
Homogeneous visible light-driven hydrogen evolution by the molecular molybdenum sulfide model [Mo2S12]2−†
Ashwene
Rajagopal
a,
Felix
Venter
a,
Timo
Jacob
bc,
Lydia
Petermann
a,
Sven
Rau
a,
Stefanie
Tschierlei
 a and
Carsten
Streb
*acd
a and
Carsten
Streb
*acd
aInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89091 Ulm, Germany. E-mail: carsten.streb@uni-ulm.de
bInstitute of Electrochemistry, Ulm University, Albert-Einstein-Allee 47, 89091 Ulm, Germany
cHelmholtz Institute Ulm for Electrochemical Energy Storage, 89081 Ulm, Germany
dKarlsruhe Institute of Technology, P. O. Box 3640, 76021 Karlsruhe, Germany
First published on 30th October 2018
Abstract
The visible light-driven hydrogen evolution reaction by the molecular molybdenum sulfide anion [Mo2S12]2− is reported. When coupled with the photosensitizer [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) and the electron/proton donor ascorbic acid, turnover numbers > 1600 and turnover frequencies > 700 h−1 are observed. Comparison with the prototype HER catalyst [Mo3S13]2− shows remarkable reactivity differences which are explored by initial mechanistic studies under reaction conditions.
The production of “solar hydrogen” by light-driven water splitting is a cornerstone of modern energy research.1 Over the past decades, significant research has gone into designing catalysts capable of performing the light-driven hydrogen evolution reaction (HER).2–5 While initially, noble metals were prime catalysts due to their high reactivity, to-date, the field is focused on earth-abundant catalysts, owing to their technological importance for large-scale deployment. One of the most promising HER catalyst classes are amorphous molybdenum sulfides (MoS2+x) which have been developed as high-performance (photo-)electrochemical solid-state HER catalysts.2–5 However, understanding the complex mechanisms which control reactivity and stability in these compounds is still limited, as their exact structure is often not known.6,7 Different mechanisms for the MoS2+x HER activity are currently discussed7 and reactive sites based on protonated disulfide ligands,8–11 molybdenum oxo groups12–14 or MoV-hydride species14–16 have been proposed.7
To address fundamental mechanistic questions, molecular molybdenum sulfides, so called thiomolybdates17 can be employed as molecular models to understand and correlate structure and reactivity in Mo–S HER catalysts.18–20 In addition, thiomolybdates are amongst the most active molecular HER catalysts,18 so that understanding their reactivity can form the basis for developing advanced homogeneous and heterogeneous Mo–S catalysts.8,11,12,20–22 Recent research has focused on the Müller-type thiomolybdate [Mo3S13]2− (={Mo3}, Fig. 1),17 which is of particular importance, as Artero, Tran et al. recently reported that amorphous molybdenum sulfides are composed of polymeric {Mo3} chains.14,23,24 For this and related prototypes, most early studies were focused on heterogenized systems, where the thiomolybdates were deposited on (photo-)electrodes, giving heterogeneous catalysts for electrocatalytic8,11,21,22,25,26 or photoelectrocatalytic27–29 HER.
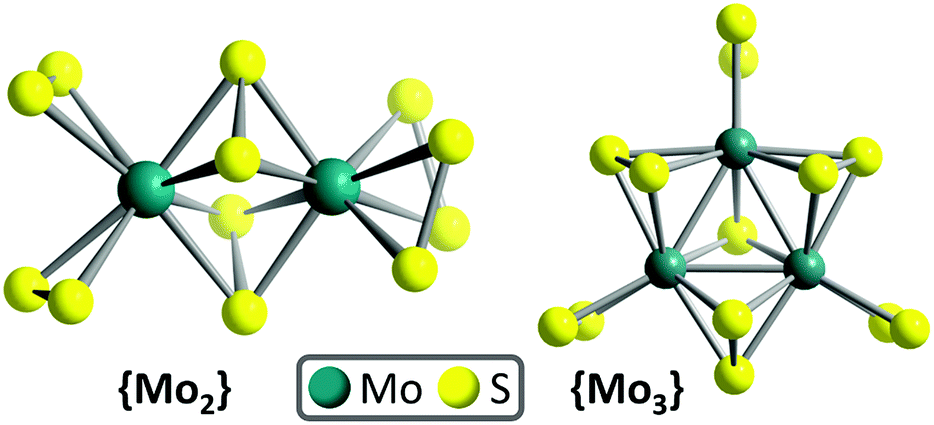 | ||
| Fig. 1 Molecular molybdenum sulfide HER catalysts: {Mo2} ([Mo2S12]2−) and {Mo3} ([Mo3S13]2−). | ||
In contrast, homogeneous, light driven HER catalysis for molecular molybdenum sulfides is essentially unexplored and was first reported in 2018:30,31 we have recently reported that {Mo3} is a highly efficient homogeneous light driven HER catalyst (TONs ∼ 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, TOFs > 150 min−1) and in solution shows highly dynamic ligand exchange behaviour on the minute-timescale, so that catalyst reactivity is controlled by the type of terminal ligand present. Specifically, we showed that partial exchange of the three terminal disulfides of {Mo3} with aquo ligands giving ([Mo3S13−x(H2O)x](2−x)−, x = 2, 4) leads to species with higher catalytic activity compared with the native {Mo3} (where x = 0). In contrast, complete exchange of all terminal disulfides (x = 6) leads to a species with decreased catalytic activity (see ESI, Fig. S4b†). Therefore, we hypothesized that thiomolybdates with different metal-sulfide coordination environments might show varied ligand exchange behaviour. This principle could in future be used to control HER catalytic reactivity.
