
Heterogeneous degradation of carbamazepine by Prussian blue analogues in the interlayers of layered double hydroxides: performance, mechanism and toxicity evaluation†
Hanxuan
Zeng
a,
Lin
Deng
 *ab,
Zhou
Shi
a,
Jinming
Luo
b and
John
Crittenden
b
*ab,
Zhou
Shi
a,
Jinming
Luo
b and
John
Crittenden
b
aKey Laboratory of Building Safety and Energy Efficiency, Ministry of Education, Department of Water Engineering and Science, College of Civil Engineering, Hunan University, Changsha, Hunan 410082, PR China. E-mail: lindeng@hnu.edu.cn
bBrook Byers Institute for Sustainable Systems, School of Civil and Environmental Engineering, Georgia Institute of Technology, 828 West Peachtree Street, Atlanta, Georgia 30332, USA
First published on 26th November 2018
Abstract
Prussian blue analogues (PBA) are efficient catalysts for peroxymonosulfate (PMS) activation. However, the longevity and recovery ability of PBA are very poor, which limits their practical applications. In this study, for the first time, we successfully incorporated a nanosheet-like PBA into the interlayers of layered double hydroxides (LDH) (PBA-LDH). The morphology and physicochemical characteristics of the as-synthesized PBA-LDH were well characterized using SEM, TEM, XRD, FTIR and XPS. PMS activation via PBA-LDH toward carbamazepine (CBZ) degradation was systematically examined. The activation of PMS via PBA alone could only achieve a maximum of 31.8% degradation efficiency for CBZ within 15 min. However, complete degradation of CBZ was achieved via a PBA-LDH-activated PMS system within a wide pH range of 3.0 to 9.0. ESR measurements and radical scavenging experimental results revealed that SO4˙− is the most active species in CBZ degradation. XPS analysis confirmed a reversible valence equilibrium between the metal ions and the recovery of surface-adsorbed oxygen, ensuring stable catalytic activity. Notably, the CBZ degradation in the PBA-LDH activated system remained as high as 95.7% after six consecutive runs. Meanwhile, only 8.1 μg L−1 cobalt ion and 5.4 μg L−1 iron ion were leached from PBA-LDH after the degradation reaction; thus, it is non-toxic to aqueous environments. In addition, a possible pathway for CBZ degradation was proposed, and an eco-toxicity assessment of the oxidation intermediates was also performed.
1. Introduction
Over the past few decades, the presence of pharmaceutical compounds in aquatic ecosystems has become one of the most important environmental challenges.1 As a widely used pharmaceutical for treatment of epilepsy, bipolar disorder and trigeminal neuralgia, carbamazepine (CBZ) is ubiquitously detected in wastewater treatment plant effluents, surface water, groundwater, and drinking water.2 It is believed that CBZ has detrimental effects on living beings, including bacteria, invertebrates, algae and fish.3 CBZ has an olefinic double bond on its central heterocyclic ring; thus, it is difficult to degrade through biodegradation or direct photodegradation.4,5 As mentioned above, serious negative environmental impacts may occur due to the low biodegradability of CBZ.4 Hence, it is necessary to propose efficient treatment technology for the degradation of CBZ in aqueous solution.Advanced oxidation processes (AOPs) are drawing increasing attention to remove various organic pollutants in view of their short treatment times and high removal efficiency. To date, hydroxyl radical (˙OH) and sulfate radical (SO4˙−)-based AOPs have been the most commonly employed processes in the degradation of organic pollutants. However, compared to hydroxyl radical (˙OH)-based AOPs, sulfate radical (SO4˙−)-based AOPs are being increasingly investigated due to their higher oxidative capabilities, longer half-life periods, and greater adaptive capabilities in a wide pH range from 2 to 8.6–8 It is generally accepted that SO4˙− radicals can be produced from decomposition that is activated by different techniques.9–12 Peroxymonosulfate (PMS) is an easily obtained and environmentally friendly strong oxidant.13,14 Also, PMS, with its asymmetric structure (HO–O–SO3−) and a longer superoxide (O–O) bond, is believed to be more readily dissociated by catalysts to produce SO4˙− than peroxydisulfate (PDS),15 which has been widely applied in sulfate radical (SO4˙−)-based AOPs.
Many previous studies have demonstrated that Co2+ ions are the most efficient homogeneous catalysts for PMS activation in the degradation of organic pollutants.12,16–18 However, due to the toxicity of discharged cobalt ions in aqueous solution, researchers are shifting their foci to heterogeneous PMS activators. Cobalt oxides (e.g., Co3O4) are the most common heterogeneous catalysts; they exhibit high activity towards PMS activation. However, these cobalt-based materials tend to aggregate, which diminishes the catalytic activity of cobalt.19 Moreover, cobalt leakage is still a concern.20,21 Hence, overcoming these deficiencies of cobalt-based materials and exploring novel cobalt-based activators of PMS is highly significant and necessary.
Bimetallic oxides have recently become an active research topic due to their excellent catalytic properties.22–24 As a class of crystalline metal–organic frameworks (MOFs), Prussian blue analogues (PBA) with the formula MII3[MIII(CN)6]2·nH2O (MII = Fe, Ni, Co, Mn, Zn and Cu, MIII = Fe, Co) can be constructed from metal ions bridged by cyanide ligands. Also, PBA possess evident advantages compared to other nanostructure materials, including (i) a unique coordination structure, (ii) different valence states of metal species, and (iii) vacancies of balancing charges. Thus, PBA are intriguing catalysts for many applications, including wet chemical oxidation,25,26 electro-oxidation,27 electro-reduction,28 decomposition and reduction of hydrogen peroxide.29 Because PBA are generally synthesized from reactions of metal ions with hexacyanometalates in solution, the resulting PBA are typically nanoscale particles.30 Even though these nanoscale PBA exhibit high specific surface areas, they are relatively difficult to use due to their poor recovery properties from aqueous solutions.31 Therefore, there is still an urgent demand for developing substrate-supported PBA which are catalytically effective, convenient to prepare, simple to use and easy to recover, especially for aqueous catalytic reactions.
Layered double hydroxides (LDHs) are important two-dimensional materials consisting of positively charged host layers and counter-anions in the interlayer space.32,33 The interactions between the metal hydroxide layers are much weaker than the intralayer bonding, leading to swelling of the unit cell c-parameter (perpendicular to the layers) with adsorption of energetically favorable species. LDH interlayers thus serve as excellent hosts for a broad range of organic and inorganic charge-balancing anions and a variety of neutral chemical species; therefore, these materials are useful in applications including environmental remediation, catalysis, electrochemistry, medicine, and molecular separation.34–36
In this study, for the first time, we intercalate the [Fe(CN)6]3− anion into the interlayers of LDH using an anion exchange method. Due to the intercalation of [Fe(CN)6]3− anion, the LDH becomes available for the construction of a PBA in the interlayer space. The incorporation of a PBA in a confined structure is expected to improve the catalytic performance of the catalyst. To our best knowledge, no studies have been performed to in situ grow PBA nanoparticles in LDH interlayers or to investigate the catalytic properties of as-prepared PBA-LDH. In this study, the physical and chemical properties of PBA-LDH were thoroughly investigated. Furthermore, CBZ degradation under various conditions via PMS activation was comprehensively determined by batch-type experiments. Moreover, a possible degradation mechanism of the PBA-LDH activated PMS system was determined. In addition, the toxicities of the oxidation intermediates were estimated using the USEPA Estimation Program Interface (EPI) Suite.
2. Experimental
2.1. Preparation of PBA-LDH
PBA was prepared through a coprecipitation method according to the literature with some modifications.37 Specially, 0.03 mol of cobalt chloride hexahydrate (CoCl2·6H2O) and 0.045 mol of trisodium citrate dihydrate (Na3C6H5O7·2H2O) were dissolved in deionized water (100 mL) and then mixed with potassium ferricyanide (K3[Fe(CN)6], 0.02 mol) dissolved in 100 mL of deionized water. After continuous stirring for 5 minutes, the obtained mixture was maintained at room temperature and aged for 20 h. The precipitates were collected, washed several times with deionized water and ethanol, and dried for further use.MgAl-LDH was synthesized according to our previous work.38 The synthesis schematic of PBA-LDH is depicted in Fig. S1.† Typically, 1 g of MgAl-LDH was first thoroughly dispersed in 100 mL of K3[Fe(CN)6] (0.02 mol) with continuous stirring for 24 h. After that, the solid was collected and washed several times with deionized water to remove any residual reactants. The obtained product was then mixed with 0.03 mol of CoCl2·6H2O and 0.045 mol of Na3C6H5O7·2H2O and magnetically agitated for 20 h. The resultants were collected, washed several times with deionized water, and dried at 60 °C overnight.
