Phase selective organogel from an imine based gelator for use in oil spill recovery†
Received
9th October 2018
, Accepted 21st November 2018
First published on 23rd November 2018
Abstract
Marine oil spills create a serious problem for the environment by destroying ecosystems, and the adulteration of fuel oils is also a serious problem, as the optimum performance of an engine is affected by damaging its parts. To save the environment and prevent adulteration, a number of Schiff base-derived organogelators (OG-1, OG-2 and OG-3) are synthesized with different pendant groups containing anthracene, pyrene and cyclohexane moiety, respectively. They exhibit thermo-reversible gelation behavior in organic and mixed solvents. Only OG-1 produces gels in most organic solvents and also in fuel and edible oils, indicating it to be a smart gelator. It exhibits thixotropic behavior with very high yield stress, while the other gels do not exhibit thixotropic properties. Spectroscopic and X-ray studies indicate the concomitant presence of H-bonding, π-stacking and van der Waals interactions in the OG-1 gel; however, the OG-2 and OG-3 gels do not exhibit all these interactions, making weaker gels. Due to its superior gelling property, the OG-1 gel is tacitly used to detect fuel oil contamination mainly in diesel and petrol using fluorescence behavior, and the phase selective gelation of OG-1 promotes it as a successful recyclable material for oil spill recovery from oil–water mixtures, even in the presence of salts, acids and bases.
Introduction
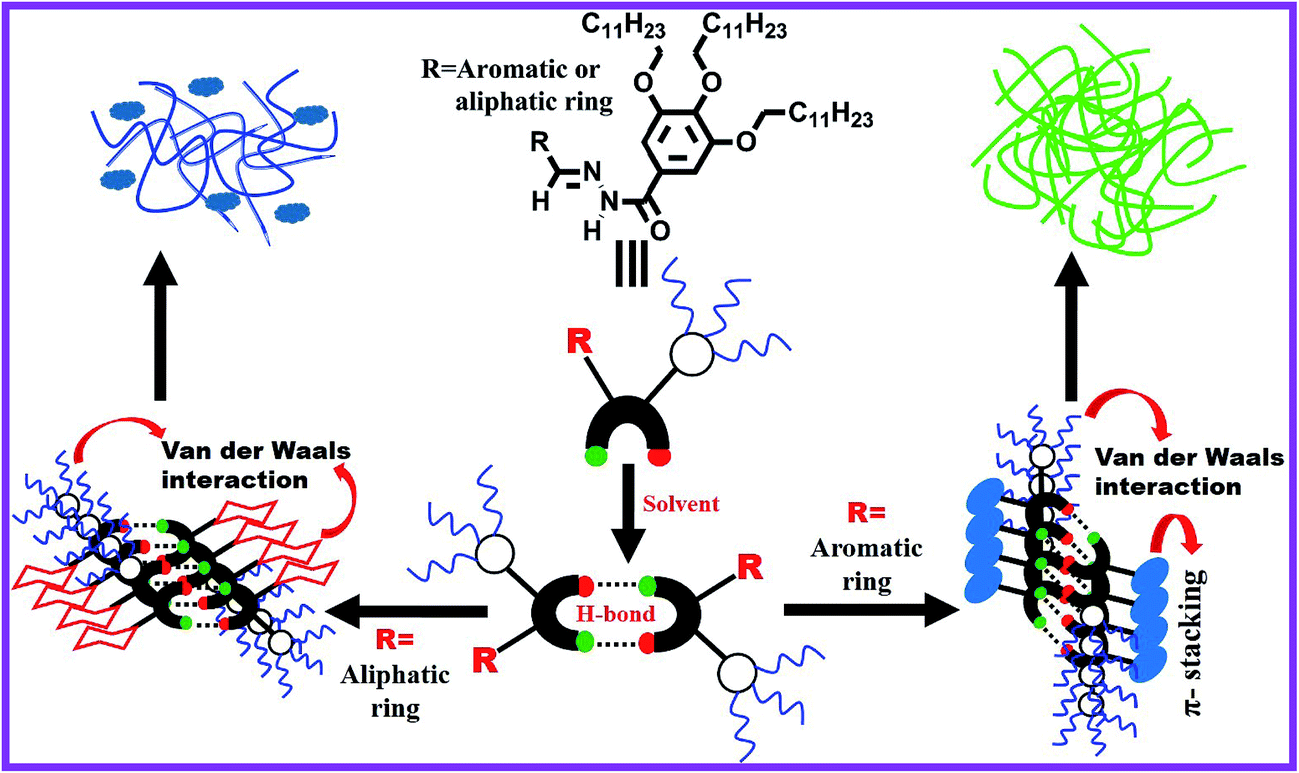
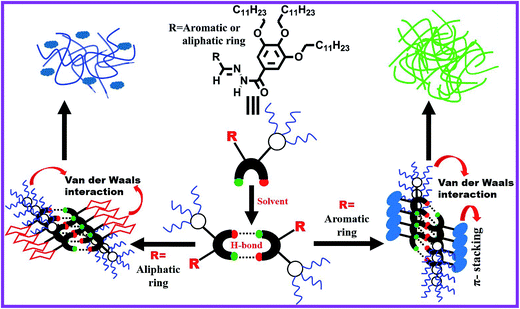
Low molecular weight organogels (LMOGs) are a most important class of soft materials and they are studied with great interest, because of their unique physical properties and classic architectures.1–4 Supramolecular organogels are formed through self-aggregation of small gelator molecules in an organic solvent. During gelation the gelator molecules in the solution first form a supramolecular assembly by non-covalent interactions (primary structure), which self-assemble to form fibrils (secondary structure) that entangle into a three-dimensional network structure (tertiary structure) encapsulating a large amount of solvent within the network.5–8 Since the fibrillar network involves weak non-covalent interactions, therefore one can manipulate its thermo-reversible, mechano-responsive (thixotropic) behavior9 and its gelation properties by using external stimuli such as heat,10,11 light,12 ultrasound,13 pH,14 anions,15 metal cations,16 and solvent.1,17 In the past few decades, a wide variety of LMOGs have been developed for various applications including cosmetics,18 sensors,19,20 template materials,21 catalysts,22 imaging,23 photoresponsive materials,24,25 dye removal,26,27 dye-sensitized solar cells28,29 and soft optical devices.30,31 Currently, the field of LMOGs is developing greatly; nevertheless, it is still a very challenging task to predict the gelation properties of a molecule from its molecular structure. In some cases, researchers have serendipitously discovered gelation properties of an organic molecule. Now it is believed that there are some fundamental structural motifs, such as long alkyl chains that facilitate van der Waals interactions, planar aromatic rings that promote π–π stacking interactions, and functional groups that enable multiple hydrogen-bonding interactions, that must be required for a particular molecule to be a good gelator.32,33 Apart from these parameters, solvent selection is also an important factor for the preparation of molecular gel.34 A solvent that cannot easily solubilize a gelator at room temperature, but can do so after heating or sonication, can be considered as a good solvent for gelation.17 Some gelators can form gels in mixed solvents either organic–organic or aqueous–organic mixtures.35 A phase selective organogelator (PSG) is capable of forming a gel in one solvent preferentially among two immiscible solvents, and this type gelator is very useful for application to separate a particular phase.36
Marine oil spills due to leakage of crude oils and petroleum products (refined fuel oils) create a serious problem for the environment, especially for marine life by destroying marine ecosystems. Over several decades, a number of accidents have occurred throughout the world involving marine oil spills.37,38 Therefore it is very necessary to develop some smart materials and techniques that can manage this type of environmental pollution immediately and save our environment ecosystems and mitigate socioeconomic losses. In this perspective, a large number of different methods have been developed such as bioremediation,39,40 dispersants,41 solidifiers,42–44 sorbents,45–47 and skimmers.48–50 However, these methods have some drawbacks; for example, in the bioremediation process, the complete conversion of a hydrocarbon into carbon dioxide and water with the help of bacteria takes a longer time. In the dispersants technique, the materials used for water–oil separation are majorly toxic for marine life. On the other hand, polymers are used in the solidifier technique that converts oil into solid or semi-solid phase, and recovery of the oil from that solid or semi-solid phase is very difficult and also needs a longer time.
Nowadays, chemists study PSGs with great interest because of their excellent ability to separate oil from an oil–water biphasic system.36 An amino acid amphiphile based PSG was reported in 2001, since then different types of PSGs have been reported in the literature.51–58 However, all the developed soft materials have some limitations for practical applications. Some fundamental requirements must be fulfilled by a gelator molecule to be a good PSG; these are: (i) the gel should be formed from gelator molecules selectively, efficiently and instantly in the oil phase at room temperature, (ii) the production cost of the synthesized gelator should be low, (iii) recovery of the oil from the gel phase should be easy and (iv) finally the gelator should be a recyclable and reusable material. Sugar-based gelators can selectively form a gel in various oils and can be used in the recovery of oil but it takes longer time to form a strong gel.59 But instant gelation at ambient temperature in oils is of utmost necessity to minimize the spreading of the oil onto seawater.
