS-ZnS at 0.5C is much inferior to that of LS-ZnS@WCF, with a discharge capacity of 330 mA h g−1 and 391 mA h g−1, respectively, after 300 cycles. Both of them exhibit extremely high fading decay of over 0.2% per cycle. The corresponding charge–discharge profiles of each cell after 300 cycles at 0.5C are shown in Fig. S13, ESI.† Evidently, LS-ZnS@WCF displays longer charge–discharge voltage plateaus with a narrower voltage gap than LS-ZnS and LS-WCF, demonstrating that the fast reaction kinetics remained intact in LS-ZnS@WCF after long cycles.
The sluggish reaction kinetics of the LiPS intermediates in ZnS or the WCF, as mentioned before, result in an irreversible sulfur loss and limited sulfur utilization, and further leads to fast capacity degradation. The morphology of ZnS@WCF after cycling was also investigated, and as shown in Fig. S14a,† the wrinkled-structure remained intact and most of the ZnS spheres were still coated by a thin carbon layer. Meanwhile, almost no bulk-like Li2S can be found on the interlayer, which indicates the uniform distribution of solid products as we discussed in Fig. 5. EDS results in Fig. S14b† also show the uniform distribution of elements Zn, S and C. ZnS@WCF interlayers with different thicknesses were also prepared, as shown in Fig. S15,† and the interlayer with 6 μm thickness showed a lower specific capacity than 12 μm and 20 μm, although it showed outstanding cycling stability. Meanwhile, the interlayer up to 20 μm delivered a slightly lower capacity than 12 μm. On this basis, we consider that it is a better choice to coat the separator with an interlayer thickness of around 12 μm. To illustrate the enhanced capacity retention provided by the ZnS@WCF interlayer, a long-term cycling test of LS-ZnS@WCF was operated at 1C. As shown in Fig. 6h, LS-ZnS@WCF delivers an initial discharge capacity of 954 mA h g−1. After 600 cycles at 1C, a reversible discharge capacity of 685 mA h g−1 can still be maintained with an extraordinarily high coulombic efficiency of 96.15%. The corresponding capacity decay rate is calculated to be 0.045% per cycle, much superior to that of some hybrid interlayer-based Li–S batteries in previous studies (Table S2, ESI†). When the cathode areal sulfur loading increases to 3.5 mg cm−2, LS-ZnS@WCF exhibits a high initial specific capacity of 910 mA h g−1 at 0.1C and its capacity retention is 62% after 100 cycles (Fig. S14, ESI†). Owing to both the structural and chemical synergistic effects, the ZnS@WCF modified separator comprises the merits of polar ZnS and the conductive 3D WCF, which brings about abundant sites for LiPS restriction and Li2S deposition. Thus, such an impressive electrochemical performance of LS-ZnS@WCF can be achieved.
4. Conclusions
We have designed and prepared a novel ZnS@WCF heterarchical structure as a multifunctional interlayer for Li–S batteries via a facile hard template method. ZnS nanospheres have a great number of adsorbent sites to trap solvable LiPS intermediates through Zn–S* and Li–S interactions and have an extremely low surface polysulfide migration barrier. Meanwhile, the graphene-like WCF exhibits outstanding electron and ion conductivity, and such a multifunctional hybrid interlayer material can not only block polysulfide shuttling but can also act as a recycle bin for polysulfides. Based on the combination of our theoretical calculations and experimental results, the ZnS@WCF hybrid interlayer can establish a conversion pathway with fast kinetics from sulfur to LiPSs and to solid Li2S, and it further exhibits excellent rate capacity and remarkable cycling stability after integrating with the CB/S cathode. This study can provide a novel design concept for functional interlayer materials and push forward the application of long life, fast kinetics Li–S batteries.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 51772164)References
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- X. Chen, T. Hou, K. A. Persson and Q. Zhang, Mater. Today, 2019, 22, 142–158 CrossRef CAS.
- B. Liu, R. Fang, D. Xie, W. Zhang, H. Huang, Y. Xia, X. Wang, X. Xia and J. Tu, Energy Environ. Mater., 2018, 1, 196–208 CrossRef.
- J. Park, S.-H. Yu and Y.-E. Sung, Nano Today, 2018, 18, 35–64 CrossRef CAS.
- H. Wang, W. Zhang, J. Xu and Z. Guo, Adv. Funct. Mater., 2018, 28, 1707520 CrossRef.
- J. Zhang, H. Huang, J. Bae, S.-H. Chung, W. Zhang, A. Manthiram and G. Yu, Small Methods, 2018, 2, 1700279 CrossRef.
