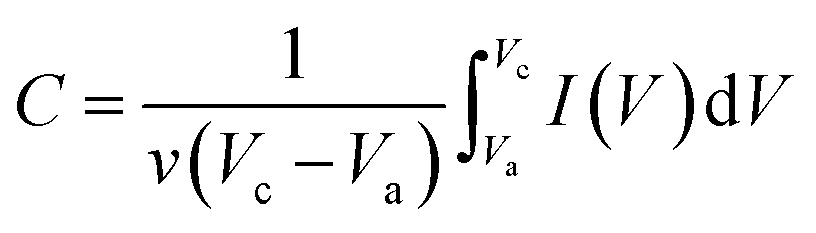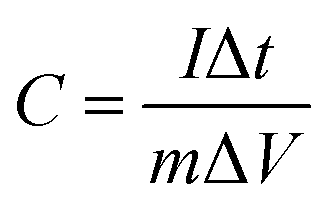Metallic CoO/Co heterostructures stabilized in an ultrathin amorphous carbon shell for high-performance electrochemical supercapacitive behaviour†
Received
9th October 2018
, Accepted 3rd December 2018
First published on 3rd December 2018
Abstract
As a highly electrochemically active transition-metal oxide (TMO), cobalt monoxide (CoO) has been extensively investigated for applications in lithium-ion batteries and electrochemical oxygen evolution. However, its capacitive performance has been rarely studied due to the intrinsic low conductivity and poor stability. Here, we report an asymmetric supercapacitor with excellent electrochemical capacity based on an ultrathin carbon shell entrapped Co-doped CoO heterostructure (CoO/Co@C). The metallic conductivity and mesoporous configuration make the as-prepared CoO/Co@C deliver a dramatic specific capacitance of 2165.7 F g−1 at a scan rate of 10 mV s−1. Besides the contribution to double-layer capacitance, more importantly, the amorphous carbon shell effectively prevents the CoO/Co heterostructure from further oxidation, thus resulting in a significantly improved long-term storage stability in air. An asymmetric supercapacitor cell fabricated using CoO/Co@C and active carbon achieves a maximum energy density of 146.3 W h kg−1 at a power density of 1800 W kg−1, and the maximum of 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 W kg−1 can be obtained with a remaining energy density of 63.0 W h kg−1. The easy preparation, high performance, and excellent cycling stability of the CoO/Co@C nanocomposite make it a promising material for catalyst and battery applications.
000 W kg−1 can be obtained with a remaining energy density of 63.0 W h kg−1. The easy preparation, high performance, and excellent cycling stability of the CoO/Co@C nanocomposite make it a promising material for catalyst and battery applications.
Introduction
Supercapacitors, as high-performance and environment-friendly energy storage devices, have attracted much attention in the past few decades.1,2 Compared to rechargeable batteries, supercapacitors exhibit many advantages such as a longer life cycle, higher power density, and faster charge–discharge rate and thus have shown great potential in the application of energy storage devices in the future.3 According to the charge storage mechanism, supercapacitors can be classified into two types: double-layer capacitors and pseudocapacitors. A double-layer capacitor based on non-faradaic electrode charge storage processes is composed of carbon materials (e.g., graphene, carbon nanotubes, active carbon (AC)).4–6 However, owing to charge accumulation in the electrical double layer, most of the carbon materials exhibit a poor specific capacitance (∼100 F cm−3). Pseudocapacitors that undergo faradaic redox reactions during charging/discharging provide a higher energy density and higher theoretical specific capacitance than carbon materials.7,8 A metal oxide such as RuO2 is the gold standard for supercapacitor materials at present; RuO2 has a theoretical specific capacitance of 1400 to 2200 F g−1.9 However, the high cost of RuO2 is the key factor that limits the bulk commercial application of RuO2.
To date, a series of transition-metal oxides (TMOs) such as MnO2, MoO3, NiO, and Co3O4 have been widely explored as supercapacitive performance materials because of their low cost and high theoretical specific capacitance.10–15 Nevertheless, TMOs often exhibit specific capacitance far below their theoretical limits. This can be attributed to (i) a poor conductivity of TMOs,16 (ii) sluggish faradaic process of pseudocapacitor,17 and (iii) narrow potential windows.18 Poor conductivity and narrow potential windows can be typically overcome by constructing asymmetric supercapacitors in which TMOs and AC work together in the same electrolyte.19 The sluggish faradaic reaction observed only at very low scan rates is the intrinsic drawback of these pseudocapacitor materials. Control of electrode materials as thin films or use of porous or layered structures is an effective strategy to solve this problem;20 a shorter interlayer distance results in a reduced charge-transfer resistance compared with the resistance of bulk materials.17,21 For example, Hu and co-workers showed that the specific capacitance of NiO porous structure can approach the theoretical value of NiO.10 Pang et al. reported that the porous cubic Mn3O4/Co3O4 nanocomposites has a specific capacitance of 500 F g−1 at 0.75 A g−1.22 Very recently, we also reported the multilayered carbon nanosphere/MnO2 supercapacitors with a specific capacitance of 1134 F g−1.23
The desired capacitor material should have a high charge storage capacity and excellent conductivity, and while offering a satisfactory durability during the storage and application. As a low-oxidized cobalt compound, to date, CoO has been successfully applied as Li-ion battery anodes and showed satisfactory Li-ion storage capacities.24–26 Notably, the theoretical specific capacitance of CoO is 4292 F g−1.27 This is higher than that of NiO (2573 F g−1),28 MnO2 (1370 F g−1),29 and Co3O4 (3560 F g−1).30 However, the practical application of CoO is severely hindered by its easily oxidized character (CoO is oxidized to Co3O4 after exposure in air) and the poor electrical conductivity (intrinsic semiconducting or insulating property). Until now, to the best of our knowledge, only a few studies focused on the fabrication of CoO nanostructures and investigated their pseudocapacitive performance.31–42 Most of these CoO nanostructures were integrated with graphitized carbon,33–36 graphene nanosheets,37 carbon nanotubes,38 foam nickel,39–41 or polypyrrole nanowires42 to improve the electrical conductivity, thus achieving satisfactory electrochemical performances. Nevertheless, only a few electrodes could achieve the specific capacitance higher than 1000 F g−1.33,35,42 No study reported a capacitance of 2000 F g−1 at a higher scanning rate, e.g., 5 mV s−1 or more or at a larger current density.
