
Tailoring the composition of a one-step electrodeposited Co,Ni/Co,Ni(OH)2 composite coating for a highly active hydrogen evolution electrode†
Karolina
Kordek
 ,
Ewa
Lorenc-Grabowska
and
Piotr
Rutkowski
*
,
Ewa
Lorenc-Grabowska
and
Piotr
Rutkowski
*
Department of Polymer and Carbonaceous Materials, Faculty of Chemistry, Wrocław University of Science and Technology, Gdańska 7/9, Wrocław 50-344, Poland. E-mail: piotr.rutkowski@pwr.edu.pl; Fax: +48 71 320 65 06; Tel: +48 71 320 64 55
First published on 16th October 2019
Abstract
Low hydrogen evolution overpotential, along with high stability during operation and ease of preparation from inexpensive raw materials, is a crucial characteristic of an effective electrode for large-scale hydrogen generation by alkaline water electrolysis. Herein, we present a method of preparation of a low-cost electrode, applying as a substrate a carbon fiber textile derived from a technical carbon fiber reinforced polymer. Noble-metal free composite films composed of cobalt–nickel alloy and amorphous Co,Ni(OH)2 phases are obtained by a one-step electrodeposition process. Deposition is optimized in terms of the applied potential as well as metal ion concentration and Co/Ni ratio in an electroplating bath. The experimental results indicate that the hCT-Co0.4Ni0.6 electrode possesses the lowest HER overpotential of 150 mV at a current density of 10 mA cm−2 in 1.0 M KOH, with negligible activity loss within 100 h of galvanostatic operation. A significant decrease of the overpotential and electrode stability boost are achieved thanks to the precise control of the bimetallic electrode composition, which affects the morphology, electrocatalytically active surface area and electronic properties of the material.
Introduction
The storage of electrical energy has become a critical issue in recent years.1 Due to the high energy density per unit weight and possibility of its emission-free expenditure, H2 fuel is a promising candidate for an energy carrier.2,3 Among the methods of its production, achieved without emitting pollutant gases, alkaline water electrolysis is currently the most advanced technology.4,5 Hydrogen produced this way is also highly pure; therefore it can be directly applied for driving hydrogen-fuel-cell vehicles. The reaction overpotentials of hydrogen and oxygen evolution make a high contribution to the overall energy consumption in electrolyzers.6 It can be, however, reduced by application of appropriate electrocatalysts.7 Nonetheless, the high cost and scarcity of the state-of-the-art noble-metal based electrocatalytic materials limit the large-scale use of this technology.8,9 Thus, a sustainable hydrogen production by means of electrolysis requires the advancement of an electrocatalyst offering low hydrogen evolution overpotential along with robust stability and possibility of simple production from earth-abundant materials.10Over the past few years many efforts have been devoted to exploration of new, noble-metal free electrocatalytic materials for the hydrogen evolution reaction (HER).11 Because of their proper binding ability with hydrogen,12 numerous cobalt and nickel-based materials, such as their phosphides,13–16 sulfides,17,18 or metallic nanoparticles embedded in carbonaceous matrixes,19–22 have been developed and have shown distinct electrocatalytic activities in an acidic medium. However, in alkaline solutions the HER performance of those catalysts is often hindered because of the additional reaction step of water dissociation H2O + e− = Hads + OH− (Volmer process), which initiates the HER process. The reaction is subsequently accomplished through one of the processes of hydrogen desorption: Hads + e− = H2 + OH− (Heyrovsky process) or Hads + Hads = H2 (Tafel process).2,10,23 This pathway implies that the electrocatalytic activity in an alkaline medium is dependent on the binding ability of both H and OH− species and dissociation energy of water.24
On the other hand, cobalt and/or nickel oxides/hydroxides were widely studied as oxygen evolution reaction electrocatalysts,25–30 while their use as HER electrocatalysts was much rarer, due to insufficient activity. Nonetheless, their ability to catalyze the breaking of the bonds in OH− during the OER can be utilized during H–O–H dissociation in the Volmer step of the HER. Previous research has shown that composite materials, containing metal phases, which effectively catalyze hydrogen atom recombination to H2, and metal oxide/hydroxide phases, active in the Volmer process, constitute electrocatalysts with high HER electrocatalytic performance in alkaline solution.23,31–34 Still, their application is hindered because of the complicated preparation procedures and material stability issues.2
Recently, growing the active material directly on the electrode surface has emerged as an active approach to further minimize the cost of electrocatalysts, thus avoiding the use of polymer binders, usually necessary to attach powder electrocatalysts to the electrode surface.35 In binder-free electrodes, the electric contact between the composite components is improved.36 Moreover, the attachment of the deposit is enhanced, minimizing the possibility of active material delamination from the surface during vigorous hydrogen evolution. Several two-step methods have been applied to obtain free standing cobalt/cobalt (hydr)oxide electrocatalysts, including deposition from the solution followed by calcination,32 and electrodeposition combined with plasma treatment.34 In a recent study,35 composite materials, containing the Ni–Co–Ti alloy and Co,Ni(OH)2 phases, were prepared by one-step electrodeposition on titanium foil, forming an electrode with high HER catalytic performance in alkaline solution. Nonetheless, all the aforementioned electrodes suffer from limited stability, with the reported galvanostatic performance at −10 mA cm−2 stable for no longer than 20 h or with a noticeable activity loss within the first 30 h of operation.
The aim of our work was to prepare a highly active and robust HER electrocatalytic electrode by one-step electrodeposition of a mixed Co,Ni-alloy/Co,Ni hydroxide on a cheap substrate of carbon fiber cloth. Carbon fiber cloth is a widely applied electrode substrate, because of its high conductivity, low cost and flexibility.37 In our work, in order to boost the cost-effectiveness of the electrode, the applied carbon fiber cloth was derived from a technical grade carbon fiber reinforced polymer. We present the applicability of this textile, which is not directly dedicated for electrochemical purposes, as an electrode substrate material. An electrode with high HER activity was developed by optimization of the one-step electrodeposition process in terms of metal concentration and cobalt to nickel ratio in solution for electrodeposition, as well as the deposition potential. The HER activity of the electrode exhibiting the highest performance can be attributed to the optimal composition, ensuring the highest electrochemically active surface area and synergy of the activities of two co-deposited metals and their hydroxides. Notably, the electrocatalytic performance of the electrode is highly stable with no activity loss for 100 hours, which is longer than that generally reported for transition-metal based HER electrocatalysts. The work opens new pathways for the preparation of electrocatalytic electrodes for large-scale application.