To solve the intriguing problem, in this work, we have synthesized a number of Schiff base-derived organogelators (Scheme 1) and have studied their gelation behavior in a wide variety of organic solvents, mixed organic solvents, fuel oils and edible oils. Petrol, diesel and kerosene are refined fuel oils consisting of long-chain hydrocarbons such as alkanes, cycloalkanes and aromatics. The gelators presented in Scheme 1 are designed to have long alkyl chains and large aromatic structures imparting an ability to form gels quickly in most aliphatic and aromatic hydrocarbons, including fuel oils through H-bonding, van der Waals and π–π stacking interactions. For phase selective gelation, a strong mutual interaction between gelator and solvent molecules is also a crucial factor for designing a molecular gelator and this is expected from the structure of gelators. OG-1 showed a light-induced E–Z isomerization across the imine bond in the gel state and this has been elucidated from 1H NMR spectroscopy.11 The isomerization transition in its different states (sol, gel and xerogel) upon photo-irradiation occurs at different rates.60 Here we show that amongst the Schiff base-derived gelators, OG-1 is suitable to exhibit selective, efficient and instant gelation at ambient temperature in the oil phase, and gelator molecules can be recovered by simple distillation without any structural change of the gelator. When a solution of OG-1 in THF is added to a water–oil mixture, gelator OG-1 forms a gel instantly, selectively and efficiently in the oil part of the water–oil mixture within 60 seconds at room temperature (30 °C). We have also studied contamination in fuel oils, usually brought about by mixing cheaply available kerosene oil in petrol and diesel, using fluorescence spectroscopy with the OG-1 gel.
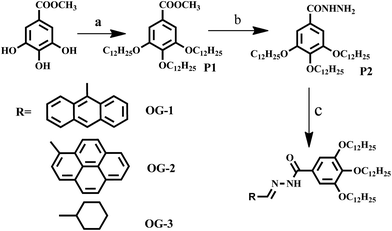 |
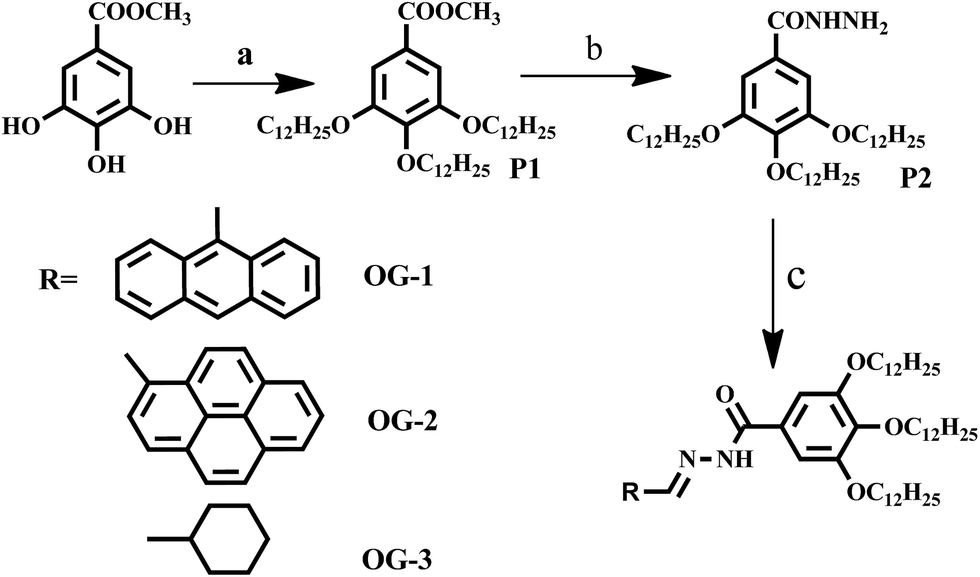
| | Scheme 1 Synthetic scheme for producing different Schiff base organogelators. (Reaction conditions: (a) C12H25Br, K2CO3, KI, dry DMF, 80 °C, 48 hours; (b) NH2NH2·H2O, MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (1:1), 75 °C, 24 hours; (c) aldehyde derivatives, THF, RT, 2 hours). :THF (1:1), 75 °C, 24 hours; (c) aldehyde derivatives, THF, RT, 2 hours). | |
Experimental
Synthesis
The synthesis procedure applied for producing the three Schiff base organogelators is summarized in Scheme 1 and the details of the synthetic procedure and characterization of each product are presented in ESI.†
Preparation of organogel
10 mg of synthesized compound was taken in a 5 ml vial, and 1 ml solvent was added to it. The solution mixture was then sealed, sonicated, and heated until a homogeneous solution was prepared. When the resultant solution was cooled to room temperature, an organogel was produced. By using the above procedure, organogels were prepared from compounds OG-1, OG-2 and OG-3. For making gels of OG-1 in fuel oils, 10 mg of OG-1 and 1 ml of kerosene/diesel/petrol were taken in a 5 ml vial. The mixture was then sealed and only sonicated for 4–6 minutes until the solution mixture was transformed into a semisolid gel. In another technique the gels were produced by the addition of a minimum amount (50 to 100 μL) of solvent (a solvent in which the solubility of OG-1 is very high) to a vial containing 10 mg of compound OG-1 and a clear solution was obtained by shaking. Immediately a transparent gel was produced after addition of the above solution to a vial containing 0.9 ml of fuel oil.
Thermal study
Thermal study of the organogels (OG-1, OG-2 and OG-3) was performed by using differential scanning calorimetry (DSC; Perkin Elmer, Diamond) using large-volume capsules (LVC) fitted with O-rings under nitrogen atmosphere. The instrument was calibrated with indium. The gel samples were kept in the LVC pan which was hermetically sealed with a rubber O-ring. It was equilibrated at 0 °C for 10 minutes and was heated to 130 °C (depending on gel samples) with a heating rate of 10 °C per minute. It was then cooled at a cooling rate of 5 °C per minute to 0 °C. The melting point was determined from the computer attached to the instrument using Pyris software.
Rheology
The mechanical properties of the organogels were investigated using an advanced rheometer (AR 2000, TA Instruments, USA) using cone plate geometry on a Peltier plate. The diameter of the plate was 40 mm and cone angle was 4° with a plate gap of 121 μm.
Diffraction study
A wide-angle X-ray scattering (WAXS) study of xerogels and powders of compound OG-1, OG-2 and OG-3 was performed using a Bruker AXS diffractometer (model D8 Advance) fitted with a Lynx Eye detector. The instrument was operated at 40 kV voltage and at 40 mA current. Samples were pasted on glass slides and were scanned at a scan rate of 0.5 s per step with a step width of 0.02° in the range of 2θ = 4–50°.
Microscopy
SEM images of the samples were obtained with a FESEM instrument (JEOL, JSM 6700F) operating at 5 kV after platinum coating. Small portions of organogels OG-1 (in kerosene oil), OG-2 (ethanol and CHCl3 mixture) and OG-3 (ethanol and methanol mixture) were placed on glass coverslips and were dried in open air at 30 °C for one day and finally in a vacuum for 3 days. The morphologies of the OG-1, OG-2 and OG-3 gels were investigated by transmission electron microscopy (TEM; JEOL, model 2010EX). Small portions of organogels OG-1 (in petrol oil), OG-2 (ethanol and CHCl3 mixture) and OG-3 (ethanol and methanol mixture) were diluted and drop-cast on carbon-coated copper grid (300 mesh). The samples were dried in open air at 30 °C and finally kept in a vacuum for one day at 30 °C before the experiment.
Spectroscopy
1H NMR data of synthesized compounds were collected with a Bruker spectrometer (1H: 400 MHz). The FTIR spectra of the compounds (OG-1, OG-2 and OG-3) were recorded in their powder and xerogel states using KBr pellets of the samples with an FTIR-8400S instrument (Shimadzu). The UV-visible spectra in the sol or gel state of samples OG-1, OG-2 and OG-3 were recorded with a Hewlett-Packard UV-visible spectrophotometer (model 8453) using a cuvette of 1 mm path length. The fluorescence study was performed with the gel states of OG-1, OG-2 and OG-3. The samples were prepared in a sealed cuvette, and scanned using a Horiba Jobin Yvon Fluoromax 3 instrument. Each gel sample in a quartz cell of 1 cm path length was excited at a specific wavelength and the emission scans were recorded in the range of 290–725 nm with an increment of 1 nm having an integration time of 0.1 s.
MALDI-TOF
Mass spectra of OG-1, OG-2 and OG-3 were recorded using a MALDI TOF Ultraflextreme (Bruker Daltonics) instrument by using dithranol and 2,5-dihydroxybenzoic acid as matrix.
Results and discussion
In this work, we have synthesized a number of gelator molecules including our previously reported gelator molecule (OG-1) by applying simple alkylation, substitution and Schiff base reaction method (Scheme 1). The details of the synthetic procedure and characterization of the compounds are given in the ESI.† The gelation studies of compounds OG-1, OG-2 and OG-3 were carried out in a number of organic solvents, mixed organic solvents, fuel oils and edible oils. The gels obtained from these gelators exhibit thermo-responsive behavior. The details of the gelation studies in different solvents with their critical gelation concentration are presented in Table S1.† Most of the OG-1 gels in different solvents possess fluorescent, thixotropic and thermo-responsive properties (Fig. 1).