In this study, we report a CoO-based energy storage material to address these challenges by elaborately tailoring both the electrical conductivity and material configuration. For these goals, a cobalt-doped CoO (denoted as CoO/Co) heterostructure is designed to improve the electrical conductivity; an ultrathin layer of amorphous carbon is casted on the surface of heterostructure (denoted as CoO/Co@C) to prevent further oxidation of CoO/Co. The metallic Co, acting as a current “expressway”, can effectively improve the electrical conductivity of the composite, thus accelerating the electron transport. The mesoporous structure offers an optimized ion diffusion pathway. Especially, the amorphous carbon shell serves as a surface stabilizer to hinder further oxidation of Co/CoO, thus sustaining good stability of supercapacitor. As a result, the CoO/Co@C composites exhibit a remarkable supercapacitor performance of 2165.7 F g−1 at a scanning rate of 10 mV s−1. Even more importantly, such a composite also showed a good cycling ability and improved long-term storage stability.
Results and discussion
Synthesis of materials
Without glycerol (the precursor of carbon), the as-formed Co-based precursor is a flower-like structure (diameter of ∼ 4 μm, Fig. S1a and b†), composed of composed of Co(OH)2 and CoC2O4 (Fig. S2†). Fig. 1 shows the SEM images of Co-based precursor under different magnifications after annealing at 450 °C in Ar atmosphere. The annealed product maintains the initial structure of precursor (Fig. 1a); only the petal surface became rough (Fig. 1b). As one of the product of hydrolysis of HMT, a number of HCHO molecules formed and then absorbed on the surface of flower-like spheres in the hydrothermal process, affording the Co-based precursor.43 These HCHO molecules subsequently decomposed and generated reductive CO and H2 when the Co-based precursor was annealed at 450 °C in Ar, thus triggering the following reactions:
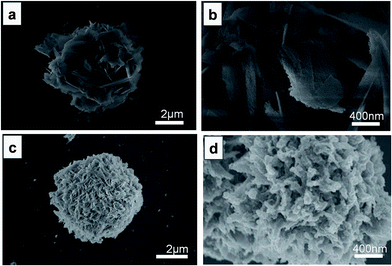 |
| | Fig. 1 SEM images of CoO/Co composites (a and b) and CoO/Co@C composites (c and d). | |
Clearly, the CoO species originate from the decomposition of Co(OH)2 (Reaction (2)). The formation of metallic Co species is related to the added HMT (Reaction (3)) and oxalic acid (Reaction (4)). To achieve better electrochemical performance, the CoO/Co preparation procedure is optimized with respect to HMT concentration (Fig. S3a†), oxalic acid concentration (Fig. S3b†), annealing temperature (Fig. S3c†), and annealing atmosphere (Fig. S3d†), as outlined in the ESI.† The best electrochemical response is achieved for CoO/Co prepared using 4 mM HMT and 0.2 g oxalic acid during the hydrothermal process, followed by treatment in Ar atmosphere at 450 °C. To verify the composition and crystalline structure of product, X-ray powder diffraction (XRD) patterns are collected as shown in Fig. 2a. The diffraction peaks located at 2θ of 44.4°, 51.6°, and 76.1° can be perfectly indexed to the (111), (200), and (220) planes of Co (JYCPS card no. 00-001-025), respectively. The diffraction peaks located at 2θ of 36.6°, 42.6°, and 61.8° can be perfectly indexed to the (111), (200), and (220) planes of CoO (JYCPS card no. 01-075-0533), respectively. No other peaks indicate the composition of CoO/Co.
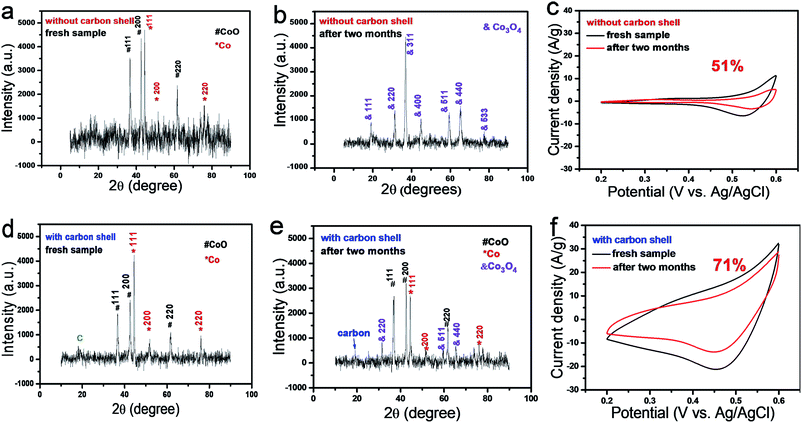 |
| | Fig. 2 Characterization of CoO/Co@C and CoO/Co samples before and after the storage in air at room temperature for two months. XRD patterns of CoO/Co@C sample before (a) and after (b) two months of storage, and the corresponding electrochemical performance in 1 M KOH (c). XRD patterns of CoO/Co@C sample before (d) and after (e) two months of storage, and the corresponding electrochemical performance in 1 M KOH (f). | |
On the other hand, if the precursor is prepared with glycerol, the glycerol molecules are calcined at 450 °C in Ar. In Fig. 2d, all the peaks in the XRD pattern for CoO/Co@C are consistent with those obtained from both CoO and Co in position, except the peak at 2θ = 19.0° obtained from the amorphous carbon. Notably, the relative intensity of Co (111) in the carbon containing sample (Fig. 2d) is higher than that in the CoO/Co sample (Fig. 2a) prepared without using glycerol as the carbon precursor. The samples showed a larger electrochemical capacitive performance when 4 mL DI water is replaced with glycerol (Fig. S4a†). The SEM images show that with the addition of glycerol, the Co-based precursor presents a microsphere morphology (diameter ∼ 5.5 μm) with lots of nanoneedles on the surface (Fig. S1c and d†). After the annealing treatment, the resulting CoO/Co@C microsphere shows a hydrangea-like structure with a diameter ∼ 5.0 μm (Fig. 1c and d) and ∼7.7 wt% carbon (Table S1†). Electrochemical impedance spectroscopy (EIS) measurements are further used to investigate the electrical conductivity and ion transfer in the as-prepared sample (Fig. S4b†). The vertical line of Nyquist plots at lower frequencies parallel to the imaginary axis suggests an ideal behaviour, representative a better ion diffusion in the samples, providing an ideal pathway for ion transport with less kinetic limitations. As the semi-circle corresponding to constant phase element and charge transfer resistance completely disappears, the Nyquist plots can be fitted using a modified Randles circuit (inset of Fig. S4b†), which consists of a series resistance (Rs) and modified mass-transport impedance (Ma).44 The fitting results (Table S2†) show that all these samples have low Rs, which can contribute to good electrochemical performance.