2.2. Catalyst characterization
The morphologies and microstructures of the prepared samples were characterized by scanning electron microscopy (SEM, Hitachi s-4800, Japan) and transmission electron microscopy (TEM, FEI TECNAI G2 F30, USA) with 300 kV acceleration voltage. X-ray diffraction (XRD) of the materials was carried out at room temperature using a D8 Advance X-ray diffractometer employing Ni-filtered Cu Kα radiation at a scan rate (2θ) of 0.02° s−1. The FT-IR spectra were recorded in the wavenumber range of 4000 to 400 cm−1 by a Nicolet 6700 spectrometer (Thermo, USA) using KBr pellets. The chemical compositions and states of the samples were determined using X-ray photoelectron spectroscopy (XPS) using an ESCALAB 250 apparatus (Thermo Fisher Scientific) at 3 × 10−10 mbar using Al Kα X-ray beams (1486.6 eV). Radicals in the filtrate of the reaction solution were identified by ESR spectroscopy (Bruker model ESP300E, Germany) operated at a 3514 G center field and a 9.85 GHz microwave frequency. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was used as the spin trap compound in the ESR analysis.2.3. Experimental procedures
PMS activation by PBA-LDH was evaluated using batch-type experiments of CBZ degradation at different pH values (3, 5, 7 and 9), PMS doses (0.25, 0.5, 1.0 and 2.0 mM) and catalyst doses (0.1, 0.2 and 0.4 g L−1). Typically, before the addition of PMS, 10 mg catalyst was dispersed in 50 mL CBZ solution (20 mg L−1), which was stirred for 30 min to achieve adsorption equilibrium. Then, the degradation reaction started after the desired amount of PMS was added to the above suspension. At a given time interval, a water sample was withdrawn and poured into a centrifuge tube with a few drops of Na2S2O3 as a reaction quenching agent. Then, the solution was filtered through a 0.45 μm membrane filter and analyzed. To determine the effects of radical inhibitors, methanol (MeOH) or tert-butyl alcohol (TBA) was first added to the CBZ solution; then, PMS and catalyst were added to the solution. The recyclability test was carried out by collecting the used catalysts via centrifugation and then adding them to another batch experiment containing fresh CBZ. All the experiments were conducted at least twice. The relative standard deviations (RSD) for the different batches were usually less than 5%.2.4. Analytical methods
The concentrations of carbamazepine, oxcarbazepine, atrazine, bisphenol A, and tetracycline were analyzed via a high-performance liquid chromatograph (HPLC, Agilent 1260, USA) equipped with a XDB-C18 column (150 × 4.6 mm, i.d., 5 μm particle, Waters, USA) and a VWD detector. Detailed information regarding the HPLC analyses is listed in Table S1.† The concentrations of metal ions leached from the catalyst into the solution were determined using an inductively coupled plasma mass spectrometer (ICP-MS, Agilent 7700, USA). The transformation products of CBZ degradation were identified by high performance liquid chromatography-electrospray ionization-tandem mass spectrometry (HPLC-ESI-MS/MS) in positive ionization mode using TSQ Quantum Access MAX equipment (Thermo, USA) equipped with a reverse-phase C18 analytical column (100 × 2.1 mm i.d., 5 μm particle, Thermo, USA). The detailed test conditions are given in the ESI.†3. Results and discussion
3.1. Catalyst characterization
The morphologies of the samples were characterized by SEM and TEM. As shown in Fig. 1a, the PBA microcubes are highly uniform, with an average size of ca. 400 to 500 nm. This observation was also confirmed by the TEM image (Fig. 1b). The SEM image of PBA-LDH exhibits sheet-like shapes with rough surfaces as well as granular aggregates (Fig. 1c). To examine the morphology of the PBA formed in the interlayers of LDH, delamination of the layered double hydroxides was carried out using a solvent mixture of glycine/formamide according to the method reported by T. Hibino and W. Jones.39 TEM and corresponding EDS elemental mappings were performed to study the morphologies and chemical compositions of the exfoliated products. Surprisingly, unlike the reported regular cubic morphology of PBAs, the PBA in this study exhibits a nanosheet-like structure, as shown in Fig. 1d. Moreover, the EDS elemental mapping (Fig. 1e) clearly manifests the presence of PBA particles and the homogeneous distribution of the existing elements (Co, Fe, N and C). The elemental composition of the as-prepared PBA is consistent with those of reported PBA. The formation of the nanosheet structure of the PBA was attributed to the electrostatic forces existing between the very confined regions of the LDH layers, which restricted the morphology and particle size growth of the PBA.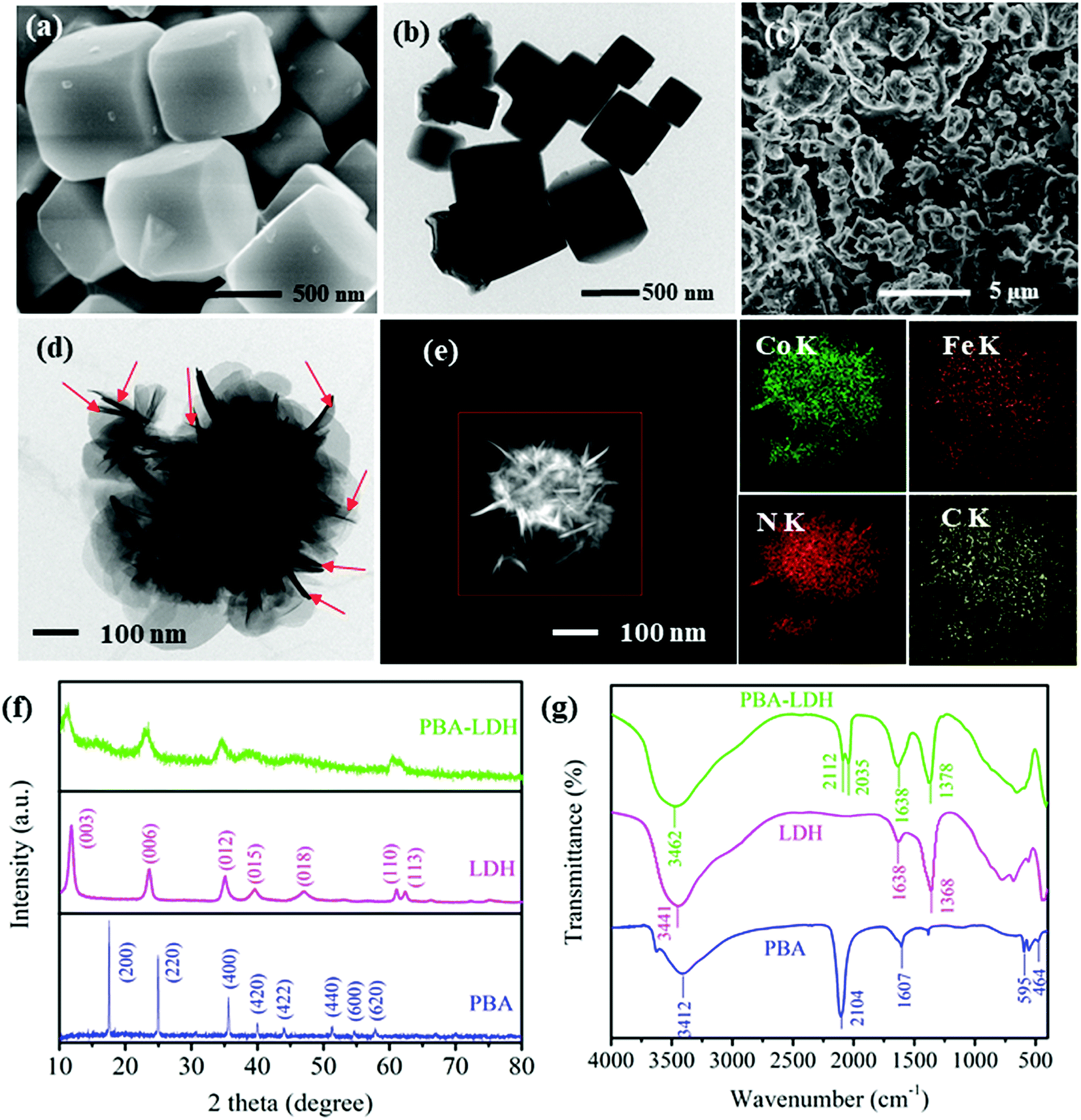 | ||
| Fig. 1 (a) SEM image and (b) TEM images of the PBA, (c) SEM image of PBA-LDH, (d) TEM images and (e) corresponding EDS elemental mappings (Co, Fe, N, and C) of the PBA in the interlayers of LDH, (f) XRD diffraction patterns and (g) FTIR spectra of the PBA, LDH and PBA-LDH. | ||