Experimental section
Materials
The carbon fiber reinforced polymer was purchased from Carbotec GmbH. KOH (85%) was supplied by Merck Chemicals. CoCl2·6H2O (98%), NiCl2·6H2O (98%) and Pt (10% on carbon dry) were purchased from Alfa Aesar. (NH4)2SO4 (98.5%) was supplied by Chempur. All reagents were used without further purification. In all experiments Milli-Q water with a resistivity of 18.2 MΩ was used.Preparation of the carbon fiber substrate
Pieces of the carbon fiber reinforced polymer (1 × 2 cm2), denoted as CT-raw, were washed in sequence with acetone, ethanol and water. Next, in order to remove the insulating polymer matrix, the pieces were placed in alumina crucibles and subjected to heating in a muffle furnace at 500 °C for 2 hours. The resulting carbon fiber substrates were denoted as hCT.Electrodeposition of the Co,Ni-based films
The Co,Ni-based films were prepared by electrodeposition onto hCT in a three-electrode system, composed of hCT as a working electrode, a carbon rod as a counter electrode and Ag/AgCl as a reference electrode. The 1 × 1 cm2 area of the working electrode was immersed in a water solution of CoCl2·6H2O and/or NiCl2·6H2O. The potentiostatic electrodeposition was carried out for 30 minutes at room temperature using an ATLAS 0531 Electrochemical Unit. The total metal ion concentration and Co/Ni molar ratio in solution used during electrodeposition as well as electrodeposition potential were optimized. First, the electrodeposition was carried out for different CoCl2 concentrations in solution for electrodeposition (1, 2, 10, 20 and 40 mM) at a constant potential of −1.2 V. Next, the electrodes were prepared by applying different potentials (−1.0; −1.2; −1.4; −1.6 V vs. Ag/AgCl) with a constant cobalt to nickel ratio of x = 0.4 and total metal ion concentration of 20 mM. Finally, the hCT-CoxNi1−x electrodes were prepared with different molar ratios of cobalt to nickel (for x = 0; 0.25; 0.4; 0.5; 0.6; 0.75; 1.0), maintaining a total concentration of metal ions of 20 mM and a deposition potential of −1.2 V. After each deposition, the electrode was removed from the solution, thoroughly washed with water and dried at 40 °C overnight.Material characterization
The surface morphology of the electrodes was evaluated by Field Emission Scanning Electron Microscopy (FE-SEM, Magellan 400L, UC Technology). Transmission Electron Microscopy images of fresh samples were acquired using an FEI Tecnai G2 F20, while post-performance TEM images were acquired using an FEI Tecnai G2 20 X-TWIN. The crystalline structure of the electrodes was evaluated by X-ray diffraction (Rigaku Ultima IV diffractometer with CuKα radiation of λ = 0.1542 nm). Raman and FT-IR spectra were acquired by using a Bruker Optic GmbH spectrometer with a Senterra module for a Raman Tensor 27 module for FT-IR characterization. X-ray Photoelectron Spectroscopy (XPS) measurements were performed using a Microlab 350 spectrometer (Thermo Electron) with an X-ray source of Al Kα (1486.6 eV). The elemental compositions of the electrodes were evaluated with an Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES) Agilent 720.Electrochemical measurements
The electrochemical measurements were performed with an ATLAS 0531 Electrochemical Unit in a three-electrode system in 1.0 M KOH. The carbon cloth-based electrode, graphite rod and Hg/HgO were applied as working, counter and reference electrodes respectively. Before the measurement of HER activity, the working electrode was pre-activated by several cyclic voltammetry (CV) scans at a scan rate of 50 mV s−1, until repeatable plots were recorded. Polarization curves were obtained with the Linear Sweep Voltammetry (LSV) technique with a scan rate of 5 mV s−1. The potentials were converted to a reversible hydrogen electrode (RHE) following the equation: ERHE = EHg/HgO + 0.059 × pH + 0.098 and corrected for the iR compensation. The stability of the electrode performance in the HER was assessed by chronopotentiometry at the current densities of 10 and 100 mA cm−2. The double layer capacitances of the electrodes were measured by cyclic voltammetry in the potential range between −0.20 and 0.20 V vs. Hg/HgO at the scan rates of 5, 10, 15, 20, 25 and 30 mV s−1. The capacitive currents measured at 0.0 V vs. Hg/HgO were plotted against the scan rate and the specific capacitances were acquired by the linear fitting of the plots. The electrochemical impedance spectra (EIS) were acquired in a frequency range from 200 kHz to 10 mHz and an amplitude of 5 mV at a HER overpotential of 275 mV. The faradaic yield of hydrogen evolution was determined by comparison of the gas volume generated during potentiostatic electrolysis at −0.1 V vs. RHE with the theoretical (calculated) value.Results and discussion
Substrate material characterization
In order to obtain a cheap, metal-free conductive substrate for the electrocatalytic electrode, a piece of the commercial carbon fiber reinforced polymer composite (CT-raw) was subjected to a treatment aimed at removal of the polymer matrix coating. The applied thermal treatment at 500 °C for 2 h has resulted in exposure of fibers due to decomposition of epoxy resin. The obtained carbon fiber material was denoted as hCT.In previous work it was shown that the properties of substrates for electrodeposition of metal compounds influence the growth of the electrodeposited films, and therefore affect the morphology of the product.38 Consequently, the properties of the carbon fiber surface have a strong impact on the electrocatalytic properties of the entire electrode. Therefore, the substrate carbon fiber material used in this work was characterized in terms of morphology and surface composition. The SEM image shows the fibers with a diameter of ca. 5 μm and clear longitudinal grooves on the surface (Fig. 1a). Little bright spots are visible, which might constitute pieces of the residual insulating polymer matrix. The high magnification image shows a rather smooth surface of the fibers (Fig. S1†).
 | ||
| Fig. 1 (a) The SEM image and high-resolution XPS spectra of the hCT electrode in (b) C 1s, (c) O 1s and (d) N 1s regions. | ||
The clear change in the composition of the material surface before and after the thermal treatment can be distinguished based on the XPS survey spectra, presented in Fig. S2.† Both spectra indicate the presence of only carbon, oxygen and nitrogen elements in CT-raw and hCT. Their atomic concentrations are compared in Table S1.† The results demonstrate that the concentration of oxygen has significantly decreased from 19.0 to 10.4 at% as a result of almost total removal of the oxygen-rich epoxy resin during the thermal treatment. At the same time, the nitrogen concentration increased from 2.13 to 8.86 at%, evidencing that the exposed carbon fibers are rich in nitrogen.
The high-resolution spectra of the hCT sample in the C 1s, O 1s and N 1s regions were further analyzed. The C 1s peak was fitted into 6 components (Fig. 1b). The signals located at 284.7 and 285.2 eV correspond to delocalized and localized C–C respectively. The signals located at higher binding energies can be ascribed to carbon atoms bound to heteroatoms in functional groups, such as: C–OH (285.9 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N (286.9 eV) and COOH (288.3 eV).39 The intensity of the delocalized C–C, corresponding to 42.0 at% of the entire C 1s signal, reveals that the contribution of the graphitic lattice is not predominant on the surface of the fiber.