To evaluate the advantage of carbon introduction on the electrochemical supercapacitive behaviour, the I–V curves of CoO/Co and CoO/Co@C samples are recorded in 1 M KOH at 10 mV s−1. The potential window is confined between 0.2 V and 0.6 V to avoid O2 reduction at lower potentials and O2 evolution at higher potentials (Fig. S5†). The effect of carbon shell in view of electrochemical response is plotted in Fig. S6.† Without carbon shell decoration, CoO/Co sample shows a small hysteresis loop. For CoO/Co@C sample, it can be noted that the I–V curves become a typical triangle, and there is a clear increase in specific capacitance compared to CoO/Co sample. Both the I–V curves exhibit one pair of wide redox peaks, indicating a typical pseudocapacitive behaviour of CoO. The related faradaic redox reaction is as follows:
| | | CoO + OH− ↔ CoOOH + e− | (5) |
Based on the I–V curves shown in Fig. S6,† the capacitance was evaluated in term of specific capacitance. According to eqn (6) (details are offered in Experimental section), the as-formed CoO/Co spheres show a specific capacitance of 231.3 F g−1 at a scan rate of 10 mV s−1. In contrast, the CoO/Co@C exhibits a drastic specific capacitance of 2165.7 F g−1 at a scan rate of 10 mV s−1. This high capacitive performance of CoO/Co@C can be ascribed to the unique ultrathin carbon shell (allowing good connection to an electron collector), mesoporous structure (allowing the electrolyte transport and contact to maintain sufficient OH− for faradaic reactions even at a high scan rate), negligible charge transfer (metallic Co, acting as a current “expressway”, effectively improves the electrical conductivity of the composite) and a high surface area. Particularly, it should be mentioned that C@CoO/C maintained ∼71% of the original specific capacitance after storage in air at room temperature for two months (Fig. 2f). In contrast, CoO/Co sample exhibited a lower specific capacitance retention rate of only 51% (Fig. 2c). To understand the factors that lead to this obvious difference in specific capacitance retention, XRD patterns of CoO/Co and CoO/Co@C samples were collected again (Fig. 2b and d). For CoO/Co sample, as shown in Fig. 2b, all the diffraction peaks originated from CoO and Co completely disappear after two month of storage (Fig. 2b). Instead, all the new diffraction peaks coincide with Co3O4 in position (Co3O4 is also the product obtained by annealing the Co-based precursor in air at 450 °C as demonstrated in Fig. S7†). These results indicate that the Co and CoO species in CoO/Co heterostructure are not stable, and they can be definitely oxidized into Co3O4 during the storage. For CoO/Co@C sample, some weak peaks of Co3O4 are observed in the XRD pattern after two months of storage (Fig. 2e), indicating that a small part of Co and CoO species are oxidized into Co3O4 by O2 in air. More importantly, all the peaks originated from CoO and Co are still present in the XRD pattern of CoO/Co@C sample, even though some decrease in peak intensity is observed for CoO and Co. These results indicate that this ultrathin carbon shell has the following two functions: (i) distribution in the increase of surface area and decrease of pore size (Fig. S8†), and (ii) protective cap to isolate metallic CoO/Co from air, thus slowing down the oxidation speed of metal (Co) or metal oxide (low chemical state, i.e., CoO), and maintaining the storage stability of supercapacitor materials. In addition, compared with CoO/Co, the as-synthesized CoO/Co@C electrode has a better cycling life (Fig. S9†). The cycling stability shows that the CoO/Co@C electrode can retain 87.1% of the initial capacitance after 1000 cycles, whereas CoO/Co only retains 82.1% of the initial capacitance after 1000 cycles. The increase in electrochemical stability is also related to the double-layer capacitive contribution of carbon shell. It is generally accepted that double-layer capacitors have more electrochemical stability than pseudocapacitors.