The crystal structures of both the individual (i.e., LDH and PBA) and grafted (i.e., LDH-PBA) materials were analyzed by XRD. Fig. 1g displays the XRD patterns of the PBA, LDH and PBA-LDH over the 2θ range of ∼10 to 80°. The diffraction peaks of the PBA at 17.5°, 24.9°, 35.6°, 40.0°, 44.0°, 51.3°, 54.7° and 57.9° correspond to (200), (220), (400), (420), (422), (440), (600) and (620), respectively, which is in good agreement with the standard values for face-centered-cubic lattice Prussian blue (JCPDS 73-0687).40 No additional peaks from impurities were detected, indicating the high purity of the product. The XRD pattern of LDH shows characteristic diffraction peaks of hydrotalcite at (003), (006), (012), (015), (018), (110), and (113) (JCPDS 48-0593).41 The XRD pattern of PBA-LDH displays the characteristic peaks of LDH, and no diffraction peaks of the PBA were observed; this is due to the low intensity of the XRD pattern of the PBA, as shown in Fig. S2.† The extent of penetration of the PBA into the layers is unknown. It is also possible that the PBA is sticking out from the interlayers and connecting with the PBA in other layers to form particles covering the edges of the LDH crystallites.42 Moreover, the d spacing value (d003) of pristine LDH was calculated to be 0.71 nm from the 2θ value of 11.64° using Bragg's equation (i.e., d = λ/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ).43 Particularly, the d003 spacing value of PBA-LDH increased from 0.71 to 0.82 nm. The increased d003 spacing provides additional evidence of the intercalation of the PBA in the interlayers of LDH.44
sinθ).43 Particularly, the d003 spacing value of PBA-LDH increased from 0.71 to 0.82 nm. The increased d003 spacing provides additional evidence of the intercalation of the PBA in the interlayers of LDH.44
FTIR spectra of the PBA, LDH and PBA-LDH were also acquired (Fig. 1h). As we can see, the sharp peak at 2104 cm−1 can be assigned to the characteristic absorption peak of Fe2+–CN–Co2+ in the PBA. The OH stretching vibration and bending vibration of water molecules can be observed in the peaks around 3412 and 1607 cm−1, respectively. The peaks at 595 and 464 cm−1 are related to the Fe–CN and Co–CN flexural vibrations, respectively.45 The FTIR spectrum of LDH shows three typical absorption peaks. The absorption peaks at around 3441 and 1638 cm−1 are ν(OH) stretching models, representing an interlayer of water in the LDH layers and physically adsorbed water. The peak at 1368 cm−1 is carbonate (CO32−) in the interlayer of LDH, and the peaks in the 400 to 800 cm−1 region can be ascribed to the M–O and O–M–O (M = Mg, Al) vibrations.32 The FTIR spectrum of PBA-LDH confirms the presence of the O–H (3462 and 1638 cm−1) and CO32− (1378 cm−1) functional groups of LDH. Also, the appearance of two new bands at 2035 and 2112 cm−1 are related to ν(CN), which is derived from [Fe(CN)6]3− intercalated into the interlayers of LDH. This observation is consistent with previously reported ferricyanide-intercalated LDH.46 In addition, the CO32− groups detected at 1378 cm−1 are related to the CO2 pollution in the air or the incomplete ion exchange between CO32− by [Fe(CN)6]3− during the catalyst preparation.
Based on these results, it can be concluded that PBA particles with a nanosheet morphology were successfully intercalated into the interlayers of LDH.
3.2. Catalytic performance evaluation of the as-synthesized samples
The degradation of CBZ in different systems under the same experimental conditions is displayed in Fig. 2a. The concentration of CBZ follows these trends for 15 min of reaction time: (1) when PMS alone is present in solution, very little degradation of CBZ is observed (this can be attributed to the low oxidative power of PMS (E0 = +1.82 V per SHE)). (2) In the absence of PMS, PBA-LDH removes a small amount of CBZ (less than 2%), which is primarily attributed to surface adsorption. (3) 31.8% of CBZ is degraded in the PBA-activated PMS system. (4) 20 mg L−1 of CBZ solution is completely degraded in 15 min by the PBA-LDH activated PMS system. The incorporation of PBA in the interlayers of LDH is expected to improve the catalytic performance of the catalyst. The results demonstrate the superior catalytic performance of PBA-LDH compared to PBA. This phenomenon was also reported in previous literature, in which the oxidation of L-cysteine in L-cysteine-intercalated LDH by Br2 was higher than that in the bulk reaction between L-cysteine and Br2.47 Moreover, as illustrated in Fig. S3,† PBA-LDH can be more readily separated and recovered from solutions than PBA.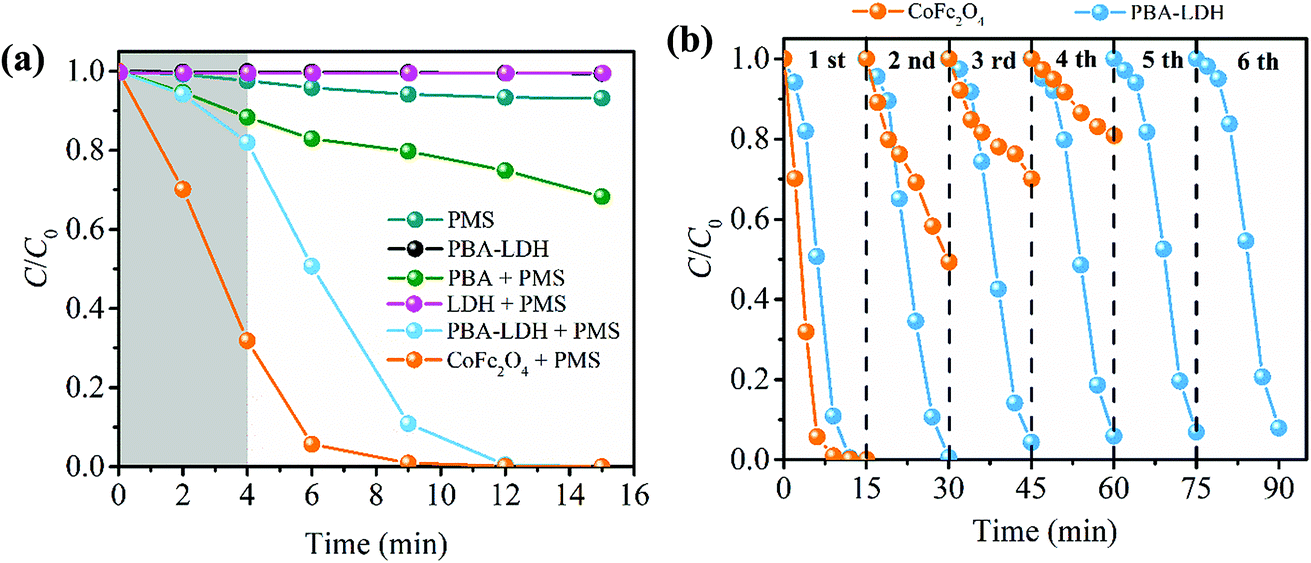 | ||
| Fig. 2 (a) Comparison of CBZ removal in different systems; (b) recyclability of CoFe2O4 and PBA-LDH for activating PMS for CBZ degradation (catalyst dose 0.2 g L−1, PMS dose 0.5 mM, CBZ concentration 20 mg L−1). | ||
Spinel ferrites have been shown to be efficient catalysts for PMS activation for organic pollutant degradation.16,22,48 Thus, in this study, the degradation of CBZ by CoFe2O4-activated PMS was also carried out for comparison. It is interesting to note that the CBZ degradation curve of the PBA-LDH-activated PMS system is different from that of the CoFe2O4-activated PMS system (Fig. 2a). In the PBA-LDH-activated PMS system, the degradation is relatively slow at the initiation phase, and only about 18.1% of CBZ is degraded after 4 minutes of reaction time. Similar trends of CBZ degradation are also shown in Fig. 3 and 4b. Meanwhile, in the CoFe2O4 activated PMS system, almost 70% of the CBZ in solution can be removed after 4 min; this degradation efficiency trend is consistent with that in PMS systems activated by reported heterogeneous catalysts (i.e., mesoporous MnxCo3−xO4 nanocages,2 cobalt-based nanosheets,7 nano-Co3O4,21 Co2SnO4 nanoparticles,23 NixFe3−xO4,49 CoFe2O4-GO composite,50 and LaCo1−xCuxO3 perovskites51). On the other hand, as shown in Fig. S4,† the experimental data for CBZ degradation in both the PBA and CoFe2O4-activated PMS systems are described by a pseudo-first-order kinetic model, with R2 = 0.993 and 0.987, respectively (ln(C/C0) = −kobst, where C0 and C are the CBZ concentrations at reaction times of t = 0 and t, respectively, and kobs is the observed pseudo-first-order rate constant).49 However, the experimental data for CBZ degradation in the PBA-LDH-activated PMS system fail to fit with the pseudo-first-order kinetic model, and the R2 value is only 0.826.