- X. Chen, H.-J. Peng, R. Zhang, T.-Z. Hou, J.-Q. Huang, B. Li and Q. Zhang, ACS Energy Lett., 2017, 2, 795–801 CrossRef CAS.
- X. Liu, J. Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef PubMed.
- H. J. Peng, G. Zhang, X. Chen, Z. W. Zhang, W. T. Xu, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12990–12995 CrossRef CAS PubMed.
- Y. Tao, Y. Wei, Y. Liu, J. Wang, W. Qiao, L. Ling and D. Long, Energy Environ. Sci., 2016, 9, 3230–3239 RSC.
- Z.-W. Zhang, H.-J. Peng, M. Zhao and J.-Q. Huang, Adv. Funct. Mater., 2018, 28, 1707536 CrossRef.
- H. Yuan, H.-J. Peng, J.-Q. Huang and Q. Zhang, Adv. Mater. Interfaces, 2019, 6, 1802046 CrossRef CAS.
- L. Jiao, S. Wu, Y. Tao, W. Lv, C. Zhang, G. Ling and Q.-H. Yang, Energy Storage Materials, 2019, 23, 112–136 CrossRef.
- X. Xiang, J. Y. Wu, Q. X. Shi, Q. Xia, Z. G. Xue, X. L. Xie and Y. S. Ye, J. Mater. Chem. A, 2019, 7, 9110–9119 RSC.
- X. Han, Y. Xu, X. Chen, Y.-C. Chen, N. Weadock, J. Wan, H. Zhu, Y. Liu, H. Li, G. Rubloff, C. Wang and L. Hu, Nano Energy, 2013, 2, 1197–1206 CrossRef CAS.
- Z. Xiao, Z. Yang, L. Wang, H. Nie, M. Zhong, Q. Lai, X. Xu, L. Zhang and S. Huang, Adv. Mater., 2015, 27, 2891–2898 CrossRef CAS PubMed.
- N. Hu, X. Lv, Y. Dai, L. Fan, D. Xiong and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 18665–18674 CrossRef CAS PubMed.
- H. Tang, S. Yao, S. Xue, M. Liu, L. Chen, M. Jing, X. Shen, T. Li, K. Xiao and S. Qin, Electrochim. Acta, 2018, 263, 158–167 CrossRef CAS.
- N. Kaisar, S. A. Abbas, J. Ding, H. A. Chen, C. W. Pao, K. M. Boopathi, A. Mohapatra, Y. T. Chen, S. H. Wu, J. Fang, S. Jou and C. W. Chu, Nanoscale, 2019, 11, 2892–2900 RSC.
- Z. A. Ghazi, X. He, A. M. Khattak, N. A. Khan, B. Liang, A. Iqbal, J. Wang, H. Sin, L. Li and Z. Tang, Adv. Mater., 2017, 29, 1606817 CrossRef PubMed.
- B. Qi, X. Zhao, S. Wang, K. Chen, Y. Wei, G. Chen, Y. Gao, D. Zhang, Z. Sun and F. Li, J. Mater. Chem. A, 2018, 6, 14359–14366 RSC.
- J. He, Y. Chen and A. Manthiram, Energy Environ. Sci., 2018, 11, 2560–2568 RSC.
- L. Zhang, X. Chen, F. Wan, Z. Niu, Y. Wang, Q. Zhang and J. Chen, ACS Nano, 2018, 12, 9578–9586 CrossRef CAS PubMed.
- Y. Guo, Y. Zhang, Y. Zhang, M. Xiang, H. Wu, H. Liu and S. Dou, J. Mater. Chem. A, 2018, 6, 19358–19370 RSC.
- S. Choi, D. H. Seo, M. R. Kaiser, C. Zhang, T. van der laan, Z. J. Han, A. Bendavid, X. Guo, S. Yick, A. T. Murdock, D. Su, B. R. Lee, A. Du, S. X. Dou and G. Wang, J. Mater. Chem. A, 2019, 7, 4596–4603 RSC.
- Z. Chang, H. Dou, B. Ding, J. Wang, Y. Wang, X. Hao and D. R. MacFarlane, J. Mater. Chem. A, 2017, 5, 250–257 RSC.
- Y. Zhong, D. Chao, S. Deng, J. Zhan, R. Fang, Y. Xia, Y. Wang, X. Wang, X. Xia and J. Tu, Adv. Funct. Mater., 2018, 28, 1706391 CrossRef.