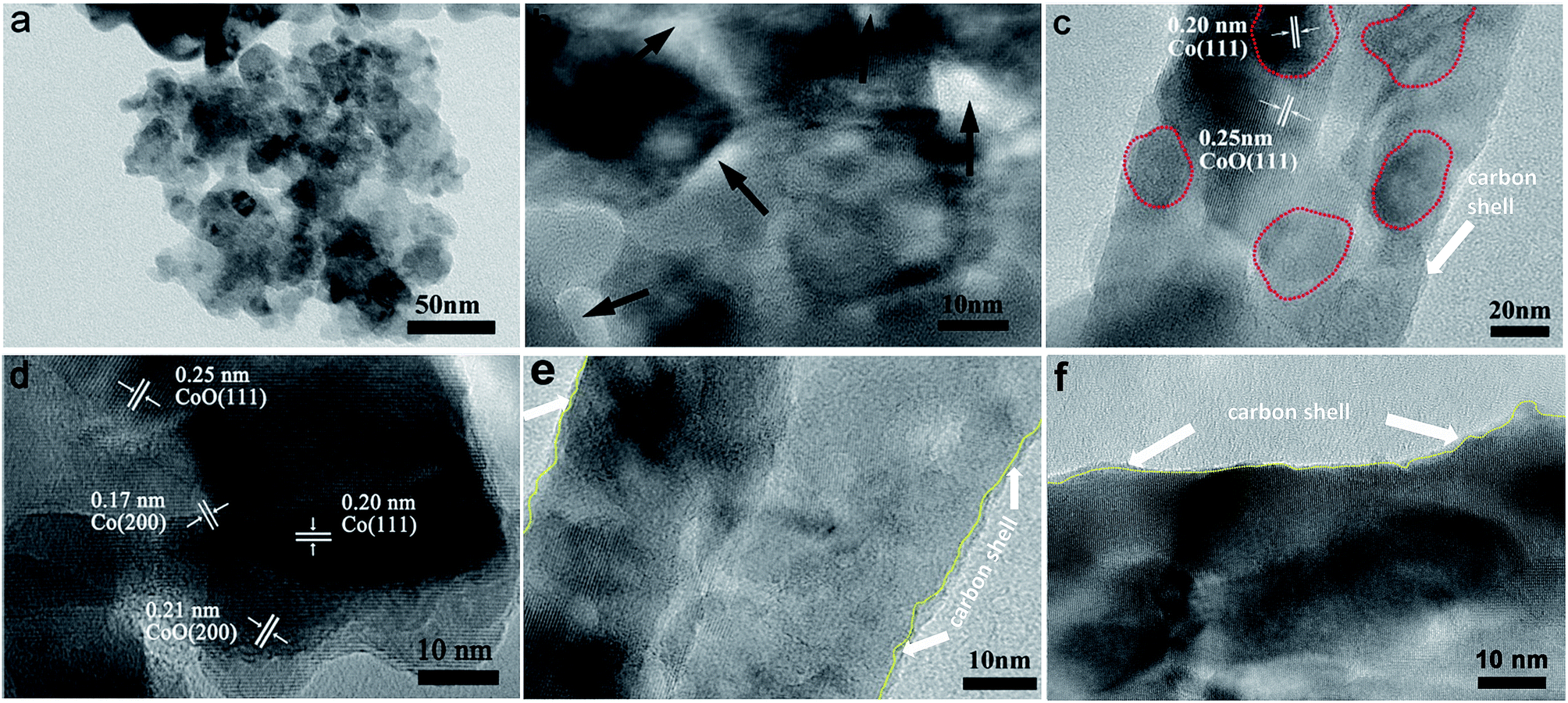
To further understand the structural configuration of CoO/Co@C spheres for the high-performance capacitive behaviour, we characterized the CoO/Co@C spheres by TEM and high-resolution TEM (HRTEM) analysis, as shown in Fig. 3. The hydrangea-like CoO/Co@C microspheres are composed of numerous nanosheets (Fig. 3a), and many pores ranging from 2 nm to 10 nm appear on the surface of these nanosheets (Fig. 3b). It is well known that a porous structure is crucial for improving the electrochemical performance in energy storage electrodes by providing sufficient OH− ions for the faradaic charge transfer reaction, beneficial for achieving a high performance in electrochemical capacitance. The nitrogen adsorption and desorption isotherms of the sample show the pore size distribution at ∼3.93 nm (Fig. S8b†), verifying that the pores are mainly in the region of mesopores (2–50 nm, Fig. S9b†). Moreover, the HRTEM images further confirm the existence of a CoO/Co heterostructure in the CoO/Co@C spheres. In Fig. 3c, the clear grain boundaries are apparent between CoO and Co compositions, and the Co cores are probably surrounded by CoO. In Fig. 3d, the distinct lattice fringes of 0.20 and 0.17 nm are assigned to the (111) and (200) planes of Co, respectively. The spacing of 0.25 and 0.21 nm can be attributed to the (111) and (200) planes of cubic phase CoO, respectively. In addition, as outlined in Fig. 1e and f, a layer of amorphous carbon with a thickness of ∼1 nm is present on the outer side of these nanosheets. These TEM results confirm that the as-prepared CoO/Co@C is a well-crystallized heterostructure with an ultrathin carbon shell wrapping. For the evaluation of thermal stability and Co/CoO amounts, thermogravimetric analysis (TGA) analysis was carried out. Compared to CoO/Co, more weigh increase is observed on the CoO/Co@C sample from room temperature to 800 °C (Fig. S8e and f†), which can be attributed to the higher amount of Co and CoO in the CoO/Co@C sample.45
 |
| | Fig. 3 TEM images of CoO/Co@C composites: nanopetals (a), porous structure (b), and heterostructures (c) on nanopetals. HRTEM images showing the details of characteristic substructures of CoO/Co@C (d–f), the carbon shell is the outer part of the yellow lines. | |
To investigate the composition and chemical bonding states of each element in the CoO/Co@C hybrid, the CoO/Co@C spheres are analyzed by XPS. In Fig. 4a, the survey spectrum shows the presence of Co, C, and O elements in the sample. The presence and nature of carbon layer are evident from the C 1s peak at 284.6 eV (Fig. S10†). Fig. 4b shows the detailed peaks of Co 2p signal. The peaks at 780.4 eV and 796.8 eV correspond to Co 2p3/2 and Co 2p1/2, respectively. The two satellite peaks at 786.5 eV and 802.6 eV are the characteristic features of Co(II) oxide. The small peak at 778.9 eV can be attributed to metallic Co.22 These XPS data verify that the hydrangea-like spheres are composed of CoO, Co, and carbon, consistent with the TEM (Fig. 3) and XRD (Fig. 2d) results.