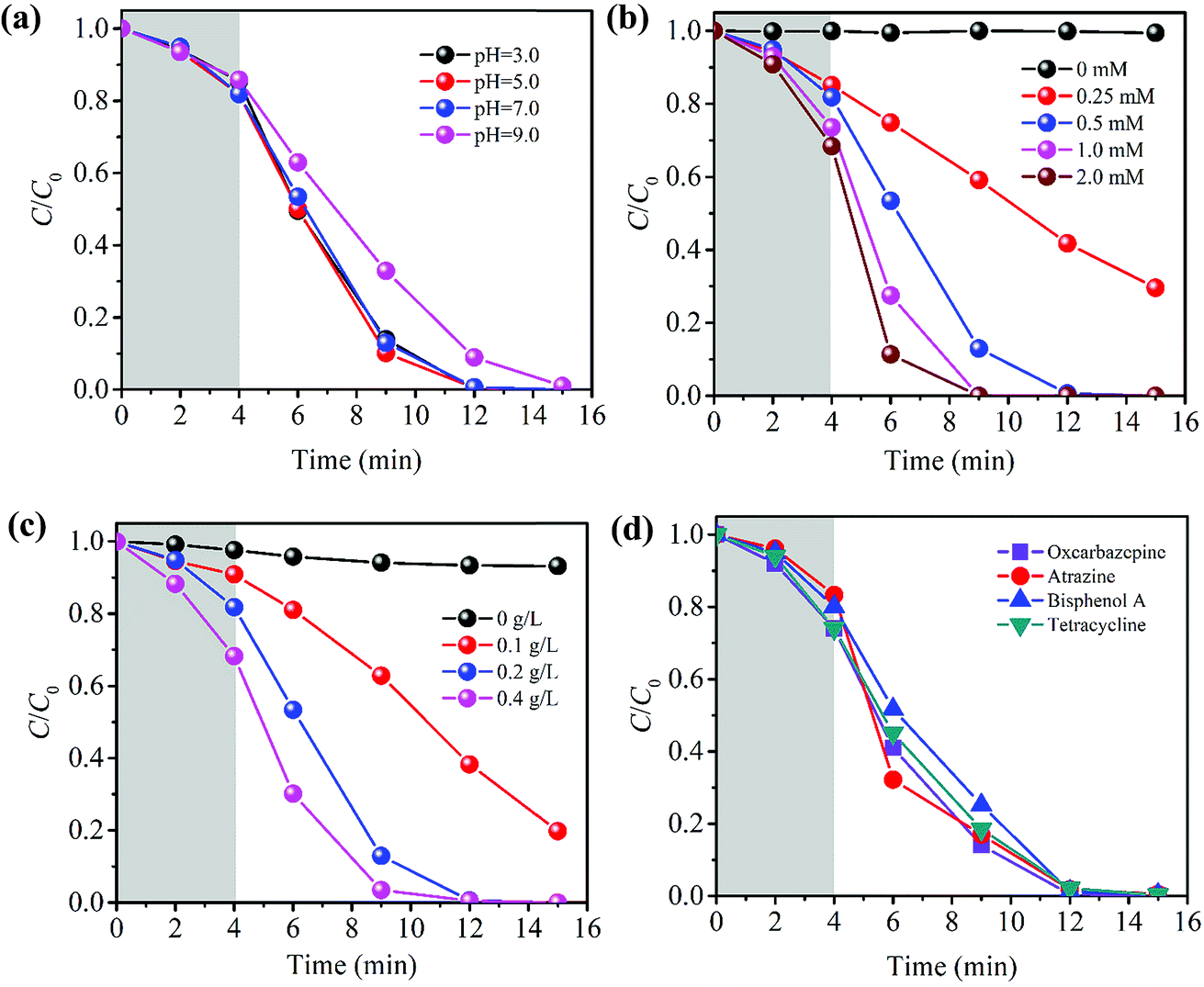 | ||
| Fig. 3 PBA-LDH-activated PMS system for CBZ removal: (a) effects of solution pH (catalyst dose 0.2 g L−1, PMS dose 0.5 mM, CBZ concentration 20 mg L−1), (b) effects of PMS dose (catalyst dose 0.2 g L−1, CBZ concentration 20 mg L−1, pH 7.0), and (c) effects of catalyst dose (PMS dose 0.5 mM, CBZ concentration 20 mg L−1, pH 7.0); (d) PBA-LDH-activated PMS for removal of different pollutants (catalyst dose 0.2 g L−1, PMS dose 0.5 mM, pollutant concentration 20 mg L−1). | ||
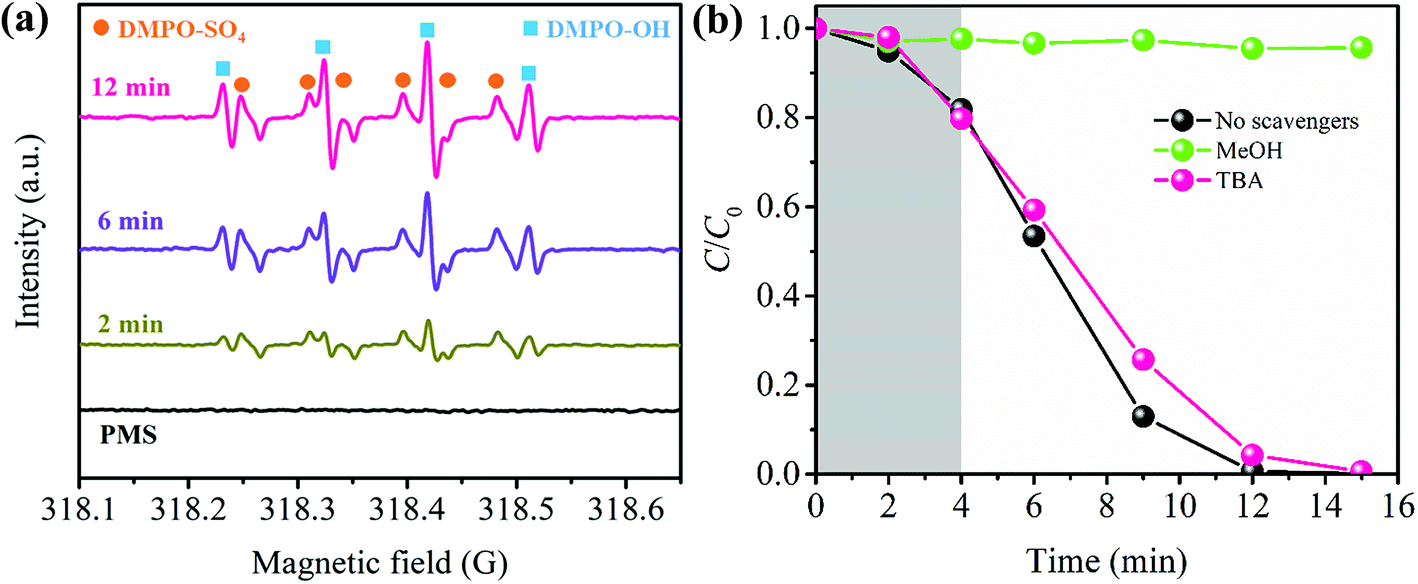 | ||
| Fig. 4 (a) ESR spectra using DMPO as a spin-trapping agent at different time intervals. (b) Effects of MeOH and TBA on CBZ degradation (PBA-LDH dose 0.2 g L−1, PMS dose 0.5 mM, CBZ concentration 20 mg L−1, pH 7.0, DMPO dose 100 mM). | ||
Based on the catalyst characterization, the as-synthesized PBA-LDH is a typical layered double hydroxide. In previous work on heterogeneous catalyst-activated PMS systems, HSO5− molecules in solution could reach and react with active sites of the catalyst surface as soon as the catalyst was added to the solution, resulting in generation of reactive radicals and rapid initial destruction of organic pollutants. However, in this study, HSO5− must enter the interlayers of PBA-LDH to react with the active sites there, and this process is limited by mass transfer. Also, when the HSO5− concentration in the interlayer is high, sharp degradation can be observed due to the production of a large quantity of radicals. This was also confirmed by the PMS concentration profile shown in Fig. S5.† The PMS concentration in solution declines slowly in the initial phase and then decreases rapidly in the following stage, resulting in rapid CBZ removal.