O and C–N (286.9 eV) and COOH (288.3 eV).39 The intensity of the delocalized C–C, corresponding to 42.0 at% of the entire C 1s signal, reveals that the contribution of the graphitic lattice is not predominant on the surface of the fiber.
The O 1s signal, presented in Fig. 1c, can be deconvoluted into 4 peaks, located at 531.0 eV (CO), 532.1 eV (C–O–H and oxygen double bonds in OC–O), 533.3 eV (oxygen single bond in OC–O) and 534.6 eV (COOH).40 The atomic concentration of nitrogen in the sample is particularly high. The signal in Fig. 1d was fitted into 3 components: pyridinic N6 (398.6 eV), graphitic NC (400.3 eV) and quaternary nitrogen NQ (401.7 eV), among which N6 and NC possess the highest intensities.41 Previous reports have shown that carbon fibers rich in heteroatoms constitute good anchoring points for the electrodeposited films and promote uniform material growth with good attachment to the substrate.42,43 The nitrogen doping in the carbonaceous substrate further increases the interaction with transition metals.1,44,45
The material after thermal treatment was further characterized by Raman and FT-IR spectroscopies. The Raman spectrum (Fig. S3†) possesses two intense peaks located at approximately 1350 and 1599 cm−1, which correspond to D and G bands respectively, both originating from the carbonaceous structure of the carbon fibre.46 Moreover a broad peak centred at 2746 cm−1 can also be noted, which is likely the overlap of the 2D (≈2680 cm−1) and D + D′ (≈2940 cm−1) bands.47 This result confirms the defective graphitic structure of hCT. Also, the presence of abundant oxygen functional groups on the surface of hCT is confirmed by the FT-IR spectrum (Fig. S4†), in which the signals originating from hydroxyl groups (3437 cm−1) and carboxylates (1354 and 1599 cm−1) can be distinguished.48
Characterization of the deposited films
Upon removal of the epoxy resin coating and exposure of the conductive support for the free-standing electrocatalytic HER electrodes, metal-based films were grown on the hCT surface applying potentiostatic electrodeposition from a metal chloride bath. In order to obtain electrodes with optimal HER activity, the deposition parameters were modified and the performance of the electrodes was evaluated.At first, the influence of metal concentration in the solution for electrodeposition was assessed. The HER LSV plots for the obtained electrodes are presented in Fig. S5,† indicating an optimal metal concentration of 20 mM. Next, the electrodeposition potential for the mixed Co/Ni films was optimized and the resulting LSV plots are presented in Fig. S6.† As can be seen, the optimal Co/Ni electrodeposition was achieved at −1.2 V vs. Ag/AgCl. The established optimal deposition potential and metal concentration were applied during the synthesis of all electrodes further described.
A series of electrodes were prepared with different Co/Ni ratios in solution for deposition and denoted as hCT-CoxNi1−x for x ranging from 0 to 1. The electrodes were characterized in terms of morphology, composition and structure.
ICP-OES results of the actual cobalt concentration in the total metal mass of the sample versus the theoretical concentration, based on the cobalt to nickel ratio in solution for electrodeposition, are plotted in Fig. 2a. As can be seen, the real proportion is similar to the expected, with cobalt concentration slightly higher than predicted theoretically. The preferential electrodeposition of cobalt with regard to nickel is in agreement with previous reports.49 Furthermore, the total mass concentration of metal atoms (cobalt and nickel) in reference to the total weight of the electrode was similar for all samples, ranging between 4.5 and 6.0 wt% with no clear tendency in the series (Fig. 2b).
 | ||
| Fig. 2 (a) The real versus theoretical cobalt concentration in the total metal (Co + Ni) mass of the electrode and (b) the total metal percentage in the electrode mass versus x in the hCT-CoxNi1−x electrodes. | ||
Fig. 3a and b present the surface morphology of the hCT-Co0.4Ni0.6 electrode. The surface is composed of densely connected ultrathin sheets, almost vertically grown on the carbonaceous substrate. The film forms a three-dimensional porous network available for the reagents during the electrocatalytic reaction. Fig. 3c and d present the reference surfaces of hCT-Ni and hCT-Co respectively. Similarly to the mixed hCT-Co0.4Ni0.6 electrode, the films possess a flower-like morphology formed by interconnected nanosheets, which are, however, distinctly larger and thicker. Nonetheless, in the case of all electrodes the formed films are uniform and tightly cover the fibers. The obtained sheets with no clear lattice fringes are also visible in the TEM image of hCT-Co0.4Ni0.6 (Fig. 3e). The corresponding SAED pattern (Fig. 3f) presents weak and broad diffraction rings, which suggest the presence of poorly crystalline domains. The rings can be attributed to the (100) and (110) lattice planes of α-Co(OH)2.50 The results collectively illustrate that a porous nanosheet structure was formed on the surface of the composite electrodes.
 | ||
| Fig. 3 The SEM images of (a and b) hCT-Co0.4Ni0.6, (c) hCT-Ni and (d) hCT-Co electrodes. (e) TEM image of the hCT-Co0.4Ni0.6 electrode and (f) the corresponding SAED pattern. | ||
The structure of the obtained electrodes was characterized by the XRD technique. In Fig. S7† the normalized XRD diffractograms are presented for the entire hCT-CoxNi1−x series. All diffraction patterns possess a broad signal centered at 25–26°, which corresponds to the (002) reflex from the graphitic carbon.51 Furthermore, several reflexes are present between 40 and 50°, which are magnified in the high-resolution XRD diffractograms presented in Fig. 4a (without normalization). For the electrodes with a cobalt concentration lower than 0.6, two reflexes can be distinguished, located at 44.4 and 51.7°, which can be ascribed to the signals from the face centered cubic (fcc) nickel phase (JCPDS: 89-7128). With increasing cobalt concentration in the samples, the position of those reflexes does not change, suggesting the formation of a solid solution of the fcc phase. In the diffractograms of the hCT-Co0.6Ni0.4 electrode, two additional weak reflexes can be recognized, whose intensities increase with the further increase of cobalt concentration in the electrodes. The reflexes located at 41.7, 44.5 and 47.4° can be ascribed to the hexagonal close packed (hcp) ε-cobalt phase (JCPDS: 89-7094), which coexists with the fcc phase in the electrodes with higher cobalt concentrations. Similar changes in the crystalline structure depending on the cobalt–nickel alloy composition were previously reported.52–54 No XRD features of other crystalline structures can be identified in the diffractograms, revealing that the other possible phases present in the samples form very small crystalline domains or are in the amorphous form. In the entire range of concentrations, the intensity of metal reflexes (cobalt, nickel and/or alloy) clearly decrease with increasing cobalt concentration. The results evidently confirm the presence of the metallic phase(s) in the structure of all electrodes.