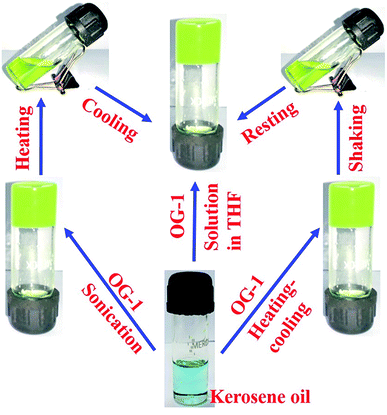 |
| | Fig. 1 Schematic representation of OG-1 gel (1% w/v) in kerosene oil, derived by heating–cooling, shaking–resting and addition of OG-1 solution in THF to the kerosene. | |
Structural analysis
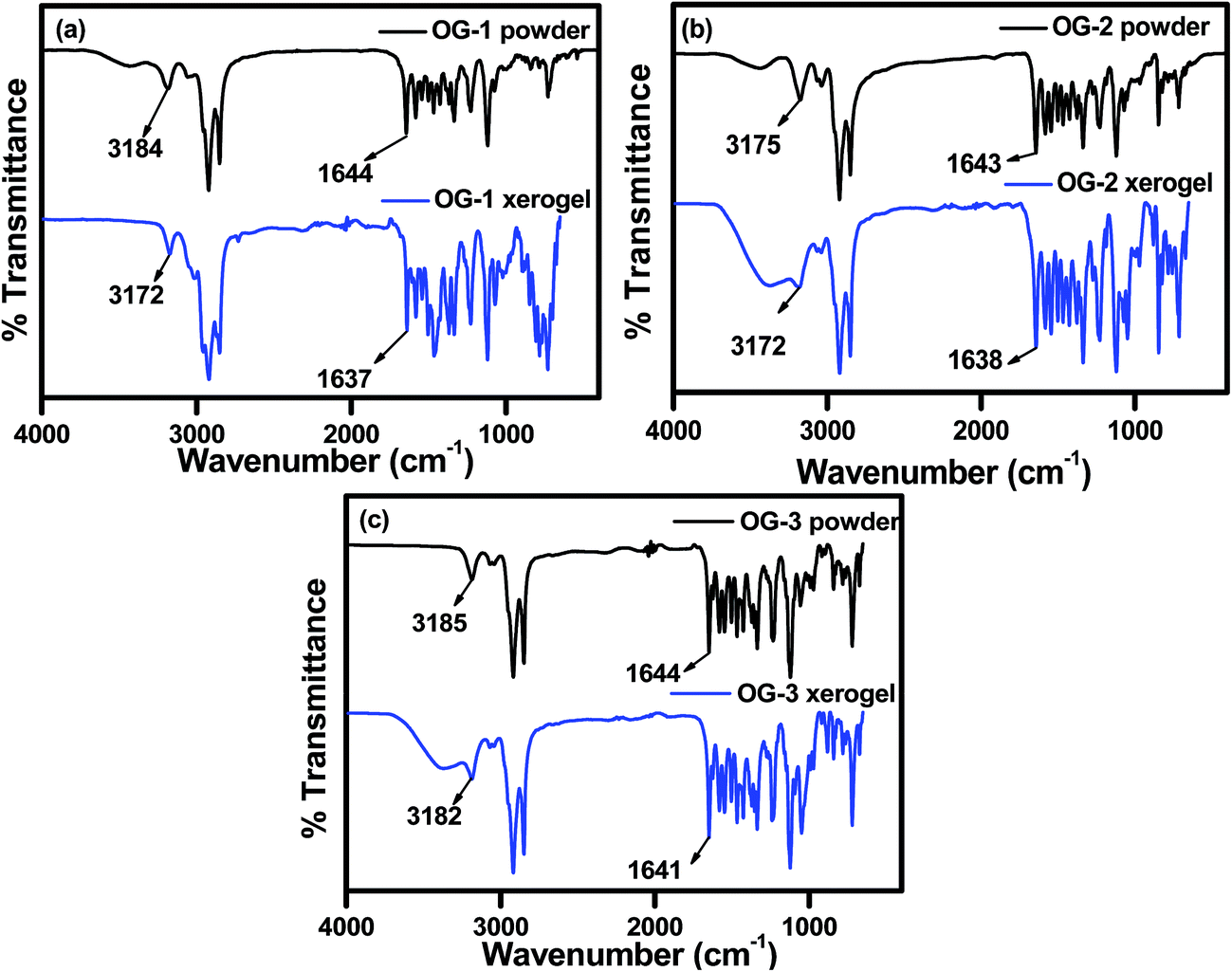
FTIR spectra of powders and xerogels of the above gelator molecules are presented in Fig. 2 to understand the hydrogen bonding interactions between the different functional groups. The –NH and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequencies at ∼3180 and ∼1644 cm−1 in the powder states of all the compounds, i.e. OG-1, OG-2, and OG-3, have shifted to lower frequencies in the xerogel states due to the formation of intermolecular hydrogen bonding between the –NH and –CO groups in the gel state (Fig. 2). In the xerogel of compound OG-1, the shifting of the –NH and –CO stretching peaks from those in the powder state is 12 and 7 cm−1 respectively; the shifting for OG-2 is 3 and 5 cm−1, respectively, and the shifts of OG-3 are 3 and 3 cm−1, respectively. Thus, OG-1 xerogel shows greater shifts than those of OG-2 and OG-3 from their powder state, which may be attributed to the formation of strong intermolecular hydrogen bonding between complementary functional groups in the gel state.3,61 From the gelation studies of gelators OG-1, OG-2, and OG-3 in different solvents (Table S1†), it is evident that gelator OG-1 has a greater ability to form a gel efficiently and instantly in a large number of different types of solvents and solvent mixtures with a minimum gelator concentration indicating that OG-1 is a very good gelator. The FTIR spectra of OG-1 also reveal strong intermolecular hydrogen bonding between the complementary functional groups of the gelator molecules, which eventually help to construct a well-defined entangled network structure leading to better gel formation than the other two.
O stretching frequencies at ∼3180 and ∼1644 cm−1 in the powder states of all the compounds, i.e. OG-1, OG-2, and OG-3, have shifted to lower frequencies in the xerogel states due to the formation of intermolecular hydrogen bonding between the –NH and –CO groups in the gel state (Fig. 2). In the xerogel of compound OG-1, the shifting of the –NH and –CO stretching peaks from those in the powder state is 12 and 7 cm−1 respectively; the shifting for OG-2 is 3 and 5 cm−1, respectively, and the shifts of OG-3 are 3 and 3 cm−1, respectively. Thus, OG-1 xerogel shows greater shifts than those of OG-2 and OG-3 from their powder state, which may be attributed to the formation of strong intermolecular hydrogen bonding between complementary functional groups in the gel state.3,61 From the gelation studies of gelators OG-1, OG-2, and OG-3 in different solvents (Table S1†), it is evident that gelator OG-1 has a greater ability to form a gel efficiently and instantly in a large number of different types of solvents and solvent mixtures with a minimum gelator concentration indicating that OG-1 is a very good gelator. The FTIR spectra of OG-1 also reveal strong intermolecular hydrogen bonding between the complementary functional groups of the gelator molecules, which eventually help to construct a well-defined entangled network structure leading to better gel formation than the other two.
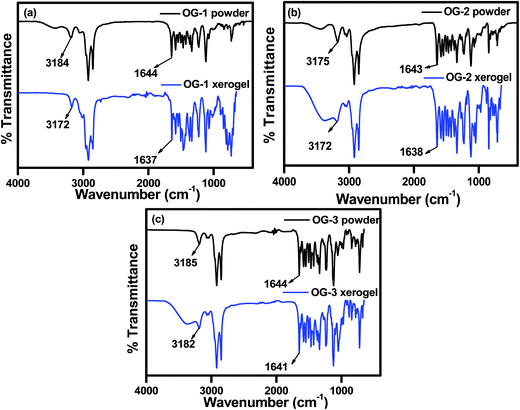 |
| | Fig. 2 FTIR spectra of (a) OG-1, (b) OG-2 and (c) OG-3 in their powder and xerogel states obtained from kerosene, ethanol/CHCl3 mixture and ethanol/methanol mixture, respectively. | |
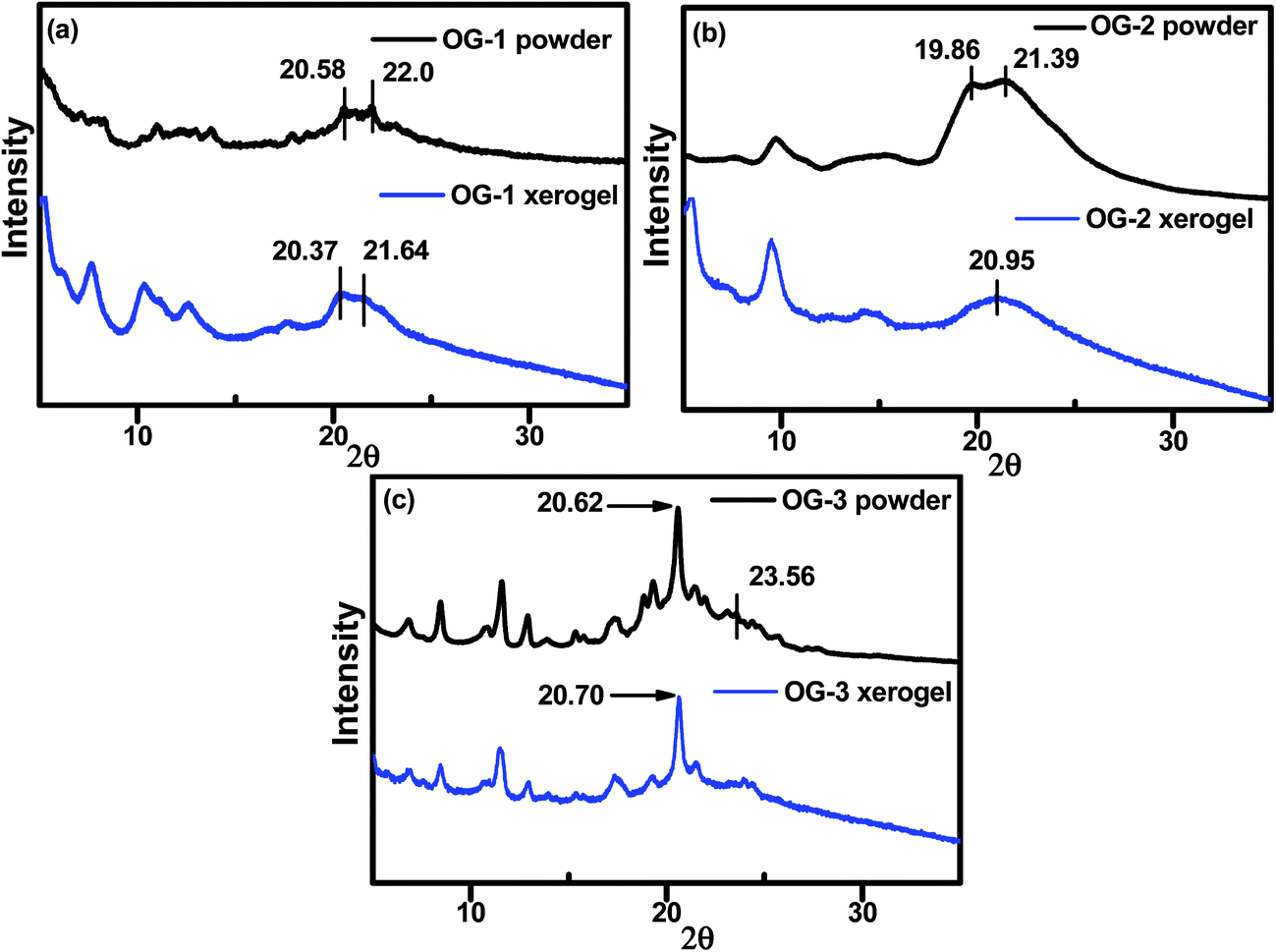
X-ray diffraction (XRD) experiments have been carried out to understand the molecular packing in self-assembled or gel state of the gelator molecules. From Fig. 3, it can be visualized that the packing nature in the xerogel state of OG-1 and OG-2 is different from that in the powder state; however, the OG-3 XRD patterns are almost similar for the powder and xerogel states. A broad peak in the range of 19.4–23.5° in the XRD pattern of OG-1 (Fig. 3a) consists of two peaks at 2θ = 20.37 (d = 4.35 Å) and 21.64° (d = 4.1 Å) in the xerogel state corresponding to the packing distance of the alkyl chains and π–π stacking distance of the aromatic rings respectively.62 These peak positions are at somewhat lower positions than those of the powder indicating more compact packing of the molecules in the xerogel state. OG-2 (Fig. 3b) exhibits two peaks at 2θ = 19.86° (d = 4.46 Å) and 21.39° (d = 4.14 Å) in the powder state due to the packing distance of the alkyl chains and π–π stacking distance of the aromatic rings respectively; but for the xerogel state of OG-2 a broad peak at 20.95° (d = 4.23 Å) is observed.