3.3. Reusability of PBA-LDH for CBZ degradation
Long-term reusability is an important aspect of the application of a catalyst. The stability of the as-prepared catalysts was evaluated by testing the degradation efficiency of CBZ for many catalytic cycles. Thus, a comparison study of multi-cyclic tests using CoFe2O4 and PBA-LDH to activate PMS for CBZ degradation was conducted, as illustrated in Fig. 2b. For CoFe2O4, complete removal of CBZ was achieved in the first cycle. However, as the catalytic cycle continued, a dramatic decrease in degradation efficiency occurred; the CBZ degradation decreased to 19.3% in the fourth cycle under identical conditions. In contrast, the degradation performance of PBA-LDH shows only a small change after six cycles, and the degradation efficiency of CBZ remained as high as 95.7% after 15 minutes of reaction time during the 6th cycle. Accordingly, the as-prepared PBA-LDH has extremely stable degradation performance and excellent repeatability. Thus, our work provides a template for developing high performance interlayer catalysts.We also monitored the leaching of metal ions from the CoFe2O4 and PBA-LDH catalysts. As shown in Fig. S6,† after 15 min of reaction, the leaching concentrations of Co and Fe for the CoFe2O4-activated PMS system were 1197.2 and 121.7 μg L−1, respectively. Large quantities of released cobalt ions may be toxic and carcinogenic, and they must be removed to avoid impacts on human health and the environment.50 We found that the amounts of leached metal ions from the as-synthesized PBA-LDH were very small. Only 8.1 μg L−1 cobalt ion and 5.4 μg L−1 iron ion were found after 15 minutes of reaction time. This is probably due to the strong intralayer bonding forces between the LDH layers and the interlayer species, which eliminates Co and Fe leaching.35 In addition, the XRD patterns of PBA-LDH before and after the catalytic reaction are shown in Fig. S7.† No obvious difference is observed from the XRD patterns, implying that the as-synthesized catalyst is structurally stable.
3.4. Effects of various solution conditions on CBZ degradation
3.5. Activation mechanism of PMS over PBA-LDH
In order to elucidate the reaction mechanism in depth, the dominant radical that contributes to CBZ degradation was identified by free radical trapping experiments. In this study, TBA and MeOH were utilized as hydroxyl radical and sulfate radical scavengers, respectively.58 As can be seen in Fig. 4b, the addition of 500 mM TBA shows little influence on the CBZ removal, while the degradation efficiency of CBZ decreases from 99.99% to 4.32% in the presence of 500 mM MeOH. According to these results, it can be concluded that CBZ degradation by the PBA-LDH-activated PMS system is primarily due to SO4˙− and, to a lesser extent, ˙OH.
:Fe ratios of the catalyst before and after reaction are the same as the as-produced ratios of Co and Fe precursors (Table S2†). This proves the excellent structural stability of the catalyst.
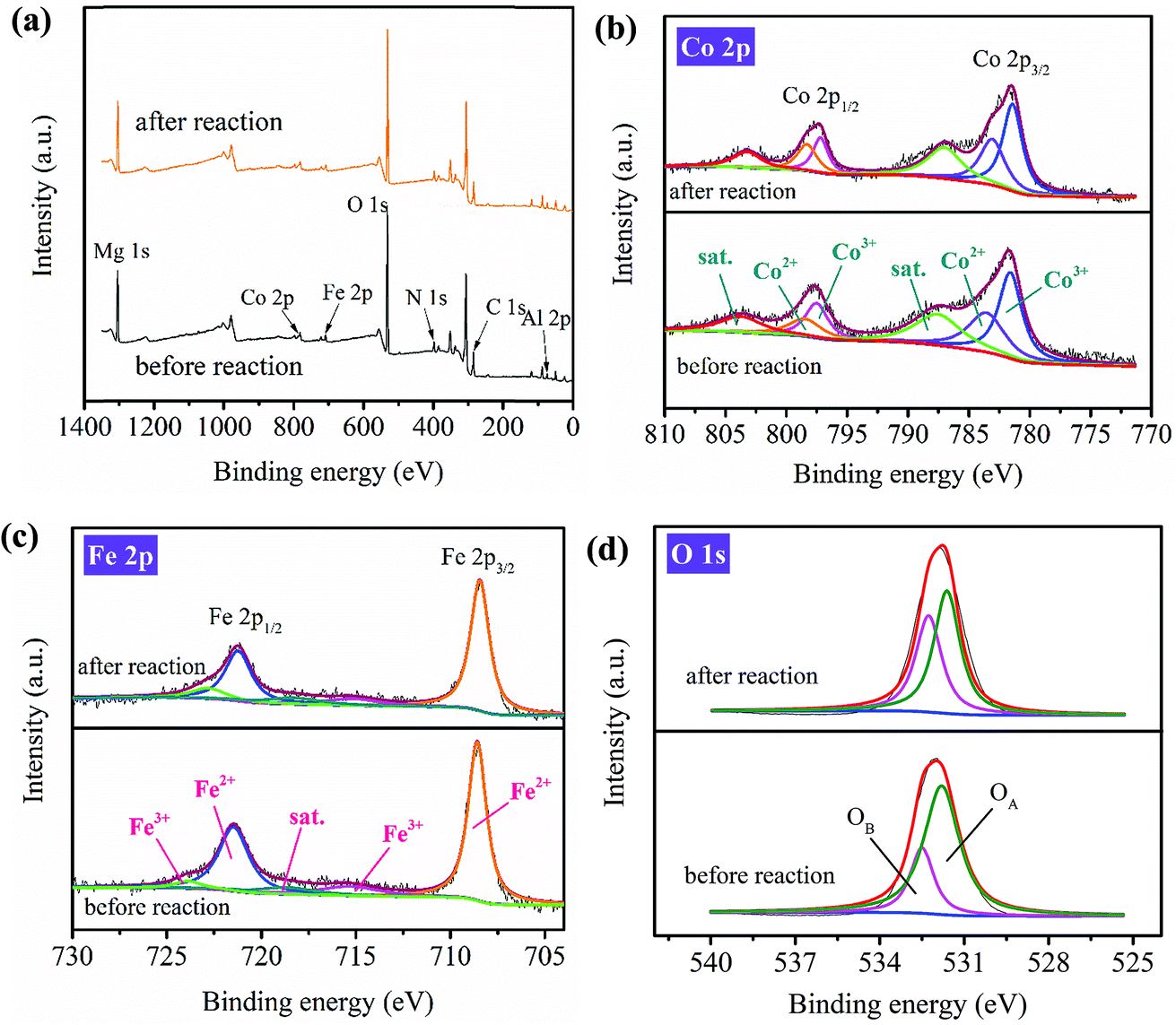 | ||
| Fig. 5 XPS spectra of PBA-LDH before and after reaction: (a) survey, (b) Co 2p, (c) Fe 2p and (d) O 1s. | ||
The high-resolution XPS spectra of Mg 1s and Al 2p before and after the reaction are identical (Fig. S10†); this suggests that Mg and Al in the catalyst do not participate in the activation process. For Co 2p (Fig. 5b), two major Co 2p1/2 peaks at 797.5 and 798.5 eV, with relative ratios to the overall Co intensity of 57% and 43% before the catalytic reaction (Table S3†), are assigned to Co3+ and Co2+, respectively; meanwhile, the relative ratios of the two peaks after the reaction changed to 50% and 50%, respectively. The increased relative ratio of Co2+ is due to the reduction of Co3+ to Co2+.51 For the Fe 2p3/2 spectrum of the fresh PBA-LDH (Fig. 5c), the relative ratios to the overall Fe intensity are about 89% for Fe2+ (at 708.6 eV) and 11% for Fe3+ (at 715.3 eV). After the reaction, the relative ratios of these two peaks became 80% and 20%. In addition, two major Fe 2p1/2 peaks were detected at 721.5 (Fe2+) and 723.9 eV (Fe3+), with relative ratios to the overall Fe intensity of 92% and 8%, respectively; meanwhile, the relative ratios of the peaks after the catalytic reaction changed to 79% and 21%, respectively. The higher ratio of Fe3+ is due to the oxidation of Fe2+ to Fe3+. The high-resolution XPS spectra of Co 2p and Fe 2p before and after the reaction indicate that both the Co3+/Co2+ and Fe2+/Fe3+ redox pairs are involved in the PMS activation.