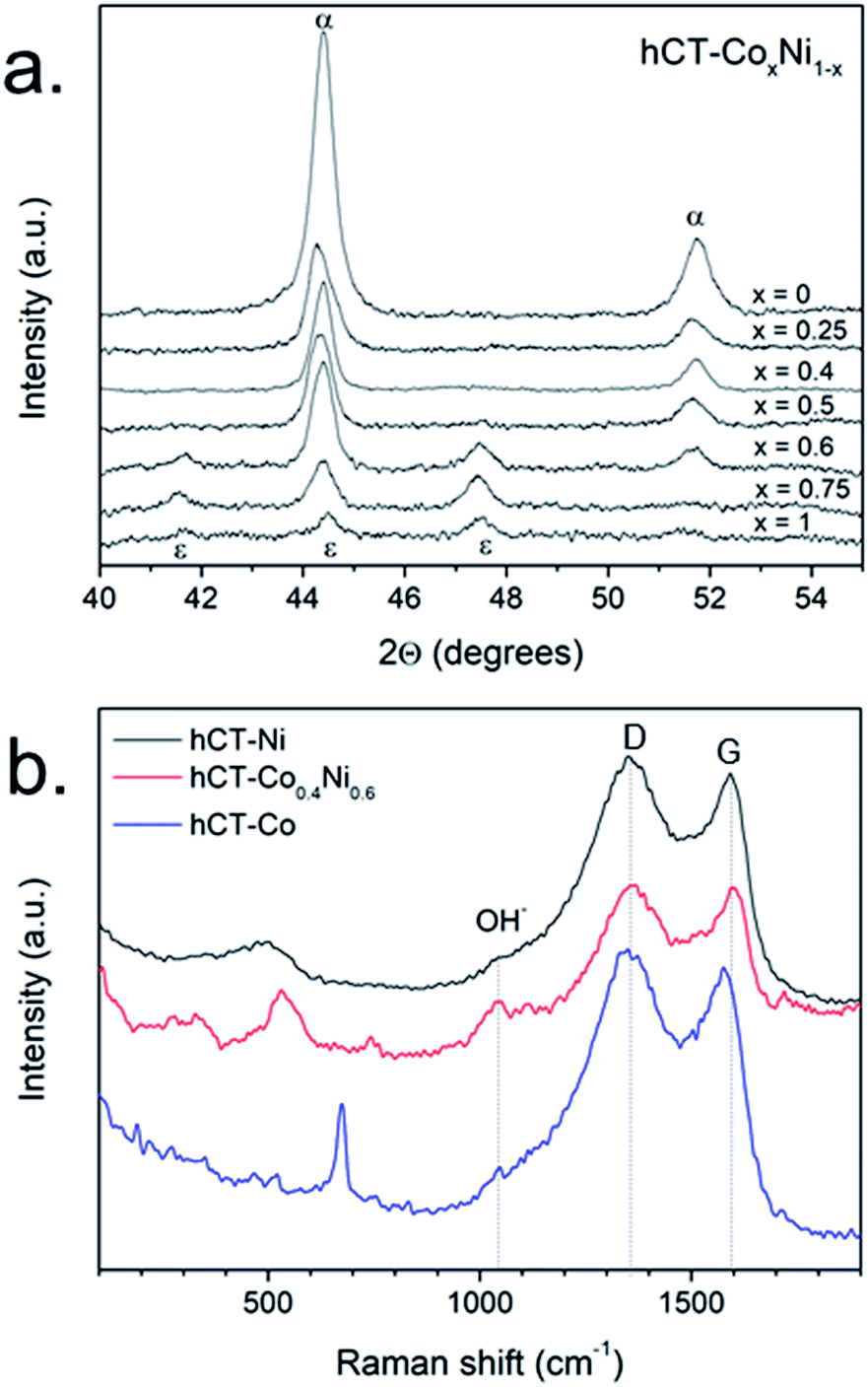 | ||
| Fig. 4 (a) XRD diffractograms of hCT-CoxNi1−x electrodes and (b) Raman spectra of hCT-Ni, hCT-Co0.4Ni0.6 and hCT-Co. | ||
In order to identify the other components of the electrodes, Raman spectra were acquired for hCT-Co0.4Ni0.6, hCT-Co and hCT-Ni electrodes and are presented in Fig. 4b.55 Besides the D and G bands, originating from the substrate, in all spectra a broad shoulder located at ∼1050 cm−1 can be distinguished, which can be ascribed to the deformations of hydroxyl groups in metal hydroxides.56 The spectrum of the hCT-Co electrode has also clear additional features characteristic of amorphous Co(OH)2, namely the bands located at 190, 467, 521 and 674 cm−1.57 In the case of the hCT-Co0.4Ni0.6 electrode, the most intense band located at 531 cm−1 with the shoulder at 450 cm−1 reveals the presence of the mixed Co,Ni(OH)2 phase.56,58 The spectrum of the hCT-Ni electrode possesses a broad feature centered at 502 cm−1. A similar signal has been reported previously as the translational vibration lattice mode of Ni(OH)2.59–61 Consequently, the presented Raman spectra demonstrate the presence of the metal hydroxide phase, which because of its largely amorphous feature is indistinguishable with the XRD technique.
In order to support the results obtained by Raman spectroscopy and uncover the valence state of the elements present on the electrode surface, high-resolution XPS spectra were acquired for the hCT-Coo.4Ni0.6 electrode. The spectrum in the O 1s region (Fig. 5a) can be fitted into 3 peaks. In comparison with the spectrum of the substrate (hCT – Fig. 1c), the relative intensities of the signals from the carbon fiber decrease (with complete disappearance of the signal located at 532.1 eV), while the intensity of the peak located at the binding energy of 531.0 eV increases significantly and shifts slightly to 531.3 eV. This signal is characteristic of oxygen hydroxyl groups in Co(OH)2 and Ni(OH)2.62 The presence of the hydroxides was further confirmed by spectra in Ni 2p and Co 2p regions. Fig. 5b presents the deconvoluted Ni 2p spectrum with two distinct peaks located at 856.3 and 873.9 eV, corresponding to 2p1/2 and 2p3/2 features respectively63 and intense satellite features at 861.9 and 878.0 eV, which can be attributed to Ni2+ in Ni(OH)2.64,65 In Fig. 5c the deconvoluted Co 2p spectrum of the electrode is presented, with two main peaks located at the binding energies of 781.6 eV and 797.2 eV, which can be attributed to Co 2p1/2 and Co 2p3/2 respectively. The weak satellite features peak at 786.2 and 803.6 eV. The spectrum can be ascribed to Co2+ in Co(OH)2.64,65 Notably, no signals are visible in the spectral regions characteristic of metallic Co (778.0 eV) or Ni (852.5 eV),62 even though the presence of metallic species in the electrode was clearly indicated by the XRD technique.