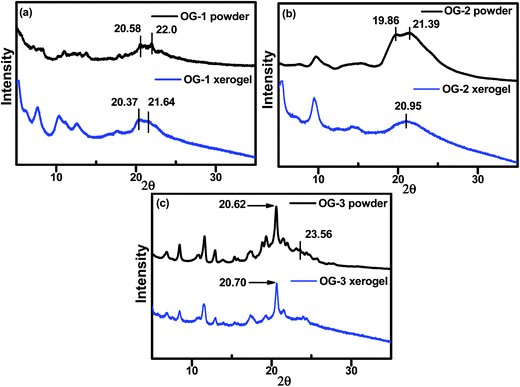 |
| | Fig. 3 Wide-angle XRD patterns of (a) OG-1 powder and its xerogel obtained from kerosene oil, (b) OG-2 powder and its xerogel obtained from ethanol/chloroform mixture and (c) OG-3 powder and its xerogel obtained from ethanol/methanol mixture. | |
Probably, this broad peak may arise for the overlapping of the packing distances of alkyl chains and π–π stacking of the aromatic rings. The XRD patterns of OG-3 in its powder and xerogel state are not much different (Fig. 3c) and the peak at 2θ = 20.62° (d = 4.3 Å) corresponds to the packing of alkyl chains in the powder state of OG-3, while in the xerogel state this peak is shifted to 20.70° (d = 4.28 Å). A weak diffraction peak at 2θ = 23.56° having a d spacing value of 3.77 Å corresponds to the π–π stacking distance of the aromatic phenyl rings in the powder state of OG-3 and this peak is absent in the xerogel state of OG-3, which indicates the π–π stacking interaction does not occur in the gel state due to lack of aromatic rings. Therefore, from the XRD observations it has been revealed that gelator OG-1 forms stronger π–π stacking interaction than OG-2, whereas OG-3 lacks such a π–π stacking interaction in the gel state.
Thermal study
The melting temperature of gels can be measured by DSC thermograms. The DSC thermograms of OG-1 gel in kerosene, OG-2 gel in EtOH:CHCl3 (9.5:0.5) and OG-3 gel in EtOH:MeOH (1:2) at a 1.5% (w/v) concentration for each gel are recorded and are shown in Fig. 4. From Fig. 4a it is evident that on heating, the OG-1 gel shows a melting peak at 80 °C and the broad nature of the melting peak indicates the polydisperse nature of the aggregates. On cooling, the OG-1 gel exhibits an exothermic peak at 28 °C indicating a large hysteresis in the gel melting and gelation temperatures, suggesting the first-order transition nature of the gelation process. For OG-2 gel, a broad melting peak occurs at 63.5 °C and on cooling an exothermic peak appears at 31.2 °C. In the case of OG-3, on heating, a comparatively sharp melting peak appears at 59.70 °C and, on cooling, there is also a sharp peak observed at 29.87 °C. The sharp peaks in the DSC thermogram of OG-3 in contrast to those of OG-1 and OG-2 may be due to the crystalline/precipitate nature in the gel state of OG-3 which is also evident from the XRD results (Fig. 2c) and mechanical studies of gels presented below.
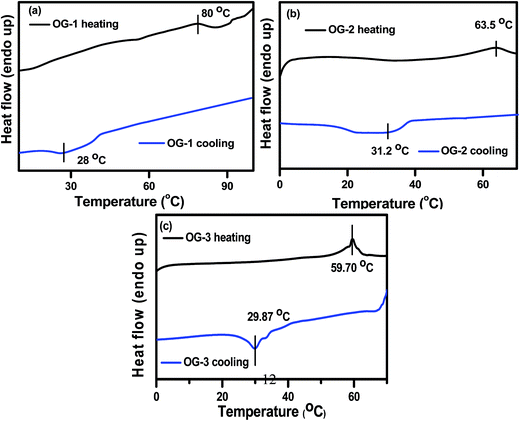 |
| | Fig. 4 DSC thermograms for gels of (a) OG-1 (1.5% w/v) in kerosene (b) OG-2 (1.5% w/v) in EtOH/CHCl3 mixture and (c) OG-3 (1.5% w/v) in EtOH/MeOH mixture. | |
Morphology
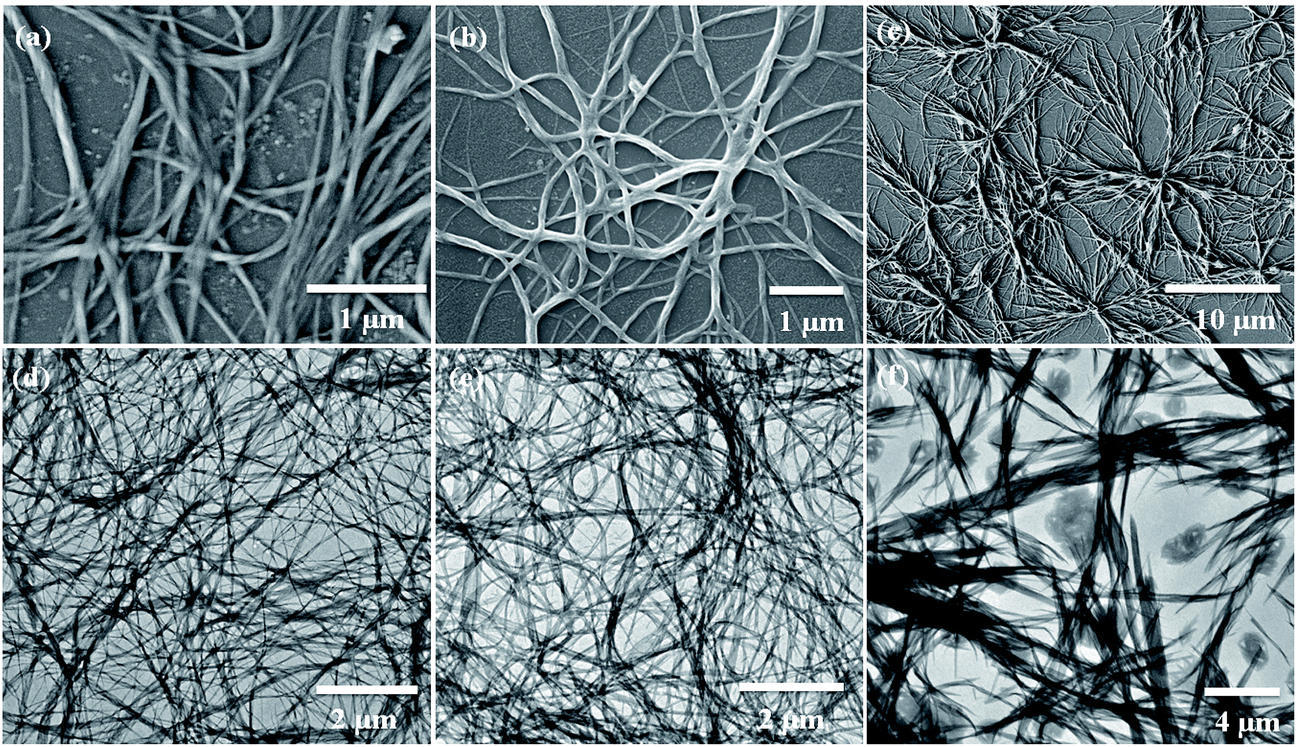
Scanning electron microscopy (SEM) is widely applied for supramolecular gel systems to extract valuable information about the morphology of the self-organized structures. From the SEM images (Fig. 5a–c) it is apparent that the xerogels of OG-1, OG-2, and OG-3 consist of fibrils which are entwined with each other. It is clearly evident from the TEM images shown in Fig. 5d–f that OG-1 and OG-2 xerogels possess a three-dimensional fibrillar network morphology, while OG-3 xerogel has a mixture of fibrillar network along with spheroidal morphology. The formation of the fibers can be understood from the intermolecular H-bonding between ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CO and –NH groups and the H-bonded complexes undergo π-stacking (OG-1 and OG-2) and van der Waals interaction through the long alkyl chains of the gelator molecules (all of three gelators). A schematic representation for the formation of the fibrillar network morphology (OG-1 and OG-2) and the fibrillar network along with spheroidal morphology (OG-3) is presented in Scheme 2. Thus growth of a supramolecular assembly occurs, producing fibres which entangle to produce a three-dimensional fibrillar network.11
CO and –NH groups and the H-bonded complexes undergo π-stacking (OG-1 and OG-2) and van der Waals interaction through the long alkyl chains of the gelator molecules (all of three gelators). A schematic representation for the formation of the fibrillar network morphology (OG-1 and OG-2) and the fibrillar network along with spheroidal morphology (OG-3) is presented in Scheme 2. Thus growth of a supramolecular assembly occurs, producing fibres which entangle to produce a three-dimensional fibrillar network.11
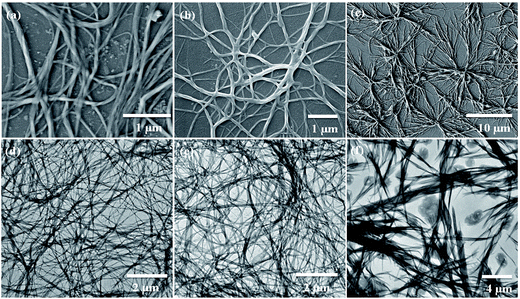 |
| | Fig. 5 SEM images of (a) OG-1 in kerosene oil, (b) OG-2 in EtOH:CHCl3 (9.5:0.5) mixture and (c) OG-3 in EtOH:MeOH (1:2) mixture. TEM images of (d) OG-1 in petrol, (e) OG-2 in EtOH:CHCl3 (9.5:0.5) mixture and (f) OG-3 in EtOH:MeOH (1:2) mixture. | |
 |
| | Scheme 2 Schematic representation of the formation of a fibrillar network (for OG-1 and OG-2) and the fibrillar network along with spheroidal morphology (for OG-3) in the presence of solvent. | |
Mechanical properties