The high-resolution O 1s spectra of PBA-LDH before and after the catalytic reaction are compared in Fig. 5d. The spectra can be resolved into two individual peaks located at 531.8 and 532.5 eV, corresponding to the surface lattice oxygen of the metal oxides (denoted as OA in the figure) and adsorbed oxygen or surface hydroxyl species (denoted as OB).59 The relative ratio of OB increases from 25% to 44%, which is ascribed to the formation of M–OH groups or O2 adsorbed on the catalyst. The decrease of lattice oxygen (from 75% to 56%) results from its oxidation to O2 coupled with the reduction of Co3+/Fe3+ to Co2+/Fe2+. This implies that O2− definitely participates in redox reactions with cobalt and iron species.51
According to the above discussion, the activation mechanism of PMS on PBA-LDH was proposed and is shown in Fig. 6. First, Co2+ activates the adsorbed HSO5− species to produce SO4˙− (eqn (1)). Also, ˙OH can also be generated viaeqn (2). Secondly, in order to maintain the charge balance on the catalyst surface, the Fe3+ in the catalyst accepts electrons from the lattice oxygen and is reduced to Fe2+ (eqn (3)). The lattice oxygen can be transformed to oxygen through eqn (4); this completes the cycle of O2−/O2 and improves the catalytic efficiency. Moreover, lattice oxygen is released in the form of 1O2 through the transfer of electrons from O2− to Co3+ and Fe2+ (eqn (5) and (6)), which was also reported for a LaCo1−xCuxO3 perovskite-activated PMS system for phenol degradation.51,60 In addition, because the standard reduction potential of Co3+/Co2+ (+1.82 V) is higher than that of Fe3+/Fe2+ (+0.77 V), the reduction of Co3+ to Co2+ by Fe2+ is thermodynamically favorable (eqn (7)).
| Co2+ + HSO5− → Co3+ + SO4˙− + OH− | (1) |
| SO4˙− + OH− → SO42− + ˙OH | (2) |
| Fe3+ + HSO5− → Fe2+ + SO5˙− + H+ | (3) |
| SO5˙− + O2− → SO4˙− + O2 | (4) |
| Co3+ + HSO5− → Co2+ + SO5˙− + H+ | (5) |
| Fe2+ + HSO5− → Fe3+ + SO4˙− + OH− | (6) |
| Co3+ + Fe2+ → Co2+ + Fe3+ | (7) |
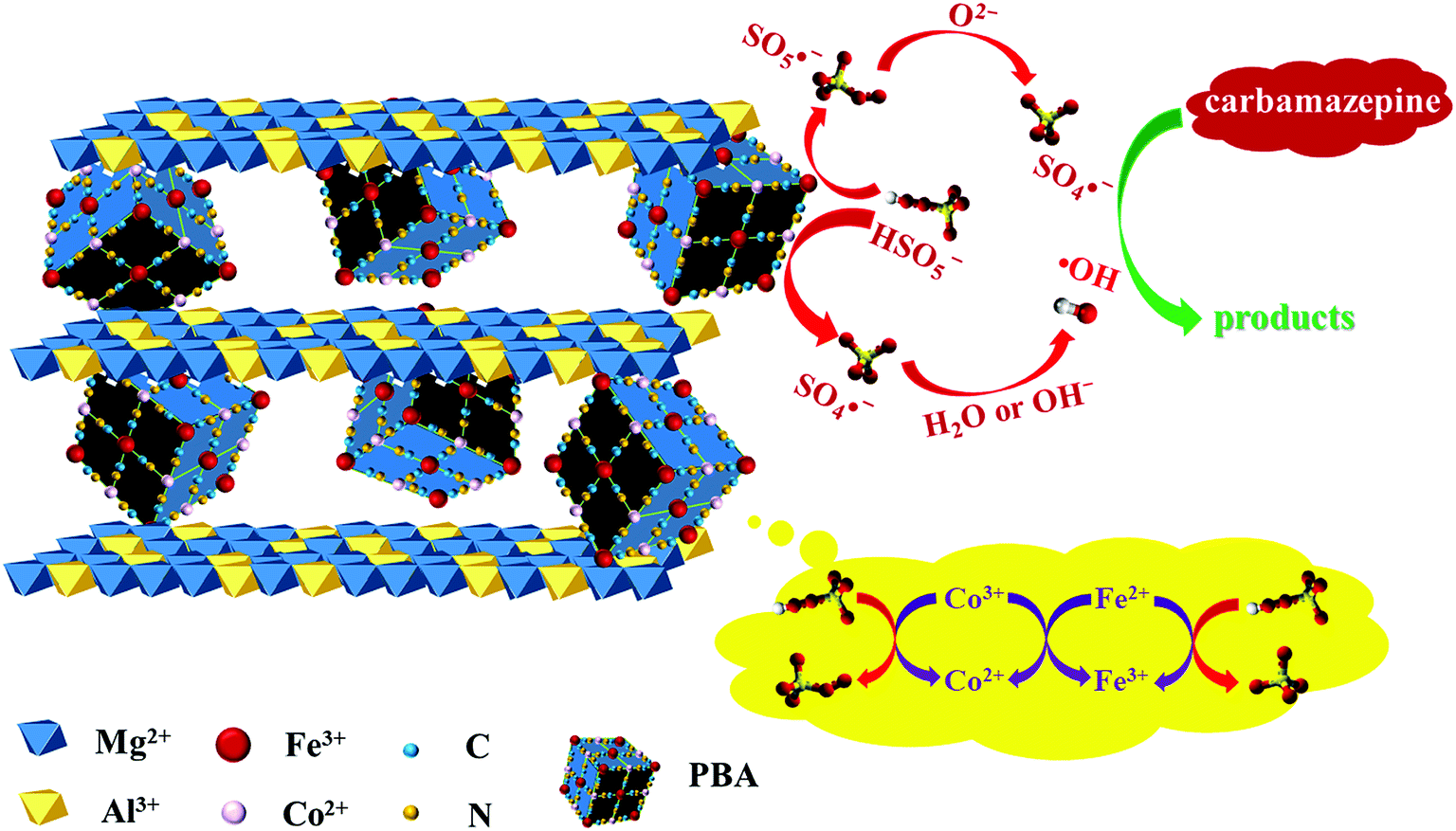 | ||
| Fig. 6 Proposed mechanism of CBZ degradation by PBA-LDH-activated PMS. | ||
3.6. Proposed degradation pathway
LC-MS/MS was employed to monitor the degradation of CBZ in the PBA-LDH-activated PMS system, and nine intermediates were detected (Table S4†). The proposed CBZ degradation pathway is illustrated in Fig. S11.† Although SO4˙− radical was determined to be the dominant active species through the radical quenching experiments and ESR analysis, no stable sulfate derivatives were detected after the attack of SO4˙−; this is due to the rapid substitution and hydrolysis of hydroxylated derivatives.Hydroxylation preferentially occurs at positions with high Frontier electron densities, indicating that the olefinic double bond on the central heterocyclic ring of CBZ is vulnerable to attack by SO4˙−. The primary identified intermediate A (m/z = 252.9000) detected in the AOPs was first reported by Doll et al.61 After this, intermediate A can be further oxidized in three different ways. Firstly, A undergoes a ring-contraction reaction to produce intermediate B (m/z = 252.9000).62 Secondly, intermediate D (m/z = 210.2000) can be formed from intermediate A due to attack by radical species.63 Finally, A can be further oxidized to C (m/z = 251.1000); C is clearly detectable in numerous wastewater samples and is considered to be an emerging pollutant.64 In an alternative pathway, CBZ hydrolyzes to yield E (iminostilbene, m/z = 193.1000), which may subsequently undergo oxidation to product F (m/z = 210.2000). Intermediates F and D then undergo hydrogen abstraction and decarboxylation to produce G (m/z = 180.1000), followed by ring cleavage to produce H (m/z = 99.0000) and I (m/z = 137.0000) by the oxidation of hydroxyl radicals or sulfate radicals.