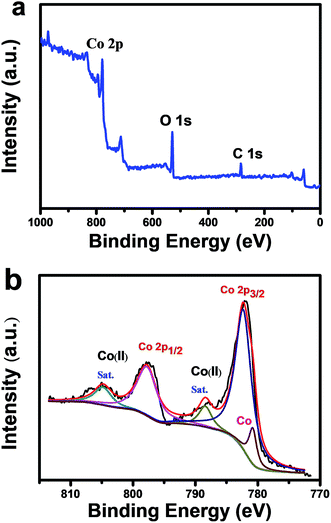 |
| | Fig. 4 XPS characterization of CoO/Co@C spheres: (a) survey spectrum, (b) corresponding Co 2p peaks. | |
Fig. 5A shows the galvanostatic charge–discharge (GCD) curves of the CoO/Co@C at various current densities. The GCD curves are approximately symmetrical demonstrating that the electrode has a high charge–discharge coulombic efficiency and low polarization. Specially, with increasing the charge–discharge current from 2 A g−1 to 50 A g−1, the comparably small IR drop for CoO/Co@C suggests the outstanding electrical conductivity of samples. According to eqn (7), the capacitance calculated from discharge curves is 1985.1 F g−1 at the scan rate of 2 A g−1. Importantly, the CoO/Co@C sample retains a remarkable value of 1101.4 F g−1 when the discharging current density reached up to 20 A g−1, demonstrating a surprising capacitive performance even in a high current density. Although the value achieved in this work was still lower than the theoretical specific capacitance of CoO (4292 F g−1), the CoO/Co@C composite prepared here are among the highest-performing alkali-based Co3O4 supercapacitors studied to date, and also higher than most of the reported CoO fabricated on graphitized carbon,33–36 graphene nanosheets,37 carbon nanotubes,38 and foam nickel.39–41 The outstanding capacitances of CoO/Co@C can be ascribed to the following reasons: (1) the Co metal in the heterostructure, which provides the material excellent electrical conductivity for electron transfer; (2) the mesoporous structure of the nanosheets, which allow more rapid electrolyte contact and transport into these spheres, and ion-diffusion lengths are thus shortened; (3) the ultrathin carbon shell encapsulating the CoO/Co heterostructure, by which each petal will be electronically connected with the neighboring petals and attached to the current collector via the carbon shell, thus decreasing the internal resistance and improving the capacitive performance; (4) the large surface area and high CoO content, which provide abundant active sites and allow maximum utilization of per CoO nanoparticle with fast and reversible faradaic processes on the electrode surfaces becomes possible. Therefore, CoO/Co@C composite has a higher capacitance and improved capacitor behavior than nake CoO/Co (Fig. S6†).
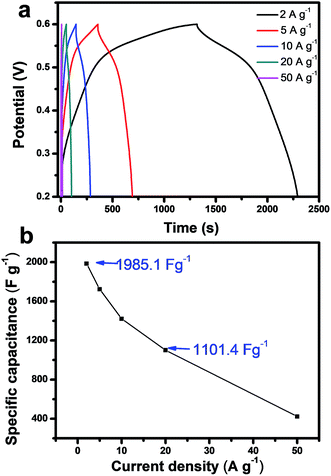 |
| | Fig. 5 (a) Charge–discharge curves for CoO/Co@C at current densities of 2, 5, 10, 20, and 50 A g−1. (b) Specific capacitance of CoO/Co@C measured at different current densities. | |
The ability of energy storage in practical application is another important goal for developing supercapacitor materials. To evaluate the electrochemical energy storage of CoO/Co@C composites, an asymmetric supercapacitor cell (ASC) is assembled using CoO/Co@C as the cathode electrode, active carbon (AC) as the anode electrode, and 2 M KOH aqueous solution as the electrolyte (denoted as CoO/Co@C//AC). Fig. 6a presents the I–V curves of CoO/Co@C (0–0.8 V) and AC (−1.1 to 0 V) at 100 mV S−1 in a three-electrode system, indicating the ASC can extend the potential window of each electrode to reach a wider overall operating potential. Fig. 6b shows the I–V curves of CoO/Co@C//AC ASC at various scan rates. The as-present full cell has a wider operating potential window from 0 V to 1.8 V. The GCD curves of CoO/Co@C//AC ASC recorded at different current densities are plotted in Fig. 6c. Based on the total mass of active materials, the specific capacitance is calculated to be 325.2, 253.6, 214.5, 190.8, 170.0, and 140.0 F g−1 at current densities of 2, 5, 10, 15, 20 and 30 A g−1, respectively. On the basis of eqn (8), the Ragone plot of the as-prepared CoO/Co@C//AC ASC is calculated and shown in Fig. 6e. The ASC delivers a high energy density of 146.3 W h kg−1 at a power density of 1800 W kg−1, and even retains 63.0 W h kg−1 at a high power density of 27000 W kg−1, suggesting a great potential for high-performance energy storage. For comparison, the power densities of some recent reported aqueous batteries using Co oxide-based cathodes are incorporated into Fig. 6c.46–52 It can be observed that the as-prepared CoO/Co@C based ASC is greater than a number of the previously reported Co oxide-based ASCs, such as CoO@NiO and graphene//AC and graphene (52.26 W h kg−1 at 1.26 kW kg−1 and 25.2 W h kg−1 at 9.53 kW kg−1),46 Co–Ni/Co–Ni oxide@carbon//AC (44.6 W h kg−1 at 0.43 kW kg−1 and 7.7 W h kg−1 at 8.50 kW kg−1),47 Ni–Co/Ni–Co LDH//AC (100 W h kg−1 at 1.50 kW kg−1 and 69 W h kg−1 at 15.00 kW kg−1)49 and core shell CoO@Co3O4//graphene (44.06 W h kg−1 at 0.80 kW kg−1 and 24.07 W h kg−1 at 12.00 kW kg−1).52 The durability of ASC is further investigated at a current density of 30 A g−1, as shown in Fig. 6f. After 15000 cycles, the ASC device retained a capacitance of ∼98.1%. The first three cycles and last three cycles within the charge–discharge test keep symmetrical with triangular shape and the same characteristics (Fig. S11†), indicating excellent cycling stability. Meanwhile, a simple application to power a light-emitting diode (LED) was demonstrated by our supercapacitor. After charging one CoO/Co@C//AC ASC to 1.8 V at 10 A g−1, it could power a green LED (working voltage 1.8–2.4 V, 20 mA) as shown in inset of Fig. 6f.