 | ||
| Fig. 5 High-resolution XPS spectra of hCT-Co0.4Ni0.6 electrodes in the (a) O 1s, (b) Ni 2p and (c) Co 2p regions. | ||
The results, acquired by XRD, Raman and XPS, collectively illustrate that the one-step electrodeposition technique made it possible to obtain the films composed of two components: the metallic phase, the presence of which has been confirmed by XRD and the cobalt and/or nickel hydroxide phase, demonstrated by Raman and photoelectron spectroscopies. The absence of Co,Ni(OH)2 reflexes in the XRD diffractograms can be explained by its amorphous feature, while the lack of the XPS signals of Co0 and Ni0 presumably results from the course of the electrodeposition process. The proposed deposition mechanism (Fig. 6) reveals that at the beginning, the Co2+ and/or Ni2+ ions are being cathodically reduced and deposited on hCT in the metallic form of Co, Ni or their alloy. The deposited metal then decreases the HER overpotential at the electrode surface in the solution for electrodeposition, thus catalyzing the formation of OH− near the electrode and locally lowering the pH. Consequently, further deposition proceeds with the formation of Co,Ni(OH)2 covering the metallic phase, which is for this reason inaccessible by the XPS technique, that allows the analysis of only the outer most 2–5 nm of the sample.66 This phenomenon enables acquiring a composite metal/metal hydroxide film by one-step electrodeposition.
 | ||
| Fig. 6 Schematic illustration of the proposed two-step mechanism of one-step electrodeposition of composite films on carbon cloth. | ||
Evaluation of electrochemical properties
The HER polarization curves of the hCT-CoxNi1−x electrodes are presented in Fig. 7a. As can be seen, a minimum overpotential of 295 mV at 100 mA cm−2 is exhibited for x = 0.4. For the higher or lower values of x a gradual increase of the overpotential can be noted, as presented in Fig. 7b, in which the overpotentials are plotted versus x. It can be noted that the slight differences in the metal concentrations, determined by the ICP-MS technique (Fig. 2b), do not correlate with the electrocatalytic activity trends for electrodes with different compositions. These results show that differences in the deposition rate from different solutions are not the origin of the variations in the electrocatalytic activities. | ||
| Fig. 7 (a) LSV plots for the series of hCT-CoxNi1−x electrodes and (b) corresponding HER overpotentials at a current density of 150 mA cm−2. | ||
The electrocatalytic activity of the hCT-Co0.4Ni0.6 electrode was compared with the activities of the control samples: hCT-Co, hCT-Ni, hCT and Pt/C (Fig. 8a) in 1.0 M KOH. As predicted, the Pt/C material exhibits the highest performance, with a low overpotential of 65 mV needed to deliver 10 mA cm−2. However, the hCT-Co0.4Ni0.6 electrode, composed of only earth-abundant elements, exhibits an overpotential of 150 mV, substantially lower than those of the single-metal containing electrodes: hCT-Co (190 mV) and hCT-Ni (235 mV) and the carbon cloth support (450 mV).
 | ||
| Fig. 8 (a) LSV plots with (b) corresponding Tafel slopes of the hCT, hCT-Co, hCT-Ni, hCT-Co0.4Ni0.6 and Pt/C electrodes in 1.0 M KOH. (c) Chronopotentiometric curves of hCT-Co0.4Ni0.6 hCT-Co and hCT-Ni at a current density of 10 mA cm−1 and (d) 100 mA cm−2 for hCT-Co0.4Ni0.6. | ||
The kinetics of the HER at the prepared electrodes was studied by analysis of the Tafel plots (Fig. 8b). Except for the Pt/C electrode, which exhibited the lowest Tafel slope of 77 mV dec−1, the bimetallic hCT-Co0.4Ni0.6 electrode has shown the most favorable kinetics among the tested samples, with a Tafel slope of 126 mV dec−1, slightly lower than 128 mV dec−1 for hCT-Co and significantly lower than 143 mV dec−1 for hCT-Ni. The reported Tafel slope values of approximately 120 mV dec−1 indicate that the step of water dissociation, which initiates the hydrogen evolution from alkaline media, is the rate determining step of the entire reaction.67,68
Transition metal-based electrodes often suffer from low stability of the electrocatalytic properties during operation.69 In order to evaluate the practical utility of the electrodes as HER electrocatalysts, a prolonged stability test was performed at a constant current density of −10 mA cm−2. The obtained chronoamperometric curves, presented in Fig. 8c, reveal that hCT-Co0.4Ni0.6 shows only a negligible overpotential increase of 5 mV during 100 h of hydrogen evolution at −10 mA cm−2, which is a current density corresponding to the energy production from the solar-to-fuel-device performing under 1 sun with about 10% efficiency.70 The HER LSV plot, depicted in Fig. S8†, was also recorded after the stability test, and presented in comparison with the LSV plot of the fresh electrode. The negligible shift of the plot supports the outstanding stability of the electrocatalytic performance of the electrode. In contrast, for the monometallic electrodes, hCT-Ni and hCT-Co, the overpotential change was much more pronounced, with an increase of 25 mV and 80 mV respectively during 100 h of operation (Fig. 8c).
Additionally, the TEM, XPS and XRD characterization of the hCT-Co0.4Ni0.6 electrode were performed after the stability test. The TEM images and SAED patterns (Fig. S9†) reveal no change in the film morphology or structure during the stability test. Also, no surface composition change can be distinguished based on XPS spectra (Fig. S10†), besides a slight decrease of the relative intensities of the oxygen peaks assigned to oxygen functional groups from the carbonaceous substrate. However, the reduction of intensity of the reflexes ascribed to the metal phase can be noticed upon comparison of the XRD diffractograms of the fresh and used electrodes (Fig. S11†). This increase can justify the slight increase of HER overpotential during the stability test. Moreover, the results show that the faradaic yield of the HER process equals 96% (Fig. S12†).
Furthermore, the performance stability of hCT-Co0.4Ni0.6 was also evaluated at a high current density of −100 mA cm−2 (Fig. 8d). No overpotential increase can be distinguished during 24 h of constant electrolysis. The high stability of the performance proves good binder-free attachment between the carbonaceous substrate and the deposited film. The material does not peel-off from the surface even during vigorous gas evolution. The comparison of the stability between the bimetallic and monometallic electrodes indicates that the incorporation of two metals in the electrode structure causes a synergic effect of stability increase.