Gels are basically viscoelastic solid-like materials comprised of an elastic cross-linked network and a solvent. A combined property of viscous and elastic nature always exists for a gel system.4 We have performed rheological experiments with 1.5% (w/v) OG-1 gel in kerosene, OG-2 gel in ETOH:CHCl3 (9.5:0.5) and OG-3 gel in EtOH:MeOH (1:2) to understand their mechanical properties. The results are presented in Fig. 6 and S1.† Dynamic frequency sweep experiments were carried out on all of the gels (Fig. 6a, S1a and c†) at a constant strain of 0.1%. From Fig. 6a, S1a and c† it is evident that the storage modulus (G′) is greater than the loss modulus (G′′) and is independent of the frequency in a wide range of frequency for all three gels, suggesting the viscoelastic nature of the materials in their gel states. When the storage modulus (G′) of a material is greater than the loss modulus (G′′) then it behaves as a solid-like material and vice versa. The stress sweep experiments (Fig. 6b, S1b and d†) have been performed on the gel states of OG-1, OG-2 and OG-3 to obtain the yield stress of the gels. G′ and G′′ of the gels cross each other for all gels upon applying oscillatory stress, which indicates the breaking of the gel to a quasi-sol state. The values of oscillatory stress at which G′ and G′′ cross each other are different for the different gels. From Fig. 6b, it is clear that for OG-1 gel the yield stress has the highest value (50 Pa) and for OG-3 gel it has the lowest value (16 Pa), whereas the value of OG-2 gel is 25 Pa, which is an intermediate value between that of OG-1 and OG-3. The greater value of yield stress indicates the gel is mechanically stronger and hence more mechanical force is needed to break the gel into a sol. Therefore, it can be revealed from the oscillatory stress results that the gelator OG-1 is mechanically more robust than the other two. The lowest value of yield stress of OG-3 gel from those of others can be explained as follows. The solubility of OG-3 in methanol is very poor but in ethanol its solubility is good, and at a higher concentration (3% w/v) it forms gel in ethanol (Table S1†). At a lower gelator concentration (1.5% w/v) it forms an opaque or turbid gel in binary solvents.63 When a saturated hot solution is kept at room temperature, three situations may arise: case-I, a well-arranged structure, i.e. crystal, may form; case-II, a precipitate may form; case-III, an intermediate of these two, i.e. a gel, may form. When a saturated hot solution of OG-3 in the binary solvent (ethanol and methanol mixture) is kept at room temperature, it forms a weak turbid gel within two hours at room temperature, which is easily broken by mechanical shaking. The weak nature of OG-3 gel is due to lack of π–π stacking interaction between the molecules making it unable to construct a well-ordered structure required for good gelation (Scheme 2). For this reason, the OG-3 gel exhibits the lowest yield stress value and it also accounts for the sharp melting and cooling peaks in the DSC thermogram (Fig. 4c). Thixotropy is an exciting viscoelastic property of supramolecular gels in which a gel phase is transformed to a sol phase with the help of mechanical force under isothermal conditions, which returns to its original state (gel state) on allowing the sample to rest. OG-1 gels in kerosene, diesel and petrol exhibit thixotropic behavior (Fig. 1). The thixotropic property of OG-1-kerosene gel is confirmed by performing time sweep experiments at a lower strain of 0.1% and at a higher strain of 50% (Fig. 6c). The figure manifests that the kerosene gel of OG-1 is quickly converted into the sol state upon applying 50% strain and it quickly returns to its original state (gel state) after releasing the high strain. At low strain of 0.1%, the value of G′ is greater than that of G′′ for the first, third and fifth steps, indicating gel nature of OG-1. On the other hand, the value of G′′ is greater than that of G′ at the higher strain of 50% (second and fourth steps), indicating the sol nature of OG-1 at the higher strain. From Fig. 6c, it is observed that the value of G′ at low strain is almost the same for all of the three steps (first, third and fifth) which indicates that the OG-1 gel almost completely returns to its original phase after releasing the mechanical force. From the rheological investigation it can be revealed that the hydrogen bonding interaction, π–π stacking interaction and van der Waals interaction co-operate together forming of a well-defined 3D network structure that leads to a robust OG-1 gel (Scheme 2). The OG-1 gel exhibits thixotropic property but OG-2 and OG-3 gels do not; this is due to gelator OG-1 containing all of the three functional motifs which concurrently operate to construct a well-defined fibrillar network leading to a mechano-responsive gel. OG-3 contains a non-aromatic cyclic ring, which fails to develop the π–π stacking interactions as in OG-1 in the gel state (Scheme 2). Although the gelator OG-2 contains all of the three functional motifs, it fails to construct a well-defined network like OG-1. The pyrene ring present in the OG-2 moiety has a strong tendency to form excimer or exciplex in the gel state, which changes structural arrangement in which all of these three non-covalent interactions might not work concurrently, and therefore it is unable to form a mechano-responsive gel like OG-1.
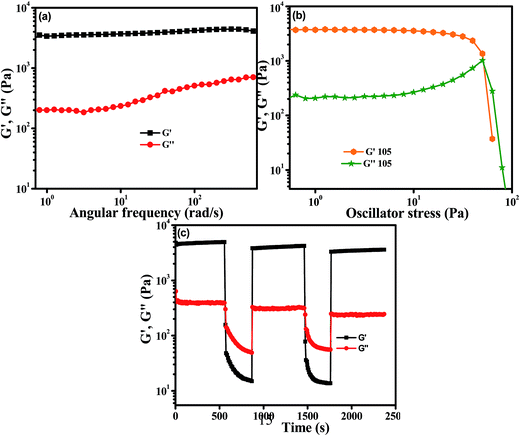 |
| | Fig. 6 Rheological properties of OG-1 gel (1.5% w/v) in kerosene: (a) angular frequency sweep, (b) stress sweep and (c) continuous-step strain measurement at alternating 50 and 0.1% strain with time. | |
Optical properties
Fig. 7 and S2† show the concentration-dependent UV-visible spectra of OG-1 in kerosene, OG-2 in EtOH:CHCl3 (9.5:0.5, v/v) mixture and OG-3 in EtOH:MeOH (1:2) mixture. In the mixed solvents, OG-2 and OG-3 are soluble at low concentration. The two peaks at 369 nm and 387 nm in the UV-visible spectra of the solution (0.01% w/v) of OG-1 are due to the π–π* and n–π* transitions of the OG-1 molecule (Fig. 7a).11 Interestingly, both of these peaks are gradually red-shifted to 373 nm and 393 nm respectively with increasing gelator concentration to 0.2% (w/v). The shifting of the peaks with increasing gelator concentration to lower energy (higher wavelength) in the absorption spectra is due to the J-type aggregation of the gelator molecules.11,64 We could not detect any significant shift of the absorption peak positions from the solution state to the gel state of the gelators OG-2 and OG-3 (Fig. 7b and S2†).