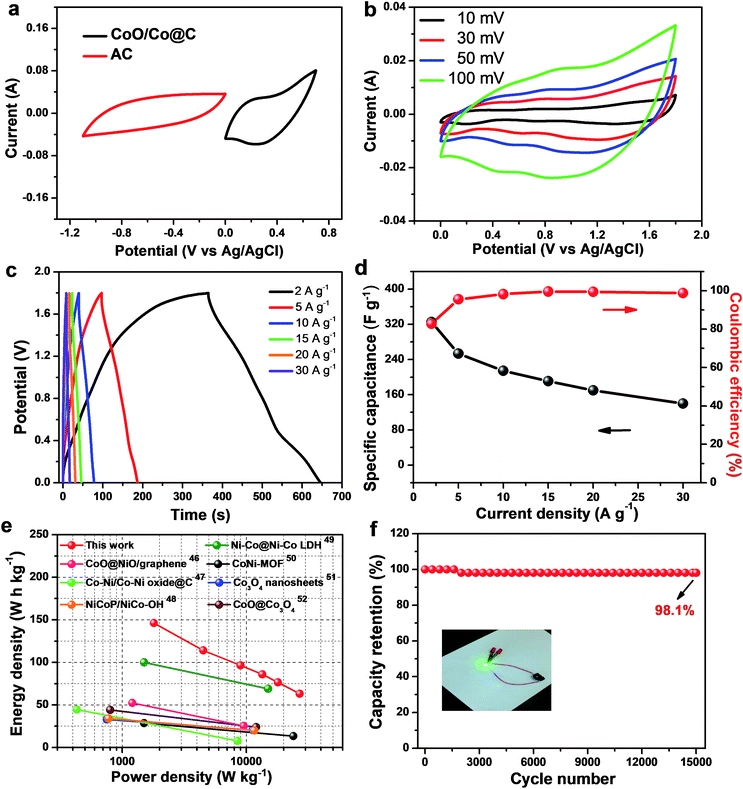 |
| | Fig. 6 (a) Comparison of I–V curves collected for CoO/Co@C and AC electrodes. Characterization of CoO/Co@C//AC ASC: (b) I–V curves at various scan rates, (c) GCD curves at various current densities, (d) the corresponding specific capacitance and coulombic efficiency, (e) the Ragone plot of Co oxide-based ASCs, and (f) cycling performance at a constant current density of 30 A g−1. Inset: photograph of a green round LED indicator powered by one of our asymmetric supercapacitors. | |
Conclusions
In summary, an ultrathin carbon shell coated CoO/Co heterostructure is synthesised via a simple hydrothermal method. Owing to the excellent electrical conductivity of metallic-Co contents and mesoporous nanostructure, the electrons can be easily transferred from the electroactive sites to the current collector. Moreover, the joining of carbon shell also provides double-layer capacitance to the simple faradaic pseudocapacitance system being a hybrid capacitor, while preventing the CoO/Co from being oxidized. The CoO/Co@C composite exhibits a significantly enhanced overall electrochemical performance with a superior specific capacitance of 2165.7 F g−1 at a scan rate of 10 mV s−1, long-term stability, and significantly improved storage stability. After assembling CoO/Co@C as positive electrode and activated carbon as the negative electrode, the aqueous ASC demonstrates energy density up to 146.3 W h kg−1 at 1800 W kg−1, which is the highest energy density achieved among the Co oxide-based supercapacitors. As CoO/Co@C can be prepared with a high yield by a facial low-cost route, it is highly expected that this composite could be a promising material for electrochemical energy storage in practical applications.
Experimental
Synthesis of materials
To prepare a CoO/Co heterostructure, the Co-based precursor was first prepared by a hydrothermal process. Briefly, 4 mM of Co(NO3)2·6H2O and 4 mM of hexamethylenetetramine (HMT) were dissolved in 20 mL deionized (DI) water at 40 °C. Then, 0.2 g of H2C2O4·2H2O was added. After stirring for 10 min, the transparent pink solution was transferred and sealed in a Teflon-lined autoclave and then kept under 180 °C for 6 h to obtain a Co-based precursor. The CoO/Co heterostructure was formed by annealing the precursor in Ar atmosphere at 450 °C for 2 h. To optimize the capacitive performance, different amounts of oxalic acid, HMT, or glycerol were added to the system before the hydrothermal process, followed by hydrothermal and annealing procedures. To prepare CoO/Co@C composite, the Co-based precursor was prepared by the optimized procedure for CoO/Co only by replacing DI water with an equal volume of glycerol. The resultant Co-based precursor was then calcined under the same condition as in the synthesis of Co/CoO.
Physical characterization
The products were characterized using field-emission scanning electron microscopy (Hitachi FE-SEM SU8000). Transmission electron microscopy (TEM, JEOL 2100FEG) was used to investigate the morphology of the as-prepared materials. Powder X-ray diffraction (XRD, PANalytical Empyrean) was performed using CuKα radiation to investigate the structure and composition of the samples. X-ray photoelectron spectroscopy (XPS, VG ESCALAB MKII) was performed by setting the peak signal corresponding to the carbon (C 1s) signal at 284.6 eV. Nitrogen adsorption–desorption isotherms were collected by Autosorb-IQ-MP-C station. The weight of active materials was measured using an analytical balance (Sartorius, maximum = 30 g; d = 0.01 mg). Nitrogen adsorption–desorption isotherms were measured by using a Quantachrome Autosorb-IQ-MP-C apparatus (America) at 77 K. The surface area was obtained by the Brunauer–Emmett–Teller (BET) method and the pore size distribution was calculated from the corresponding desorption branch of the N2 isotherm by the Barrett–Joyner–Halenda (BJH) method. Thermogravimetric analysis was carried out by using a Mettler Toledo TGA/DSC3+ analyzer (Switzerland) from room temperature to 1200 °C at a rate of 10 °C min−1 in air.