3.7. Eco-toxicity assessment of CBZ and its oxidation intermediates
The ECOSAR program is commonly used to predict the toxicity of a parent compound and its transformation intermediates. As shown in Fig. 7, the LC50 values of CBZ for fish and daphnia are 41.3 and 14.9 mg L−1, respectively, and the EC50 of CBZ for green algae is 0.26 mg L−1. The chronic toxicity values (ChVs) for CBZ were determined to be 0.901 mg L−1 for fish, 1.171 mg L−1 for daphnia, and 0.13 mg L−1 for algae. According to the standard established by the Globally Harmonized System of Classification and Labelling of Chemicals (GHS) (Table S5†), CBZ can be classified as very toxic due to its acute toxicity values to fish and green algae.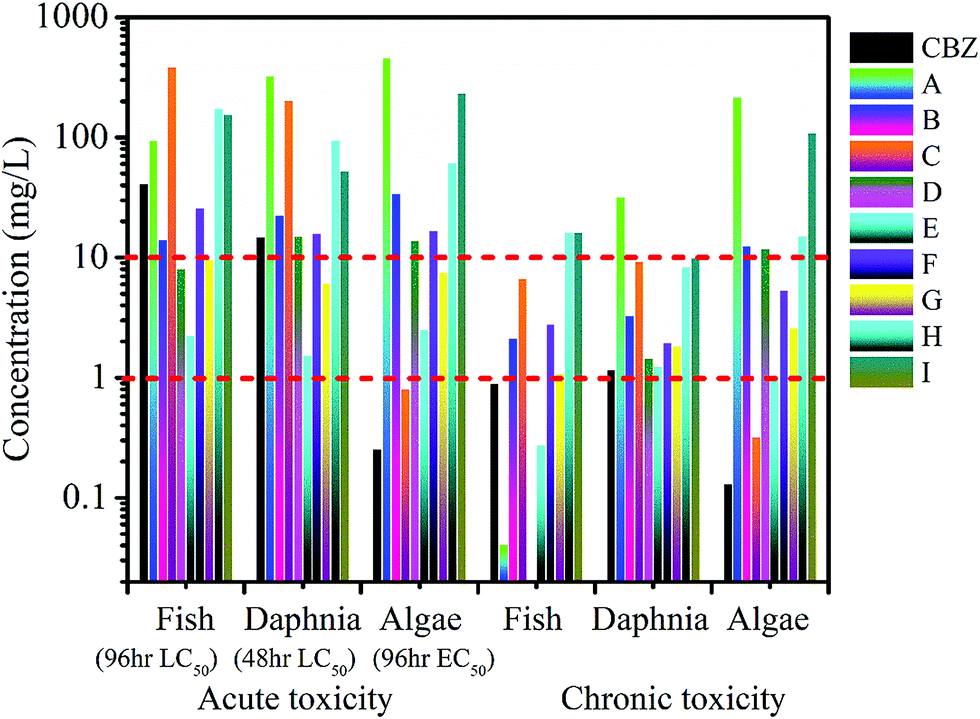 | ||
| Fig. 7 Toxicities of CBZ and its transformation products for fish, daphnia and green algae predicted by the ECOSAR program (LC50 represents the half lethal concentration and EC50 represents the half effective concentration). | ||
Compared to CBZ, the intermediates B, D, E, F, and G exhibit higher acute toxicity to fish, while intermediates E and G are more toxic to daphnia than CBZ. From the calculated EC50 values, all nine intermediates are less toxic than the parent compound. As shown in Fig. 7, fortunately, the chronic toxicities of the detected intermediates are lower than that of CBZ itself to the three aquatic organisms, except in the case of chronic toxicity towards fish, where A, D and E appear to be more toxic than CBZ. The toxicity results suggest that prolonged treatment of polluted water is better for decreasing toxicity even if CBZ is removed at an earlier time. This is because prolonged treatment can degrade toxic intermediates.
3.8. Practical application
In order to comprehensively evaluate the catalytic performance of PBA-LDH in a real aqueous environment, lake water, river water and well water were used; the samples were spiked with CBZ with a concentration of 20 mg L−1. The basic characteristics of the lake water, river water and well water are listed in Table S6.† As shown in Fig. S12,† almost complete removal of CBZ could be achieved by the PBA-LDH-activated PMS system in lake water, river water and well water, demonstrating that the as-prepared PBA-LDH has great application potential in treating actual water.4. Conclusions
PBA-LDH was successfully synthesized through in situ growth of nanosheet-like PBA in the interlayers of LDH. The as-prepared PBA-LDH exhibited excellent catalytic activity, superior stability and repeatability. CBZ could be completely degraded within 15 min in a wide pH range of 3.0 to 9.0 in the PBA-LDH-activated PMS system (PMS = 0.5 mM, catalyst dose 0.2 g L−1). Increasing the PBA-LDH and PMS doses had positive effects on CBZ degradation. Moreover, four different organic pollutants, including oxcarbazepine, atrazine, bisphenol A, and tetracycline, could be efficiently removed in the proposed catalytic system. SO4˙− played an indispensable role in CBZ degradation compared to ˙OH, which was confirmed by ESR characterization and radical scavenging experiments. Nine oxidation intermediates of CBZ were detected, and a rational degradation pathway was proposed. The activation mechanism behind the excellent catalytic activity and durability was provided. This study not only demonstrates an extremely stable and repeatable catalyst for heterogeneous activation of PMS to efficiently degrade refractory organic pollutants in water, but also provides guidance for the development of a new class of interlayer catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (Grants No. 51808206 and 51878256). They thank the reviewers for their comments. The authors also appreciate the support from the Brook Byers Institute for Sustainable Systems, Hightower Chair and the Georgia Research Alliance at Georgia Institute of Technology.References
- Y. Xu, T. Liu, Y. Zhang, F. Ge, R. M. Steel and L. Sun, J. Mater. Chem. A, 2017, 5, 12001–12014 RSC.
- J. Deng, Y.-q. Cheng, Y.-a. Lu, J. C. Crittenden, S.-q. Zhou, N.-y. Gao and J. Li, Chem. Eng. J., 2017, 330, 505–517 CrossRef CAS.
- B. t. Ferrari, N. Paxeus, R. L. Giudice, A. Pollio and J. Garric, Ecotoxicol. Environ. Saf., 2003, 55, 359–370 CrossRef CAS PubMed.
- T. A. Ternes, M. Meisenheimer, D. McDowell, F. Sacher, H.-J. Brauch, B. Haist-Gulde, G. Preuss, U. Wilme and N. Zulei-Seibert, Environ. Sci. Technol., 2002, 36, 3855–3863 CrossRef CAS PubMed.
- B. Yang, R. S. Kookana, M. Williams, J. Du, H. Doan and A. Kumar, Water Res., 2016, 100, 413–420 CrossRef CAS PubMed.
- W.-D. Oh, Z. Dong and T.-T. Lim, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS.
- K.-Y. A. Lin, H.-K. Lai and S. Tong, J. Colloid Interface Sci., 2018, 514, 272–280 CrossRef CAS PubMed.
- N. Wang, W. Ma, Z. Ren, Y. Du, P. Xu and X. Han, J. Mater. Chem. A, 2018, 6, 884–895 RSC.
- W. Chu, D. Li, N. Gao, M. R. Templeton, C. Tan and Y. Gao, Water Res., 2015, 72, 340–348 CrossRef CAS PubMed.
- O. S. Furman, A. L. Teel and R. J. Watts, Environ. Sci. Technol., 2010, 44, 6423–6428 CrossRef CAS PubMed.
- Q. Yi, L. Bu, Z. Shi and S. Zhou, Chem. Eng. J., 2016, 302, 417–425 CrossRef CAS.
- G. P. Anipsitakis, D. D. Dionysiou and M. A. Gonzalez, Environ. Sci. Technol., 2006, 40, 1000–1007 CrossRef CAS PubMed.
- J. A. Khan, X. He, N. S. Shah, H. M. Khan, E. Hapeshi, D. Fatta-Kassinos and D. D. Dionysiou, Chem. Eng. J., 2014, 252, 393–403 CrossRef CAS.