In order to estimate the electrochemically active surface area, the double layer capacitance of the series of electrodes was evaluated in the non-faradaic potential window. The CV plots at different scan rates and the corresponding linear fits of current densities versus scan rates are presented in Fig. S13,† whereas in Fig. 9a the resulting double layer capacitances are plotted against the cobalt concentration in the electrode. The results suggest that hCT-Co0.4Ni0.6 possesses the highest surface area, which gradually decreases with the change of electrode composition. Nonetheless, the tendency of the Cdl change does not directly reflect the trend in electrocatalytic HER activity. This allows us to state that the electrochemically active surface area is not the only parameter influencing the electrocatalytic activity of the electrode. However, it confirms that bimetallic films can create a higher electrochemically active surface area, which can be one of the reasons for the observed electrocatalytic activity enhancement.
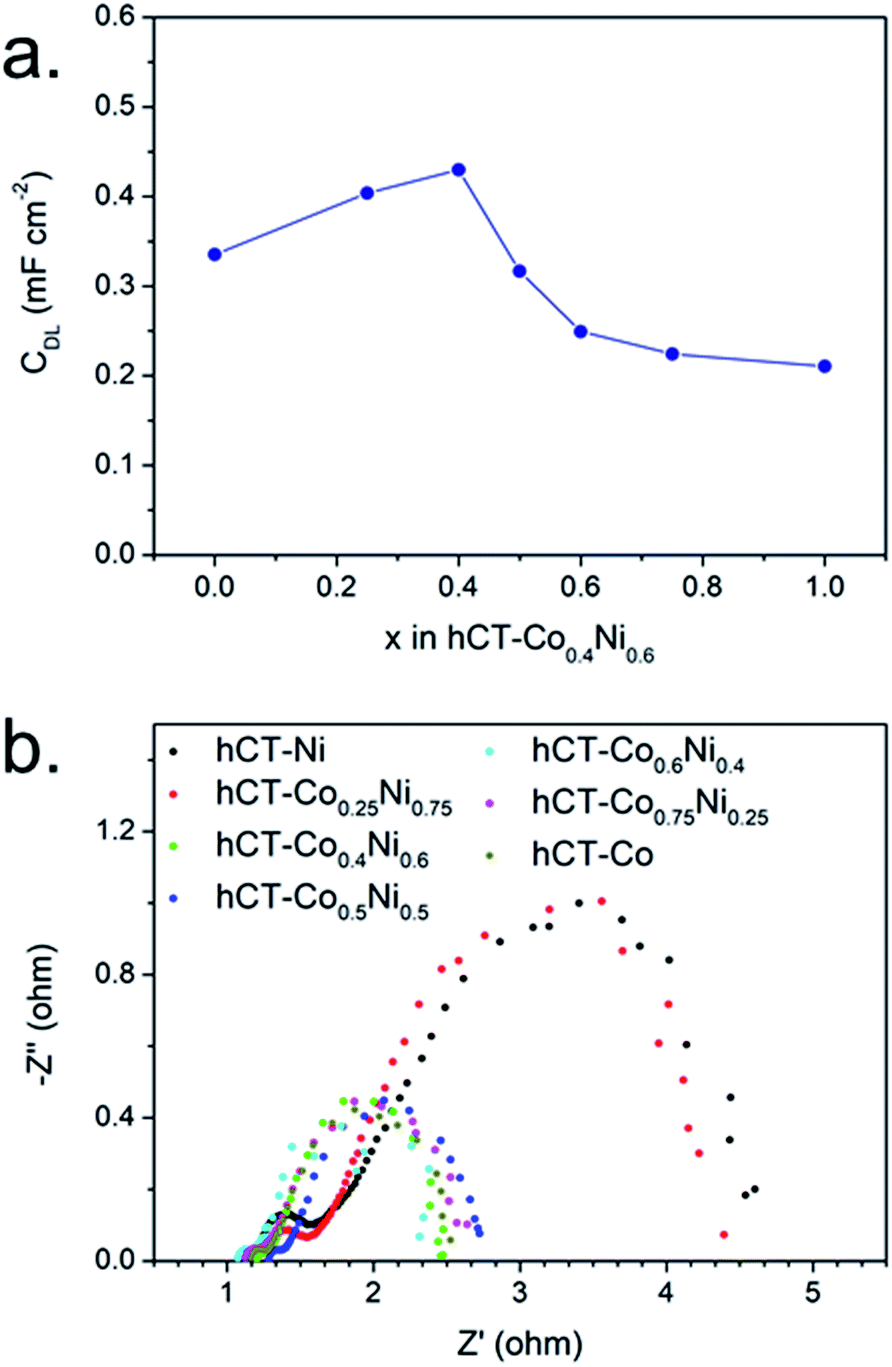 | ||
| Fig. 9 (a) Double layer capacitance and (b) electrochemical impedance spectra for the series of hCT-CoxNi1−x electrodes with different x. | ||
The electrochemical impedance spectra at an overpotential of 270 mV were acquired for the series of electrodes and are presented as Nyquist plots in Fig. 9b. For each electrode two semicircles can be distinguished. A smaller semicircle in the high-frequency region, whose diameter is independent of the applied overpotential, is indicative of the overall charge transfer resistance of the electrode (RCT). The semicircle in the low frequency region represents the resistance of the HER reaction (RHER), and thus its diameter is inversely proportional to the applied overpotential.32,58 As can be seen in the presented graph, the electrodes can be divided into two groups with similar impedance properties. The electrodes with cobalt concentration up to 25% possess both clearly lower RCT and RHER in comparison with the electrodes with a higher cobalt content. This suggests that the presence of sufficiently high cobalt concentration in the electrode increases the rate of electron transfer.31
The results illustrate that the morphology and composition of the hCT-Co0.4Ni0.6 electrode are responsible for its optimal HER performance in the series of electrodes with different cobalt to nickel ratios. The electrode possesses the most developed electrochemically active surface area, created by the most dense 3D-nanosheet structure. The tight attachment of the film to the fiber surface ensures low electrical resistivity, while the open porous structure reduces the mass transport barriers.71 Moreover, the superb activity originates from the local modulation of the electronic structure caused by the formation of the cobalt–nickel alloy phase and mixed hydroxide phase.1,58 While the hCT-Co electrode provides significantly lower HER overpotentials than hCT-Ni, the latter is characterized by much higher performance stability. Notably, the bimetallic hCT-Co0.4Ni0.6 possesses both higher activity and stability than monometallic electrodes hCT-Co and hCT-Ni, suggesting the synergic interaction of the two metals in the electrode with optimal composition.