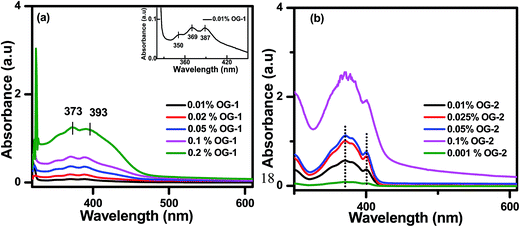 |
| | Fig. 7 UV-visible spectra of (a) OG-1 in kerosene and (b) OG-2 in EtOH:CHCl3 (9.5:0.5). | |
The peaks at 371 nm and 401 nm in the UV-visible spectrum of 0.001% (w/v) OG-2 correspond to the π–π* and n–π* transitions of OG-2 molecule (Fig. 7b). With an increase of gelator concentration the intensity of these two peaks gradually increases with intact peak positions. For the gelator OG-3, a similar observation to that of the gelator OG-2 is observed from Fig. S2.† The peaks at 219 nm and 273 nm in the UV-visible spectrum of 0.02% (w/v) OG-3 correspond to the π–π* and n–π* transitions of OG-3 molecule. From Fig. S2,† it is evident that the intensity of the peaks of OG-3 gelator gradually increases with increasing gelator concentration; however, the peak positions remain the same.
The nature of aggregation of self-assembly in the gel state can also be understood with the help of fluorescence spectroscopy. Fig. 8 and S3† show the concentration-dependent fluorescence spectra of OG-1 in kerosene, OG-2 in EtOH:CHCl3 (9.5:0.5) mixture and OG-3 in EtOH:MeOH (1:2) mixture. The concentration-dependent fluorescence spectra of OG-1 in kerosene for excitation at 370 nm are presented in Fig. 8a and it is evident that at 0.01% (w/v) OG-1, the solution exhibits an emission maximum at 481 nm, and this emission band is due to the monomer of the OG-1 molecule. The peak at 481 nm is gradually red-shifted along with a sharp rise in fluorescence intensity with increasing gelator concentration, and at higher concentration it reaches 498 nm. In general, the gelator OG-1 in kerosene behaves as a solution below a concentration of 0.15% (w/v). Therefore, it is clear from the figure that the fluorescence intensity of the OG-1 gel is nearly 8 times higher than that of its solution and also there is a 17 nm red shift of the emission peak in the gel state compared to the solution state supporting the formation of J type aggregate.11,65 For the gelator OG-2, the concentration-dependent fluorescence spectra for excitation at 360 nm are shown in Fig. 8b. The critical gelation concentration of the gelator OG-2 in EtOH:CHCl3 is found to be 0.2% (w/v); therefore, below this concentration, it behaves as a solution. The solution of OG-2 (0.01% w/v) exhibits an emission maximum at 431 nm, which is due to the monomeric peak of OG-2. When the concentration of OG-2 gelator is gradually increased from 0.01% to 2% (w/v), an interesting scenario is observed in the fluorescence spectra of OG-2 (Fig. 8b). When the gelator concentration is gradually increased from 0.01% (w/v) to 0.25% (w/v), the red shifting of the monomeric peak of OG-2 from 431 nm to 443 nm along with a sharp enhancement of the fluorescence intensity is observed (Fig. 8b).
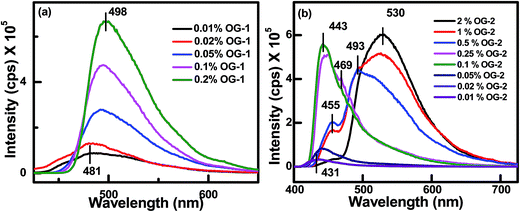 |
| | Fig. 8 Concentration-dependent fluorescence spectra of (a) OG-1 in kerosene for excitation at 370 nm and (b) OG-2 in an EtOH:CHCl3 (9.5:0.5) solvent mixture for excitation at 360 nm. | |
It is to be noted that a small hump is generated at 469 nm. Thereafter, with a further increase of gelator concentration from 0.25% to 0.5% (w/v) a red shifting of 443 nm peak to 455 nm occurs with a sharp decrease in emission intensity and a concomitant generation of a new peak occurs with a significant intensity at 493 nm. At 1% (w/v) concentration of OG-2 the 455 nm peak shows a further decrease of intensity and the new peak intensity increases significantly showing a maximum at 526 nm. For 2% (w/v) concentration, the intensity of the 455 nm peak is almost abolished and the intensity of newly generated peak further increases shifting to 530 nm (Fig. 8b). This is an interesting observation to be discussed here. The emission band at 443 nm (0.1%) in the fluorescence spectra is due to the aggregation of OG-2 monomer (emission at 431 nm), but with a further increase of OG-2 concentration, the pyrene moiety present in the OG-2 comes to closer proximity and forms an excimer complex, whose emission peak gradually increases in intensity and red-shifts from 493 nm to 530 nm at 2% concentration.66–70 For the gelator OG-3 also, an increase of fluorescence intensity is observed with increased of OG-3 concentration (Fig. S3†) due to aggregation-induced emission showing a broad nature of the peak for the gel state.
For a visual understanding of the fluorescence property, images of the gels of OG-1, OG-2, and OG-3 under normal light and UV light are shown in Fig. 9. The figure shows that the gel of OG-1 in kerosene, petrol and diesel exhibits good fluorescence under UV light, and OG-2 gel in ethanol/chloroform mixture also exhibits good fluorescence under UV light (Fig. 9). The gel of OG-3 in methanol/ethanol mixture shows violet fluorescence under UV light (Fig. 9).
 |
| | Fig. 9 Images of 0.5% (w/v) OG-1 gel (a and f) in petrol, (b and g) in diesel, (c and h) in kerosene under normal light and UV light at 365 nm wavelength respectively, 0.5% (w/v) OG-2 gel (d and i) in ethanol–chloroform mixture under normal light and UV light at a 365 nm wavelength, respectively, and 1.5% (w/v) OG-3 gel (e and j) in methanol–ethanol mixture under normal light and UV light at a 365 nm wavelength, respectively. | |
The gelation test of gelators OG-1, OG-2, and OG-3 in fuel oils and in edible oils is also shown in Fig. S4.† It is interesting that OG-1 forms gels in all edible oils as well as in fuel oils; however, OG-2 and OG-3 do not exhibit any gelation in any of the above oils.
Study of fuel oil contamination by fluorescence spectroscopy in the gel state
Adulteration of petroleum products or fuel oils (mainly diesel and petrol) is a serious problem, particularly in developing countries in recent times.71,72 The optimum performance of an engine is affected by adulteration which causes damages of the engine's parts. Fuel oils that are easily available at lower prices (e.g. kerosene) are utilized for adulteration purposes. In particular in Southeast Asia, the cheaply available kerosene oil is very much used as an adulterant in petrol and diesel fuel. Several methods have been developed for determining the adulteration of fuel oils, but these have some limitations. Recently, the fluorescence spectroscopy technique has been developed for such perspective and is widely used to determine adulteration of fuel oils sensitively.73,74 We have previously mentioned that OG-1 produces a gel in all the fuel oils (kerosene, diesel and petrol) and exhibits excellent fluorescence properties for excitation at a 365 nm wavelength (Fig. 9). Kerosene oil (blue tinged) is non-fluorescent under UV light at a 365 nm wavelength but the kerosene gel of OG-1 shows bright green fluorescence under UV light. From Fig. 10a it is evident that the emission peaks at 498, 510 and 526 nm correspond to the 0.5% (w/v) kerosene gel, diesel gel and petrol gel of OG-1. The presence of kerosene in petrol and diesel in the aggregation state of OG-1 is determined by fluorescence spectroscopy (Fig. 10b). Both in the kerosene–diesel (1:1) mixture and kerosene-petrol (1:1) mixture a prominent peak at 498 nm in the gel state of OG-1 has been observed in the fluorescence spectra. The emission peak at 498 nm in the fluorescence spectra is due to aggregation of OG-1 (0.5% w/v) in kerosene oil. Therefore, the adulteration by kerosene in diesel and petrol fuel can be easily determined by fluorescence spectroscopy in the aggregation state of gelator OG-1. A detailed study of adulteration by using OG-1 is beyond the scope of this work.