- A. Khan, H. Wang, Y. Liu, A. Jawad, J. Ifthikar, Z. Liao, T. Wang and Z. Chen, J. Mater. Chem. A, 2018, 6, 1590–1600 RSC.
- X. Duan, Z. Ao, H. Sun, L. Zhou, G. Wang and S. Wang, Chem. Commun., 2015, 51, 15249–15252 RSC.
- Q. Yang, H. Choi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 88, 462–469 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- H. Sun, X. Yang, L. Zhao, T. Xu and J. Lian, J. Mater. Chem. A, 2016, 4, 9455–9465 RSC.
- W. Guo, S. Su, C. Yi and Z. Ma, Environ. Prog. Sustainable Energy, 2013, 32, 193–197 CrossRef CAS.
- G. P. Anipsitakis, E. Stathatos and D. D. Dionysiou, J. Phys. Chem. B, 2005, 109, 13052–13055 CrossRef CAS PubMed.
- X. Chen, J. Chen, X. Qiao, D. Wang and X. Cai, Appl. Catal., B, 2008, 80, 116–121 CrossRef CAS.
- Y. Yao, Y. Cai, G. Wu, F. Wei, X. Li, H. Chen and S. Wang, J. Hazard. Mater., 2015, 296, 128–137 CrossRef CAS PubMed.
- M. B. Ali, A. Barras, A. Addad, B. Sieber, H. Elhouichet, M. Férid, S. Szunerits and R. Boukherroub, Phys. Chem. Chem. Phys., 2017, 19, 6569–6578 RSC.
- T. Zhang, H. Zhu and J.-P. Croué, Environ. Sci. Technol., 2013, 47, 2784–2791 CrossRef CAS PubMed.
- X. Li, J. Liu, A. I. Rykov, H. Han, C. Jin, X. Liu and J. Wang, Appl. Catal., B, 2015, 179, 196–205 CrossRef CAS.
- S. Pintado, S. Goberna-Ferrón, E. C. Escudero-Adán and J. R. n. Galán-Mascarós, J. Am. Chem. Soc., 2013, 135, 13270–13273 CrossRef CAS PubMed.
- M. Aksoy, S. V. K. Nune and F. Karadas, Inorg. Chem., 2016, 55, 4301–4307 CrossRef CAS PubMed.
- J. Bai, B. Qi, J. C. Ndamanisha and L.-p. Guo, Microporous Mesoporous Mater., 2009, 119, 193–199 CrossRef CAS.
- G. Uyanik and B. Pekin, J. Catal., 1970, 19, 195–203 CrossRef CAS.
- M. Hu, S. Ishihara, K. Ariga, M. Imura and Y. Yamauchi, Chem.–Eur. J., 2013, 19, 1882–1885 CrossRef CAS PubMed.
- T. Valsala, A. Joseph, J. Shah, K. Raj and V. Venugopal, J. Nucl. Mater., 2009, 384, 146–152 CrossRef CAS.
- L. Deng and Z. Shi, J. Alloys Compd., 2015, 637, 188–196 CrossRef CAS.
- C. Zhang, T. Yamamoto and Y. Einaga, Nanomater. Nanotechnol., 2016, 6, 26 CrossRef.
- L. Ma, S. M. Islam, C. Xiao, J. Zhao, H. Liu, M. Yuan, G. Sun, H. Li, S. Ma and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 12745–12757 CrossRef CAS PubMed.
- P. Padma Kumar, A. G. Kalinichev and R. J. Kirkpatrick, J. Phys. Chem. B, 2006, 110, 3841–3844 CrossRef CAS PubMed.
- S. P. Newman and W. Jones, New J. Chem., 1998, 22, 105–115 RSC.
- X. Y. Yu, L. Yu, H. B. Wu and X. W. D. Lou, Angew. Chem., 2015, 127, 5421–5425 CrossRef.
- L. Deng, Z. Shi and X. Peng, RSC Adv., 2015, 5, 49791–49801 RSC.
- T. Hibino and W. Jones, J. Mater. Chem., 2001, 11, 1321–1323 RSC.
- F.-X. Bu, M. Hu, W. Zhang, Q. Meng, L. Xu, D.-M. Jiang and J.-S. Jiang, Chem. Commun., 2015, 51, 17568–17571 RSC.
- L. Deng, H. Zeng, Z. Shi, W. Zhang and J. Luo, J. Colloid Interface Sci., 2018, 521, 172–182 CrossRef CAS PubMed.
- D. S. Robins and P. K. Dutta, Langmuir, 1996, 12, 402–408 CrossRef CAS.
- S. Yu, X. Wang, Z. Chen, J. Wang, S. Wang, T. Hayat and X. Wang, J. Hazard. Mater., 2017, 321, 111–120 CrossRef CAS PubMed.
- D. Liang, W. Yue, G. Sun, D. Zheng, K. Ooi and X. Yang, Langmuir, 2015, 31, 12464–12471 CrossRef CAS PubMed.
- Q. Wang, S. He, N. Wang, J. Zhao, J. Fang and W. Shen, New J. Chem., 2016, 40, 3244–3251 RSC.
- J. W. Boclair, P. S. Braterman, B. D. Brister, Z. Wang and F. Yarberry, J. Solid State Chem., 2001, 161, 249–258 CrossRef CAS.
- M. Wei, Z. Shi, D. G. Evans and X. Duan, J. Mater. Chem., 2006, 16, 2102–2109 RSC.
- T. Zhang, H. Zhu and J.-P. Croue, Environ. Sci. Technol., 2013, 47, 2784–2791 CrossRef CAS PubMed.
- R. Guan, X. Yuan, Z. Wu, H. Wang, L. Jiang, J. Zhang, Y. Li, G. Zeng and D. Mo, Chem. Eng. J., 2018, 339, 519–530 CrossRef CAS.
- L. Chen, D. Ding, C. Liu, H. Cai, Y. Qu, S. Yang, Y. Gao and T. Cai, Chem. Eng. J., 2018, 334, 273–284 CrossRef CAS.
- S. Lu, G. Wang, S. Chen, H. Yu, F. Ye and X. Quan, J. Hazard. Mater., 2018, 353, 401–409 CrossRef CAS PubMed.
- Y.-H. Guan, J. Ma, X.-C. Li, J.-Y. Fang and L.-W. Chen, Environ. Sci. Technol., 2011, 45, 9308–9314 CrossRef CAS PubMed.
- Y. Ding, L. Zhu, N. Wang and H. Tang, Appl. Catal., B, 2013, 129, 153–162 CrossRef CAS.
- Y.-H. Guan, J. Ma, Y.-M. Ren, Y.-L. Liu, J.-Y. Xiao, L.-q. Lin and C. Zhang, Water Res., 2013, 47, 5431–5438 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS PubMed.
- S. Yuan, P. Liao and A. N. Alshawabkeh, Environ. Sci. Technol., 2013, 48, 656–663 CrossRef PubMed.
- G. Fang, J. Gao, C. Liu, D. D. Dionysiou, Y. Wang and D. Zhou, Environ. Sci. Technol., 2014, 48, 1902–1910 CrossRef CAS PubMed.
- L. Deng, Z. Shi, Z. Zou and S. Zhou, Environ. Sci. Pollut. Res., 2017, 24, 11536–11548 CrossRef CAS PubMed.
- I. Ismail, B. Abdallah, M. Abou-Kharroub and O. Mrad, Nucl. Instrum. Methods Phys. Res., Sect. B, 2012, 271, 102–106 CrossRef CAS.
- Y. Ding, X. Xia, Y. Ruan and H. Tang, Chemosphere, 2015, 141, 80–86 CrossRef CAS PubMed.
- T. E. Doll and F. H. Frimmel, Catal. Today, 2005, 101, 195–202 CrossRef CAS.
- F. Wu, H. Huang, T. Xu, W. Lu, N. Li and W. Chen, Appl. Catal., B, 2017, 218, 230–239 CrossRef CAS.
- H. Liu, S. Bao, Z. Cai, T. Xu, N. Li, L. Wang, H. Chen, W. Lu and W. Chen, Chem. Eng. J., 2017, 317, 1092–1098 CrossRef CAS.
- A. Bahlmann, W. Brack, R. J. Schneider and M. Krauss, Water Res., 2014, 57, 104–114 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta08801b |
| This journal is © The Royal Society of Chemistry 2019 |