Furthermore, several factors characterizing the entire electrode series can be possible reasons for the excellent electrocatalytic activity, namely the composite structure, self-supporting feature and amorphous form. The composite metal/metal hydroxide structure has proven to be beneficial for the HER in alkaline media, with the metal hydroxide catalyzing the water dissociation step and metal species promoting the hydrogen atom recombination to form H2 molecules.33,34 Moreover, the bottom layer of the metal can facilitate the reaction by increasing the conductivity of the composite material.43,72 The self-supported form ensures fast electron collection and good attachment between the film and the carbon fiber substrate. The supporting fiber surface rich in hydrophilic heteroatoms promotes the formation of a well-attached catalyst layer, highly stable during operation.42 Finally, the amorphous structure is considered more advantageous for electrocatalytic materials, due to the random orientation of bonds creating numerous unsaturated active sites for electrochemical reactions.11,57,58 The activity of the hCT-Co0.4Ni0.6 electrode is among the highest reported recently for noble-metal free HER electrocatalysts (Table S2†).
Conclusions
In summary, we synthesized free-standing hCT-Co0.4Ni0.6 electrodes by electrodeposition of films on low-cost carbon fiber cloth derived from a technical CFRP material. The applied one-step electrodeposition method enabled the formation of films with a composite architecture consisting of Co,Ni-alloy and amorphous Co,Ni(OH)2 phases and exhibiting excellent electrocatalytic performance in 1.0 M KOH, with an overpotential of 150 mV required to deliver a current density of 10 mA cm−2. Furthermore, despite the application of a low-cost fiber and noble-metal free films, the durability of the electrode during operation is very high with negligible activity loss over 100 hours of operation at a current density of 10 mA cm−2. The high performance is achieved by the precise control of the optimized electrode composition. The easily synthesized high-performance electrodes can be applied for mass production. We believe that the presented electrode preparation strategy can be translated to other applications in the field of energy storage and conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wrocław University of Science and Technology and from the EU-H2020 Research and Innovation Programme under grant agreement No. 654390 having benefitted from the access provided by ICN2 in Barcelona within the framework of the NFFA-Europe Staff Members Transnational Access Activity.References
- B. Bayatsarmadi, Y. Zheng, V. Russo, L. Ge, C. S. Casari, S.-Z. Qiao, X. Sun, X. W. Lou, Y. Xie, D. J. Myers, P. Zelenay, P. M. Ajayan, D. Chen and J. M. Tour, Nanoscale, 2016, 8, 18507–18515 RSC.
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed.
- X. Du, X. Zhang, Y. Li and M. Zhao, Int. J. Hydrogen Energy, 2018, 43, 19955–19964 CrossRef CAS.
- X. F. Lu, L. Yu and X. W. David Lou, Sci. Adv., 2019, 5, eaav6009 CrossRef CAS PubMed.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS.
- X. F. Lu, L. Yu, J. Zhang and X. W. (David) Lou, Adv. Mater., 2019, 31, 1900699 CrossRef PubMed.
- W. Liu, K. Du, L. Liu, J. Zhang, Z. Zhu, Y. Shao and M. Li, Nano Energy, 2017, 38, 576–584 CrossRef CAS.
- M. Li, T. Liu, X. Bo, M. Zhou and L. Guo, J. Mater. Chem. A, 2017, 5, 5413–5425 RSC.
- S. Sarawutanukul, N. Phattharasupakun, J. Wutthiprom and M. Sawangphruk, Sustainable Energy Fuels, 2018, 2, 1305–1311 RSC.
- Z. Zhu, H. Yin, C.-T. He, M. Al-Mamun, P. Liu, L. Jiang, Y. Zhao, Y. Wang, H.-G. Yang, Z. Tang, D. Wang, X.-M. Chen and H. Zhao, Adv. Mater., 2018, 30, 1801171 CrossRef PubMed.
- P. Babar, A. Lokhande, H. H. Shin, B. Pawar, M. G. Gang, S. Pawar and J. H. Kim, Small, 2018, 14, 1702568 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- E. J. Popczun, C. W. Roske, C. G. Read, J. C. Crompton, J. M. McEnaney, J. F. Callejas, N. S. Lewis and R. E. Schaak, J. Mater. Chem. A, 2015, 3, 5420–5425 RSC.
- Y. Zhang, L. Gao, E. J. M. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS PubMed.
- J. Kibsgaard, C. Tsai, K. Chan, J. D. Benck, J. K. Nørskov, F. Abild-Pedersen and T. F. Jaramillo, Energy Environ. Sci., 2015, 8, 3022–3029 RSC.
- F. H. Saadi, A. I. Carim, W. S. Drisdell, S. Gul, J. H. Baricuatro, J. Yano, M. P. Soriaga and N. S. Lewis, J. Am. Chem. Soc., 2017, 139, 12927–12930 CrossRef CAS PubMed.
- D. Y. Chung, J. W. Han, D.-H. Lim, J.-H. Jo, S. J. Yoo, H. Lee and Y.-E. Sung, Nanoscale, 2015, 7, 5157–5163 RSC.
- L. Sun, T. Wang, L. Zhang, Y. Sun, K. Xu, Z. Dai and F. Ma, J. Power Sources, 2018, 377, 142–150 CrossRef CAS.
- H. Fei, Y. Yang, Z. Peng, G. Ruan, Q. Zhong, L. Li, E. L. G. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2015, 7, 8083–8087 CrossRef CAS PubMed.
- W. Zhou, J. Zhou, Y. Zhou, J. Lu, K. Zhou, L. Yang, Z. Tang, L. Li and S. Chen, Chem. Mater., 2015, 27, 2026–2032 CrossRef CAS.
- S. Wang, L. Zhang, Y. Qin, D. Ding, Y. Bu, F. Chu, Y. Kong and M. Liu, J. Power Sources, 2017, 363, 260–268 CrossRef CAS.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. Dong Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K.-C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 51, 12495–12498 CrossRef CAS PubMed.
- X. Wang, Y. Zhu, A. Vasileff, Y. Jiao, S. Chen, L. Song, B. Zheng, Y. Zheng and S.-Z. Qiao, ACS Energy Lett., 2018, 3, 1198–1204 CrossRef CAS.
- D. McAteer, I. J. Godwin, Z. Ling, A. Harvey, L. He, C. S. Boland, V. Vega-Mayoral, B. Szydłowska, A. A. Rovetta, C. Backes, J. B. Boland, X. Chen, M. E. G. Lyons and J. N. Coleman, Adv. Energy Mater., 2018, 8, 1702965 CrossRef.
- Y. Jiang, X. Li, T. Wang and C. Wang, Nanoscale, 2016, 8, 9667–9675 RSC.
- D. S. Dhawale, P. Bodhankar, N. Sonawane and P. B. Sarawade, Sustainable Energy Fuels, 2019, 3, 1713–1719 RSC.
- N. Cheng, L. Ren, G. Casillas, S. Zhou, J. Zhuang, L. Wang, X. Xu, S. X. Dou and Y. Du, Sustainable Energy Fuels, 2019, 3, 1757–1763 RSC.
- X. Du, Z. Yang, Y. Li, Y. Gong and M. Zhao, J. Mater. Chem. A, 2018, 6, 6938–6946 RSC.