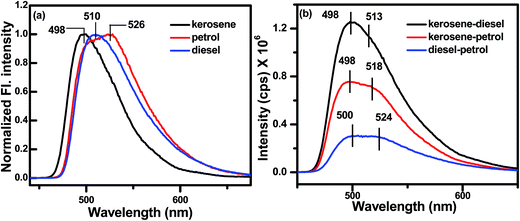 |
| | Fig. 10 Fluorescence spectra of OG-1 gel (0.5% w/v) in (a) single solvent (kerosene, petrol and diesel) and (b) a mixture of solvents (kerosene–petrol, kerosene–diesel and petrol–diesel). | |
Oil spill recovery
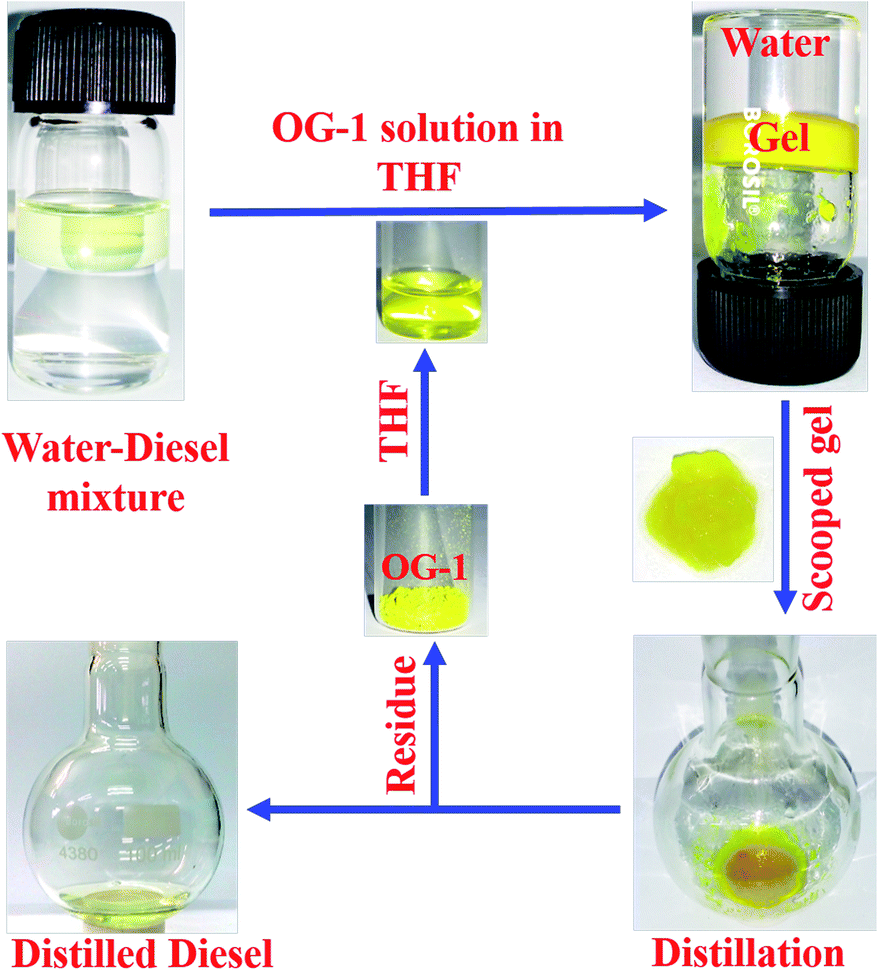
The gelator OG-1 can form a gel selectively, efficiently and instantly in a huge number of solvents including fuel oils and edible oils at ambient temperature (Fig. S4†). Therefore it can be used in the practical application of oil spill cleaning. To recover seawater from oil spills and to avoid the spreading of oil over river water or seawater, there is the need for a superior technique that can be easily applied at room temperature to overcome this serious problem. Instant gelation at ambient temperature is one of the best techniques for oil spill recovery. Our synthesized OG-1 gelator shows the phase selective gelation of the oil phase in the presence of water in an oil–water mixture. We have applied three distinct methods to prepare OG-1 gels in fuel oils, which are (i) heating–cooling, (ii) sonication and (iii) solution of OG-1 in THF is added to fuel oils. In all three techniques, OG-1 forms gels efficiently in fuel oils (kerosene, diesel, and petrol) and these gels can exhibit good fluorescence, thermoreversible and thixotropic properties (Fig. 1 and 9). We have chosen the last method, i.e. a solution of OG-1 in THF is added to fuel oils, for recovery of oil from an oil–water mixture. In this technique, OG-1 forms a gel selectively, efficiently and instantly in the oil phase in the oil–water mixture within 60 seconds at room temperature (30 °C) without application of any external energy (heat/sonication). When OG-1 solution in THF is added to the fuel oils, an instant gelation occurs. The presence of a small amount of THF does not alter the gelation ability of OG-1 gelator in diesel, petrol or kerosene. In this context, 30 mg of OG-1 has been dissolved in 300 μL THF, and the resultant solution is added to a culture tube containing water:diesel (6 ml:3 ml) mixture. The upper phase or diesel oil phase of this biphasic mixture is instantly converted to gel within 60 seconds at room temperature, keeping the aqueous phase intact. There is no displacement of the gel layer in the presence of an upper aqueous layer observed in an inverted culture tube (Scheme 3), which indicates that the gel in the culture tube is stiff enough and supports its utility on a large scale for real use. Interestingly, no trouble in the phase selective gelation has been observed in the presence of different salts (NaCl, KCl, MgSO4), acid and base in water medium, this observation being strongly encouraging for the practical application of gelator OG-1, which forms a gel in oil phase in presence of oil–salt water mixture of seawater. This is verified from stress sweep experiments of OG-1 gel in kerosene in the presence of different salts (NaCl, KCl, MgSO4), acid and base in a water medium (Fig. S5†). From Fig. S5 and S6† it is evident that the G′, G′′ and yield stress values are almost the same as those of OG-1 gel in kerosene/pure water mixture. We have also recorded the FTIR spectra of OG-1 xerogel obtained from kerosene water mixture containing different salts (NaCl, KCl and MgSO4), acid and base (Fig. S7†). From Fig. S7† it is evident that the peak positions of the –NH and CO groups of the xerogels obtained from the kerosene–water mixture in the presence of different salts, acid and base are identical to those of the pure OG-1 xerogel obtained from the OG-1 kerosene gel indicating similar intermolecular H-bonding interaction between –NH and CO groups. So rheological and FTIR studies confirm that the phase selective gelation of OG-1 in fuel oil proceeds smoothly in the presence of different salts, acid and base in the water medium.
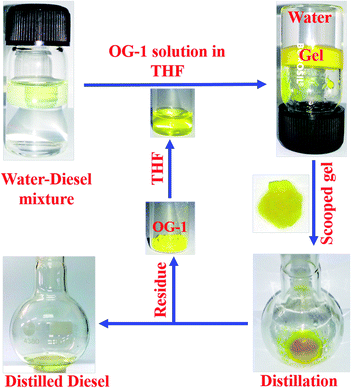 |
| | Scheme 3 Schematic representation of the gelation of OG-1 in a binary solvent mixture and vacuum distillation process. | |
Keeping in mind the importance of the reusability of a material, we recovered the gelator OG-1 by vacuum distillation in which the oil phase and gelator OG-1 (as residue) have been successfully collected. It has been found by gelation studies that the gelation ability of recovered OG-1 from gel phase by vacuum distillation is the same as in the pristine state. The whole recovery process is shown in Scheme 3. In this process, gel formed in diesel oil in the diesel–water mixture is scooped from the culture tube by a spatula and kept in a round bottom flask or in a glass Petri dish (Scheme 3). The scooped gel in the round bottom flask is vacuum distilled in order to collect OG-1 as a residue and the diesel oil as the distilled solvent. By vacuum distillation from 3 ml of OG-1 gel in diesel using a rotatory evaporator, the recovery of oil is 2.6 ml, giving a percentage recovery of 87%. The other 13% may be lost during vacuum distillation. The residue OG-1 obtained after vacuum distillation of scooped gel has been characterized by mass spectrometry (Fig. S8†) and 1H NMR spectroscopy (Fig. S9†), and does not show any structural alteration of the gelator molecules. Therefore, the recovered OG-1 gelator can be used for further studies, strongly recommending its utility for cyclic application.
Conclusions
In conclusion we have synthesized three new Schiff bases, which are proved to behave as organogelators (OG-1, OG-2 and OG-3). OG-1 containing the anthracene moiety has been found to produce gels in most organic solvents and also in fuel and edible oils. However, OG-2 containing a pyrene moiety is not so effective for gelling organic solvents, except in some alcohols and mixed solvents. OG-3 containing a cyclohexane moiety is reluctant to behave as an organogelator, gelling only in ethanol and in a few mixed solvents. Neither OG-2 nor OG-3 produce gels in fuel or edible oils. OG-1 gel exhibits thixotropic behavior with very high yield stress, while the other gels do not exhibit thixotropic property and their yield stress is also much lower. The gel melting temperature is highest for OG-1 compared to the others. OG-1 and OG-2 xerogels exhibit a fibrillar network morphology, but OG-3 xerogel has a mixture of a fibrillar network and spheroidal morphology. FTIR spectra show that hydrogen bonding between CO and –NH groups is more favorable in OG-1 compared to the other gelators. WAXS studies suggest good π-stacking interactions in OG-1, whereas OG-3 does not exhibit any π-stacking interactions. UV-visible spectra of OG-1 exhibit a redshift of the π–π* and n–π* transition in the gel state from that of the solution state, indicating J-aggregate formation, whereas other gelators do not exhibit any such shift. The concentration-dependent fluorescence spectra of OG-1 in kerosene with excitation at 370 nm exhibit shifting of emission maxima towards higher wavelengths, supporting J-type aggregate formation. Due to the superior gelling property of OG-1, it has been tacitly used to detect fuel oil contamination mainly in diesel and petrol by studying the fluorescence behavior of the produced gel in the adulterated fuel. Due to the phase selective gelation of OG-1, it has been shown that it can be easily and successfully used, with recyclability, for oil spill recovery from oil–water mixtures, even in the presence of salts, acids and bases.
Conflicts of interest
We declare that we have no conflict of interest.
Acknowledgements
We gratefully acknowledge CSIR grant no. 02(0241)/15/EMR-II for financial support. S. M. and S. D. acknowledge CSIR for fellowships.
References
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- A. Dawn and H. Kumari, Chem.–Eur. J., 2018, 24, 762–776 CrossRef CAS PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- L. E. Buerkle and S. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- T. Das, M. Häring, D. Haldar and D. Díaz Díaz, Biomater. Sci., 2018, 6, 38–59 RSC.
- A. Dawn, T. Shiraki, S. Haraguchi, S. Tamaru and S. Shinkai, Chem.–Asian J., 2011, 6, 266–282 CrossRef CAS PubMed.
- P. Chakraborty, S. Mondal, S. Khara, P. Bairi and A. K. Nandi, J. Phys. Chem. B, 2015, 119, 5933–5944 CrossRef CAS.
- D. Dasgupta, S. Srinivasan, C. Rochas, A. Ajayaghosh and J. M. Guenet, Langmuir, 2009, 25, 8593–8598 CrossRef CAS.
- S. Mondal, P. Chakraborty, P. Bairi, D. P. Chatterjee and A. K. Nandi, Chem. Commun., 2015, 51, 10680–10683 RSC.
- J. T. van Herpt, J. Areephong, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem.–Eur. J., 2014, 20, 1737–1742 CrossRef CAS PubMed.
- J. M. Malicka, A. Sandeep, F. Monti, E. Bandini, M. Gazzano, C. Ranjith, V. K. Praveen, A. Ajayaghosh and N. Armaroli, Chem.–Eur. J., 2013, 19, 12991–13001 CrossRef CAS PubMed.