Electrochemical characterization
Electrochemical tests were carried out using a CHI660e electrochemical workstation (CH Instrument, Shanghai) in 1 M KOH solution. The typical three-electrode configuration was used with a Pt wire and saturated Ag/AgCl electrode as the counter and reference electrodes, respectively. To prepare the working electrode, 10 mg of CoO/Co@C composite and 0.05% Nafion were dispersed in 1 mL DI water under sonication to form a homogeneous suspension. The suspension (2 μL) was spread on a mirror-polished glassy carbon electrode and dried in an oven at 50 °C for ∼20 min. Electrochemical impedance spectroscopy (EIS) was measured by applying an AC voltage of 5 mV amplitude in the frequency range from 0.01 Hz to 100 kHz at open circuit potential. Specific capacitance was calculated from the I–V curves using the following equation:53| | 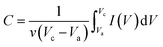 | (6) |
where C is the specific capacitance; v is the potential scan rate (mV s−1); Vc and Va are the sweep potential range (V); I (V) is the response current density (A g−1).
Based on the GCD current, the specific capacitance was calculated using the following equation:
| | 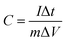 | (7) |
where
C (F g
−1),
I (A),
t (s),
V (V), and
m (g) are the specific capacitance, discharge current, discharge time, potential window, and mass of electrode material, respectively. The energy density (
E) and power density (
P) in a constant current charging/discharging process are calculated using
eqn (8):
54where
C,
V, and
t are the specific capacitance (F g
−1), discharging voltage range (V), and discharging time, respectively.
Assembly of CoO/Co@C//AC asymmetric supercapacitor cell
The CoO/Co@C//AC ASC was fabricated as follows: the cathode consists of 80 wt% of CoO/Co@C, 10 wt% of acetylene black, and 10 wt% of polyvinylidene fluoride (PVDF). The resultant powder (0.1 g) was mixed with 0.6 mL of N-methylpyrrolidinone to form a uniform slurry. The resulting slurry was coated onto a carbon cloth (CC, diameter 2 cm) and dried at 50 °C overnight in a vacuum oven. To fabricate the anode, 80 wt% AC was mixed with 10 wt% of acetylene black and 10 wt% PVDF to form a uniform powder. The following procedure was as the same as that used for the preparation of cathode. The resulting anode was combined with the cathode to assemble a full cell using 2 M KOH as the electrolyte. The mass ratio of positive and negative electrodes is based on the charge balance theory (Q+ = Q−). The specific capacitance, energy, and power densities were calculated according to the total mass of positive electrode.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 21874013, 21775016) and Fundamental Research Funds for the Central Universities (N170506005, N160502001, N170502003, N170908001).
Notes and references
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- J. Chmiola, C. Largeot, P. L. Taberna, P. Simon and Y. Gogotsi, Science, 2010, 328, 480–483 CrossRef CAS PubMed.
- S. W. Lee, B. S. Kim, S. Chen, S. H. Yang and P. T. Hammond, J. Am. Chem. Soc., 2009, 131, 671–679 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC.
- Y. X. Xu, Z. Y. Lin, X. Zhong, X. Q. Huang, N. O. Weiss, Y. Huang and X. F. Duan, Nat. Commun., 2014, 5, 4554 CrossRef CAS PubMed.
- K. R. Prasad, K. Koga and N. Miura, Chem. Mater., 2004, 16, 1845–1847 CrossRef CAS.
- R. N. Reddy and R. G. Reddy, J. Power Sources, 2003, 124, 330–337 CrossRef CAS.
- C. C. Hu, W. C. Chen and K. H. Chang, J. Electrochem. Soc., 2004, 151, A281–A290 CrossRef CAS.
- H. W. Lai, Q. Wu, J. Zhao, L. M. Shang, H. Li, R. C. Che, Z. Y. Lyu, J. F. Xiong, L. J. Yang, X. Z. Wang and Z. Hu, Energy Environ. Sci., 2016, 9, 2053–2060 RSC.
- R. B. Rakhi, W. Chen, D. Y. Cha and H. N. Alshareef, Nano Lett., 2012, 12, 2559–2567 CrossRef CAS PubMed.
- J. Yan, E. Khoo, A. Sumboja and P. S. Lee, ACS Nano, 2010, 4, 4247–4255 CrossRef CAS PubMed.
- K. Zhou, W. J. Zhou, X. J. Liu, Y. H. Sang, S. Z. Ji, W. Li, J. Lu, L. G. Li, W. H. Niu, H. Liu and S. W. Chen, Nano Energy, 2015, 12, 510–520 CrossRef CAS.
- X. H. Lu, G. M. Wang, T. Zhai, M. H. Yu, J. Y. Gan, Y. X. Tong and Y. Li, Nano Lett., 2012, 12, 1690–1696 CrossRef CAS PubMed.
- G. Zhang, W. Li, K. Xie, F. Yu and H. Huang, Adv. Funct. Mater., 2013, 23, 3675–3681 CrossRef CAS.
- G. P. Wang, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- J. Feng, X. Sun, C. Z. Wu, L. L. Peng, C. W. Lin, S. L. Hu, J. L. Yang and Y. Xie, J. Am. Chem. Soc., 2011, 133, 17832–17838 CrossRef CAS PubMed.
- T. Li, G. H. Li, L. H. Li, L. Liu, Y. Xu, H. Y. Ding and T. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 2562–2572 CrossRef CAS PubMed.
- Z. D. Gao, X. Zhu, Y. H. Li, X. M. Zhou, Y. Y. Song and P. Schmuki, Chem. Commun., 2015, 51, 7614–7617 RSC.
- Y. Y. Song, Y. H. Li, J. Guo, Z. D. Gao and Y. Li, J. Mater. Chem. A, 2015, 3, 23754–23759 RSC.
- C. L. Zhang, H. Yin, M. Han, Z. Dai, H. Pang, Y. Zheng, Y. Q. Lan, J. Bao and J. Zhu, ACS Nano, 2014, 8, 3761–3770 CrossRef CAS PubMed.
- H. Pang, J. W. Deng, J. M. Du, S. J. Li, Y. H. Ma, J. S. Zhang and J. Chen, Dalton Trans., 2012, 34, 10175–10181 RSC.
- Y. H. Li, T. T. Li, Y. X. Lu, X. J. Wang, Z. D. Gao and Y. Y. Song, J. Power Sources, 2017, 354, 108–115 CrossRef CAS.