- J. He, G. Hartmann, M. Lee, G. S. Hwang, Y. Chen and A. Manthiram, Energy Environ. Sci., 2019, 12, 344–350 RSC.
- T. Zhou, W. Lv, J. Li, G. Zhou, Y. Zhao, S. Fan, B. Liu, B. Li, F. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 1694–1703 RSC.
- H. Lin, S. Zhang, T. Zhang, S. Cao, H. Ye, Q. Yao, G. W. Zheng and J. Y. Lee, ACS Nano, 2019, 13, 7073–7082 CrossRef CAS PubMed.
- H. J. Peng, Z. W. Zhang, J. Q. Huang, G. Zhang, J. Xie, W. T. Xu, J. L. Shi, X. Chen, X. B. Cheng and Q. Zhang, Adv. Mater., 2016, 28, 9551–9558 CrossRef CAS PubMed.
- J. Xu, W. Zhang, H. Fan, F. Cheng, D. Su and G. Wang, Nano Energy, 2018, 51, 73–82 CrossRef CAS.
- R. Razaq, D. Sun, J. Wang, Y. Xin, G. Abbas, J. Zhang, Q. Li, T. Huang, Z. Zhang and Y. Huang, J. Power Sources, 2019, 414, 453–459 CrossRef CAS.
- Y. Hua, X. Li, X. Zhang, L. Zhang, Y. Shu, H. Sheng, H. Fang, H. Wei and Y. Ding, ChemElectroChem, 2019, 6, 2570–2577 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Z. A. Qiao, B. Guo, A. J. Binder, J. Chen, G. M. Veith and S. Dai, Nano Lett., 2013, 13, 207–212 CrossRef CAS PubMed.
- H. Zhang, O. Noonan, X. Huang, Y. Yang, C. Xu, L. Zhou and C. Yu, ACS Nano, 2016, 10, 4579–4586 CrossRef CAS PubMed.
- J.-L. Shi, H.-J. Peng, L. Zhu, W. Zhu and Q. Zhang, Carbon, 2015, 92, 96–105 CrossRef CAS.
- C. Luo, W. Lv, Y. Deng, G. Zhou, Z. Z. Pan, S. Niu, B. Li, F. Kang and Q. H. Yang, Small, 2017, 13, 1700358 CrossRef PubMed.
- D. Fang, S. Chen, X. Wang, Y. Bando, D. Golberg and S. Zhang, J. Mater. Chem. A, 2018, 6, 8358–8365 RSC.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef CAS PubMed.
- X. Liang, C. Y. Kwok, F. Lodi-Marzano, Q. Pang, M. Cuisinier, H. Huang, C. J. Hart, D. Houtarde, K. Kaup, H. Sommer, T. Brezesinski, J. Janek and L. F. Nazar, Adv. Energy Mater., 2016, 6, 1501636 CrossRef.
- J. He, L. Luo, Y. Chen and A. Manthiram, Adv. Mater., 2017, 29, 1700358 Search PubMed.
- S. Wang, J. Liao, X. Yang, J. Liang, Q. Sun, J. Liang, F. Zhao, A. Koo, F. Kong, Y. Yao, X. Gao, M. Wu, S.-Z. Yang, R. Li and X. Sun, Nano Energy, 2019, 57, 230–240 CrossRef CAS.
- F. Y. Fan, W. C. Carter and Y. M. Chiang, Adv. Mater., 2015, 27, 5203–5209 CrossRef CAS PubMed.
- Z. Li, Y. Zhou, Y. Wang and Y.-C. Lu, Adv. Energy Mater., 2019, 9, 1802207 CrossRef.
- Y. Song, W. Cai, L. Kong, J. Cai, Q. Zhang and J. Sun, Adv. Energy Mater., 2019, 1901075 CrossRef.
- D. Zheng, G. Wang, D. Liu, J. Si, T. Ding, D. Qu, X. Yang and D. Qu, Adv. Mater. Technol., 2018, 3, 1700233 CrossRef.
- M. Wang, L. Fan, D. Tian, X. Wu, Y. Qiu, C. Zhao, B. Guan, Y. Wang, N. Zhang and K. Sun, ACS Energy Lett., 2018, 3, 1627–1633 CrossRef CAS.
- G. Zhou, H. Tian, Y. Jin, X. Tao, B. Liu, R. Zhang, Z. W. Seh, D. Zhuo, Y. Liu, J. Sun, J. Zhao, C. Zu, D. S. Wu, Q. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 840–845 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta10560c |
| This journal is © The Royal Society of Chemistry 2020 |