- S.-Y. Hsueh, C.-T. Kuo, C.-C. Lai, Y.-H. Liu, H.-F. Hsu, S.-M. Peng, C.-h. Chen and S.-H. Chiu, Angew. Chem., Int. Ed., 2010, 49, 9170–9173 CrossRef CAS PubMed.
- H. Maeda, Chem.–Eur. J., 2008, 14, 11274–11282 CrossRef CAS.
- W. Edwards and D. K. Smith, Chem. Commun., 2012, 48, 2767–2769 RSC.
- A. R. Hirst and D. K. Smith, Langmuir, 2004, 20, 10851–10857 CrossRef CAS.
- A. Wynne, M. Whitefield, A. J. Dixon and S. Anderson, J. Dermatol. Treat., 2002, 13, 61–66 CrossRef CAS PubMed.
- S. Mondal, P. Bairi, S. Das and A. K. Nandi, Chem.–Eur. J., 2018, 24, 5591–5600 CrossRef CAS PubMed.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC.
- P. Xue, R. Lu, D. Li, M. Jin, C. Tan, C. Bao, Z. Wang and Y. Zhao, Langmuir, 2004, 20, 11234–11239 CrossRef CAS PubMed.
- Y.-M. Wang, V. Ulrich, G. F. Donnelly, F. Lorenzini, A. C. Marr and P. C. Marr, ACS Sustainable Chem. Eng., 2015, 3, 792–796 CrossRef CAS.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- E. R. Draper, R. Schweins, R. Akhtar, P. Groves, V. Chechik, M. A. Zwijnenburg and D. J. Adams, Chem. Mater., 2016, 28, 6336–6341 CrossRef CAS.
- J. H. Jung, Y. Ono, K. Hanabusa and S. Shinkai, J. Am. Chem. Soc., 2000, 122, 5008–5009 CrossRef CAS.
- Y. Yao, Y. Sun, H. Yu, W. Chen, H. Daia and Y. Shi, Dalton Trans., 2017, 46, 16802–16806 RSC.
- J. Lu, Y. Gao, J. Wu and Y. Ju, RSC Adv., 2013, 3, 23548–23552 RSC.
- S. Das, P. Chakraborty, A. Shit, S. Mondal and A. K. Nandi, J. Mater. Chem. A, 2016, 4, 4194–4210 RSC.
- S.-J. Ha, S. G. Lee, J.-W. Ha and J. H. Moon, Langmuir, 2016, 32, 7735–7740 CrossRef CAS.
- A. Vidyasagar, K. Handore and K. M. Sureshan, Angew. Chem., Int. Ed., 2011, 50, 8021–8024 CrossRef CAS.
- U. J. Kim, T. G. Kim, Y. Shim, Y. Park, C.-W. Lee, T.-H. Kim, H. S. Lee, D.-Y. Chung, J. Kihm, Y.-G. Roh, J. Lee, H. Son, S. Kim, J. Hur and S. W. Hwang, ACS Nano, 2015, 9, 602–611 CrossRef CAS PubMed.
- B. Roy, P. Bairia and A. K. Nandi, RSC Adv., 2014, 4, 1708–1734 RSC.
- A. Vintiloiu and J.-C. Leroux, J. Controlled Release, 2008, 125, 179–192 CrossRef CAS.
- Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavanc and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- A. Pal and J. Dey, Langmuir, 2013, 29, 2120–2127 CrossRef CAS PubMed.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- Y. Gong, X. Zhao, Z. Cai, S. E. O'Reilly, X. Hao and D. Zhao, Mar. Pollut. Bull., 2014, 79, 16–33 CrossRef CAS.
- Z. Liu, J. Liu, W. S. Gardner, G. C. Shank and N. E. Ostrom, Deep Sea Res., Part II, 2016, 129, 292–300 CrossRef CAS.
- B. Wang, W. Liang, Z. Guo and W. Liub, Chem. Soc. Rev., 2015, 44, 336–361 RSC.
- S. Venkatanarasimhan and D. Raghavachari, J. Mater. Chem. A, 2013, 1, 868–876 RSC.
- R. J. Fiocco and A. Lewis, Pure Appl. Chem., 1999, 71, 27–42 CAS.
- É. Pelletier and R. Siron, Environ. Toxicol. Chem., 1999, 18, 813–818 CrossRef.
- F. L. Motta, S. R. Stoyanov and J. B. P. Soares, Chemosphere, 2018, 194, 837–846 CrossRef CAS PubMed.
- D. Sundaravadivelu, M. T. Suidan, A. D. Venosa and P. I. Rosales, Chemosphere, 2016, 144, 1490–1497 CrossRef CAS PubMed.
- A. Tripathi, G. N. Parsons, O. J. Rojas and S. A. Khan, ACS Omega, 2017, 2, 4297–4305 CrossRef CAS.
- H.-M. Choi, Environ. Sci. Technol., 1992, 26, 772–776 CrossRef CAS.
- V. Singh, S. Jinka, K. Hake, S. Parameswaran, R. J. Kendall and S. Ramkumar, Ind. Eng. Chem. Res., 2014, 53, 11954–11961 CrossRef CAS.
-
N. Widiaksana, A. A. Yudiana and Y. S. Nugroho, Earth and Environmental Science, 2018, vol. 105 Search PubMed.
- S. Khandakar, Md. N. Islam, R. I. Rubel and Sk. S. Yusuf, Bitlis Eren Univ. J. Sci. Technol., 2017, 7, 115–122 CrossRef.
- A. T. Hoang, V. V. Pham and D. N. Nguyen, Int. J. Appl. Eng. Res., 2018, 13, 4915–4928 Search PubMed.
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185–186 RSC.
- T. Kar, S. Debnath, D. Das, A. Shome and P. Das, Langmuir, 2009, 25, 8639–8648 CrossRef CAS PubMed.
- S. Mukherjee and B. Mukhopadhyay, RSC Adv., 2012, 2, 2270–2273 RSC.
- A. Prathap and K. M. Sureshan, Angew. Chem., Int. Ed., 2017, 56, 9405–9409 CrossRef CAS PubMed.
- D. Wang, J. Niu, Z. Wang and J. Jin, Langmuir, 2015, 31, 1670–1674 CrossRef CAS PubMed.
- C. Ren, G. H. B. Ng, H. Wu, K.-H. Chan, J. Shen, C. Teh, J. Y. Ying and H. Zeng, Chem. Mater., 2016, 28, 4001–4008 CrossRef CAS.
- Y. Cui, M.-C. Li, Q. Wu, J. A. Pojman and D. G. Kuroda, ACS Appl. Mater. Interfaces, 2017, 9, 33549–33553 CrossRef CAS PubMed.
- A. Prathap and K. M. Sureshan, Chem. Commun., 2012, 48, 5250–5252 RSC.
- S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS PubMed.
- S. Mondal, P. Chakraborty, S. Das, P. Bairi and A. K. Nandi, Langmuir, 2016, 32, 5373–5382 CrossRef CAS PubMed.
- H.-K. Yang, H. Zhao, P.-R. Yang and C.-H. Huang, Colloids Surf., A, 2017, 535, 242–250 CrossRef CAS.
- K. K. Kartha, S. S. Babu, S. Srinivasan and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 4834–4841 CrossRef CAS PubMed.
- T. Xiao, X. Zhang, J. Wu, J. Yang and Y. Yang, ChemPlusChem, 2017, 82, 879–887 CrossRef CAS.
- V. Karunakaran, D. D. Prabhu and S. Das, J. Phys. Chem. C, 2013, 117, 9404 CrossRef CAS.
- A. Dawn, T. Shiraki, S. Haraguchi, H. Sato, K. Sada and S. Shinkai, Chem.–Eur. J., 2010, 16, 3676–3689 CrossRef CAS PubMed.
- C. Yu and V. W.-W. Yam, Chem. Commun., 2009, 1347–1349 RSC.
- I. O. Aparin, G. V. Proskurin, A. V. Golovin, A. V. Ustinov, A. A. Formanovsky, T. S. Zatsepin and V. A. Korshun, J. Org. Chem., 2017, 82, 10015–10024 CrossRef CAS PubMed.
- S. Farhangi and J. Duhamel, Macromolecules, 2016, 49, 353–361 CrossRef CAS.
- H.-S. Jang, J. Zhao, Y. Lei and M.-P. Nieh, ACS Appl. Mater. Interfaces, 2014, 6, 14801–14811 CrossRef CAS PubMed.
- D. A. V. Dyke, B. A. Pryor, P. G. Smith and M. R. Topp, J. Chem. Educ., 1998, 75, 615 CrossRef.
- A. A. Gupta, K. K. Swami, A. K. Misra, A. K. Bhatnagar and P. K. Mukhopadhaya, Hydrocarbons Technology, 1992, 15, 137–151 Search PubMed.
- S. Roy, Sens. Actuators, B, 1999, 55, 212–216 CrossRef CAS.
- D. Patra and A. K. Mishra, Anal. Chim. Acta, 2002, 454, 209–215 CrossRef CAS.
- R. Gotor, C. Tiebe, J. Schlischka, J. Bell and K. Rurack, Energy Fuels, 2017, 31, 11594–11600 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of OG-1, OG-2 and OG-3 with their NMR and mass spectra, gelation results of OG-1, OG-2 and OG-3 in different organic solvents and oils, rheological data of OG-2 and OG-3 gels, UV-visible and fluorescence spectra of OG-3 gel, gelation results of OG-1, OG-2 and OG-3 in different oils. Rheological data of OG-1 gel (1.2% w/v) in kerosene–water mixture in absence and presence of different salts, acid and base, FTIR spectra of their xerogels, MALDI-TOF mass and NMR data of extracted OG-1 from the scooped gel in diesel oil. This information is available free of charge via the internet. See DOI: 10.1039/c8ta09732a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*