- X. Du, W. Lian and X. Zhang, Int. J. Hydrogen Energy, 2018, 43, 20627–20635 CrossRef CAS.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- X. Yan, L. Tian, M. He and X. Chen, Nano Lett., 2015, 15, 6015–6021 Search PubMed.
- H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Nat. Commun., 2015, 6, 6430 CrossRef CAS PubMed.
- Q. Jin, B. Ren, D. Li, H. Cui and C. Wang, Nano Energy, 2018, 49, 14–22 CrossRef CAS.
- P. Ganesan, A. Sivanantham and S. Shanmugam, ACS Appl. Mater. Interfaces, 2017, 9, 12416–12426 CrossRef CAS PubMed.
- A. Moyseowicz, A. Śliwak, E. Miniach and G. Gryglewicz, Composites, Part B, 2017, 109, 23–29 CrossRef CAS.
- J. Tian, Q. Liu, Y. Liang, Z. Xing, A. M. Asiri and X. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 20579–20584 CrossRef CAS PubMed.
- N. Naseri, A. Esfandiar, M. Qorbani and A. Z. Moshfegh, ACS Sustainable Chem. Eng., 2016, 4, 3151–3159 CrossRef CAS.
- G. Zhang, S. Sun, D. Yang, J.-P. Dodelet and E. Sacher, Carbon, 2008, 46, 196–205 CrossRef CAS.
- U. Zielke, K. J. Hüttinger and W. P. Hoffman, Carbon, 1996, 34, 983–998 CrossRef CAS.
- P. Wiench, Z. González, R. Menéndez, B. Grzyb and G. Gryglewicz, Sens. Actuators, B, 2018, 257, 143–153 CrossRef CAS.
- K. Kordek, H. Yin, P. Rutkowski and H. Zhao, Int. J. Hydrogen Energy, 2019, 44, 23–33 CrossRef CAS.
- Y. Lv, A. Batool, Y. Wei, Q. Xin, R. Boddula, S. U. Jan, M. Z. Akram, L. Tian, B. Guo and J. R. Gong, ChemElectroChem, 2019, 6, 2497–2502 CrossRef CAS.
- N. S. K. Gowthaman, M. A. Raj and S. A. John, ACS Sustainable Chem. Eng., 2017, 5(2), 1648–1658 CrossRef CAS.
- J. Wu, P. Guo, R. Mi, X. Liu, H. Zhang, J. Mei, H. Liu, W.-M. Lau and L.-M. Liu, J. Mater. Chem. A, 2015, 3, 15331–15338 RSC.
- V. Kavimani, S. Prakash, R. Rajesh, D. Rammasamy, N. B. Selveraj, T. Yang, B. Prabakaran and S. Jothi, Appl. Surf. Sci., 2017, 424, 63–71 CrossRef CAS.
- A. Eckmann, A. Felten, A. Mishchenko, L. Britnell, R. Krupke, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 3925–3930 CrossRef CAS PubMed.
- J. Coates, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2006 Search PubMed.
- C. Fan and D. L. Piron, Electrochim. Acta, 1996, 41, 1713–1719 CrossRef CAS.
- K. Takada, T. Sasaki, Z. Liu, M. Osada and R. Ma, J. Am. Chem. Soc., 2005, 127, 13869–13874 CrossRef PubMed.
- J. Lai, S. Li, F. Wu, M. Saqib, R. Luque and G. Xu, Energy Environ. Sci., 2016, 9, 1210–1214 RSC.
- A. Karpuz, H. Kockar, M. Alper, O. Karaagac and M. Haciismailoglu, Appl. Surf. Sci., 2012, 258, 4005–4010 CrossRef CAS.
- A. Karpuz, H. Kockar and M. Alper, Appl. Surf. Sci., 2011, 257, 3632–3635 CrossRef CAS.
- L. Wang, Y. Gao, Q. Xue, H. Liu and T. Xu, Appl. Surf. Sci., 2005, 242, 326–332 CrossRef CAS.
- R. Muzyka, S. Drewniak, T. Pustelny, M. Chrubasik and G. Gryglewicz, Materials, 2018, 11, 1050 CrossRef PubMed.
- A. Adán-Más, R. G. Duarte, T. M. Silva, L. Guerlou-Demourgues and M. F. G. Montemor, Electrochim. Acta, 2017, 240, 323–340 CrossRef.
- M. A. Sayeed, T. Herd, A. P. O'Mullane, T. F. Jaramillo, M. Sastry, B. L. V. Prasad, Y. X. Tong, G. W. Yang and P. Strasser, J. Mater. Chem. A, 2016, 4, 991–999 RSC.
- A. Balram, H. Zhang and S. Santhanagopalan, ACS Appl. Mater. Interfaces, 2017, 9, 28355–28365 CrossRef CAS PubMed.
- S. Chen, J. Zhu, H. Zhou and X. Wang, RSC Adv., 2011, 1, 484 RSC.
- Z. Wu, X.-L. Huang, Z.-L. Wang, J.-J. Xu, H.-G. Wang and X.-B. Zhang, Sci. Rep., 2015, 4, 3669 CrossRef PubMed.
- S. Min, C. Zhao, Z. Zhang, G. Chen, X. Qian and Z. Guo, J. Mater. Chem. A, 2015, 3, 3641–3650 RSC.
- N. S. McIntyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS.
- H. Jiang, Y. Guo, T. Wang, P.-L. Zhu, S. Yu, Y. Yu, X.-Z. Fu, R. Sun and C.-P. Wong, RSC Adv., 2015, 5, 12931–12936 RSC.
- S. Wang, B. Y. Guan and X. W. David Lou, Energy Environ. Sci., 2018, 11, 306–310 RSC.
- J. Chen, J. Xu, S. Zhou, N. Zhao and C.-P. Wong, Nano Energy, 2016, 21, 145–153 CrossRef CAS.
- S. Oswald, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2013 Search PubMed.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- K. E. Ramohlola, G. R. Monana, M. J. Hato, K. D. Modibane, K. M. Molapo, M. Masikini, S. B. Mduli and E. I. Iwuoha, Composites, Part B, 2018, 137, 129–139 CrossRef CAS.
- R. Paul, L. Zhu, H. Chen, J. Qu and L. Dai, Adv. Mater., 2019, 1806403 CrossRef PubMed.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 1806296 CrossRef PubMed.
- A. S. Batchellor and S. W. Boettcher, ACS Catal., 2015, 5, 6680–6689 CrossRef CAS.
- Z. Yu, Z. Cheng, S. R. Majid, Z. Tai, X. Wang and S. Dou, Chem. Commun., 2015, 51, 1689–1692 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/c9se00391f |
| This journal is © The Royal Society of Chemistry 2020 |