- Y. Yu, C. H. Chen, J. L. Shui and S. Xie, Angew. Chem., Int. Ed., 2005, 44, 7085–7089 CrossRef CAS PubMed.
- X. L. Huang, R. Z. Wang, D. Xu, Z. L. Wang, H. G. Wang, J. J. Xu, Z. Wu, Q. C. Liu, Y. Zhang and X. B. Zhang, Adv. Funct. Mater., 2013, 23, 4345–4353 CrossRef CAS.
- S. L. Xiong, J. S. Chen, X. W. Lou and H. C. Zeng, Adv. Funct. Mater., 2012, 22, 861–871 CrossRef CAS.
- X. Ji, S. Cheng, L. F. Yang, Y. Jiang, Z. J. Jiang, C. J. Yang, H. Zhang and M. L. Liu, Nano Energy, 2015, 11, 736–745 CrossRef CAS.
- K. C. Liu and M. A. Anderson, J. Electrochem. Soc., 1996, 143, 124–130 CrossRef CAS.
- S. W. Lee, J. Kim, S. Chen, P. T. Hammond and Y. Shao-Horn, ACS Nano, 2010, 4, 3889–3896 CrossRef CAS PubMed.
- Q. Y. Liao, N. Li, S. X. Jin, G. W. Yang and C. X. Wang, ACS Nano, 2015, 9, 5310–5317 CrossRef CAS PubMed.
- J. C. Deng, L. T. Kang, G. L. Bai, Y. Li, P. Y. Li, X. G. Liu, Y. Z. Yang, F. Gao and W. Liang, Electrochim. Acta, 2014, 132, 127–135 CrossRef CAS.
- E. Duraisamy, H. T. Das, A. S. Sharma and P. Elumalai, New J. Chem., 2018, 42, 6114–6124 RSC.
- H. Wang, C. Qing, J. L. Guo, A. A. Aref, D. M. Sun, B. X. Wang and Y. W. Tang, J. Mater. Chem. A, 2014, 2, 11776–11783 RSC.
- M. Zhou, J. Catanach, J. Gomez, S. Richins and S. G. Deng, ACS Appl. Mater. Interfaces, 2017, 9, 4362–4373 CrossRef CAS PubMed.
- M. Cheng, S. B. Duan, H. S. Fa, X. R. Ru, Y. M. Cui and R. M. Wang, Chem. Eng. J., 2017, 327, 100–108 CrossRef CAS.
- D. N. Lan, Y. Y. Chen, P. Chen, X. Y. Chen, X. Wu, X. L. Pu, Y. Zeng and Z. H. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 11839–11845 CrossRef CAS PubMed.
- L. Jiang, R. J. Zou, W. Y. Li, J. Q. Sun, X. H. Hu, Y. F. Xue, G. J. He and J. Q. Hu, J. Mater. Chem. A, 2013, 1, 478–481 RSC.
- Y. G. Zhu, Y. Wang, Y. Shi, J. I. Wong and H. Y. Yang, Nano Energy, 2014, 3, 46–54 CrossRef CAS.
- N. Zhang, X. H. Yan, J. Lia, J. M. Ma and D. H. L. Ng, Electrochim. Acta, 2017, 226, 132–139 CrossRef CAS.
- X. Z. Wang, Y. H. Xiao, D. C. Su, L. M. Zhou, S. D. Wu, L. F. Han, S. M. Fang and S. K. Cao, Electrochim. Acta, 2016, 194, 377–384 CrossRef CAS.
- Y. B. Liu, L. Y. Lin, Y. Y. Huang and C. C. Tu, J. Power Sources, 2016, 315, 23–34 CrossRef CAS.
- C. Zhou, Y. W. Zhang, Y. Y. Li and J. P. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS PubMed.
- J. F. Xiong, H. Shen, J. X. Mao, X. T. Qin, P. Xiao, X. Z. Wang, Q. Wu and Z. Hu, J. Mater. Chem., 2012, 22, 11927–11932 RSC.
- M. Cabán-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H.-C. Chang, M.-L. Tasi, J.-H. He and J. Song, Nat. Mater., 2015, 14, 1245–1251 CrossRef PubMed.
- J. Deng, S. Li, Y. Zhou, L. Liang, B. Zhao, X. Zhang and R. Zhang, J. Colloid Interface Sci., 2018, 509, 406–413 CrossRef CAS PubMed.
- Z. Gao, N. N. Song and X. D. Li, J. Mater. Chem. A, 2015, 28, 14833–14844 RSC.
- H. M. Sun, X. J. Yang, L. S. Zhang, L. J. Zhao and J. S. Lian, J. Mater. Chem. A, 2017, 17, 8095–8107 RSC.
- X. Li, H. J. Wu, A. M. Elshahawy, L. Wang, S. J. Pennycook, C. Guan and J. Wang, Adv. Funct. Mater., 2018, 20, 1800036 CrossRef.
- Y. Liu, N. Q. Fu, G. G. Zhang, M. Xu, W. Lu, L. M. Zhou and H. T. Huang, Adv. Funct. Mater., 2017, 8, 1605307 CrossRef.
- T. Deng, Y. Lu, W. Zhang, M. L. Sui, X. Y. Shi, D. Wang and W. T. Zheng, Adv. Energy Mater., 2018, 7, 1702294 CrossRef.
- Z. Y. Xiao, L. L. Fan, B. Xu, S. Q. Zhang, W. P. Kang, Z. X. Kang, H. Lin, X. P. Liu, S. Y. Zhang and D. F. Sun, ACS Appl. Mater. Interfaces, 2017, 48, 41827–41836 CrossRef PubMed.
- M. Cheng, S. B. Duan, H. S. Fan, X. R. Su, Y. M. Cui and R. M. Wang, Chem. Eng. J., 2017, 327, 100–108 CrossRef CAS.
- D. Ghosh, S. Giri and C. K. Das, Nanoscale, 2013, 5, 10428–10437 RSC.
- Y. H. Jin and M. Q. Jia, Colloids Surf., A, 2015, 464, 17–25 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional, SEM, TEM, XPS, BET and CV tests. See DOI: 10.1039/c8ta09733j |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*