000, TOFs > 150 min−1) and in solution shows highly dynamic ligand exchange behaviour on the minute-timescale, so that catalyst reactivity is controlled by the type of terminal ligand present. Specifically, we showed that partial exchange of the three terminal disulfides of {Mo3} with aquo ligands giving ([Mo3S13−x(H2O)x](2−x)−, x = 2, 4) leads to species with higher catalytic activity compared with the native {Mo3} (where x = 0). In contrast, complete exchange of all terminal disulfides (x = 6) leads to a species with decreased catalytic activity (see ESI, Fig. S4b†). Therefore, we hypothesized that thiomolybdates with different metal-sulfide coordination environments might show varied ligand exchange behaviour. This principle could in future be used to control HER catalytic reactivity.
Here, we report, for the first time, the homogeneous, visible light-driven HER activity of the thiomolybdate [Mo2S12]2− (={Mo2}, Fig. 1).8 We show that {Mo2} is an active HER catalyst and shows notable differences in reactivity compared with {Mo3} with respect to solvent composition and the presence of competing ligands. Initial mechanistic analyses provide insights into the underlying causes for this divergent reactivity. The findings could in future be used to design new homogeneous and heterogenized HER catalysts by controlled structure modification of molecular molybdenum sulfides.
Briefly, the main structural differences between {Mo2} and {Mo3} are the different metal oxidation states ({Mo2}: MoV, {Mo3}: MoIV). Further, {Mo2} features four disulfide ligands per Mo centre (2× terminal, 2× bridging), while {Mo3} features three disulfide ligands (1× terminal, 2× bridging) and a central (apical) sulfide ligand, see Fig. 1. The role of these ligands has recently been examined under electrochemical conditions by selective thermal removal of different ligands, leading to the suggestion that the bridging disulfide ligand is the HER reactive site.20
As {Mo2} model catalyst, we used (NH4)2[Mo2(S2)6]·2H2O which was synthesized by a modification of the original synthesis reported by Müller, Krebs et al.32 The compound was fully characterized by elemental analyses, spectroscopic, diffractometric and mass spectrometric techniques (see ESI, Fig. S1–S3†). Briefly, {Mo2} is the structurally simplest thiomolybdate featuring bridging and terminal disulfide ligands (Fig. 1). {Mo2} consists of two molybdenum MoV centres linked by two bridging disulfides (μ,η2–S22−). Each Mo centres features two terminal disulfide (η2–S22−) ligands, giving the overall formula [Mo2S12]2− = [Mo2(S2,bridging)2(S2,terminal)4]2− = {Mo2}.
Based on our initial HER studies with Mo3, we employed similar reaction conditions to assess the light-driven hydrogen evolution by {Mo2}: to this end, {Mo2} (0.5 μM) was combined with [Ru(bpy)3]2+ (20 μM, bpy = 2,2′-bipyridine) as photosensitizer and ascorbic acid (0.01 M) as sacrificial electron/proton donor in degassed solvents (MeOH, H2O or mixtures thereof, see below and ESI, Fig. S4a, S5 and S6†). The solution was irradiated at room temperature with a monochromatic LED light source (λmax = 470 nm, P ∼ 40 mW cm−2), H2 evolution was measured using headspace gas chromatography (each data point was recorded in triplicate). All experiments used an established colloid detection procedure using microfiltration, dynamic light scattering (DLS) and UV-vis spectroscopy.33 No colloids were detected in the experiments reported (see ESI, Section 5.1 and 5.2, Fig. S10 and S11†). From recent experiments with {Mo3} we knew that thiomolybdate HER activity is highly solvent-dependent.31 To this end, we explored the homogeneous HER activity of {Mo2} in MeOH:H2O mixtures containing increasing amounts of H2O. As shown in Fig. 2a, light-driven hydrogen evolution is observed for all solvent compositions tested. However, significantly higher reactivity is observed at high MeOH concentrations. In consequence, after tirradiation = 6 h, we observed TONs of ∼75 for aqueous solutions, whereas TONs of ∼1630 are noted in pure MeOH, giving initial TOFs ∼ 720 h−1 (Fig. 2d). Note that under electrochemical conditions, {Mo2} shows significantly higher rates with TOFs as high as ∼12000 h−1,8 thus the rate-limiting steps might be associated with photosensitizer–catalyst interactions. Chemical linkage of both species could therefore be one promising approach to optimize reactivity.34,35
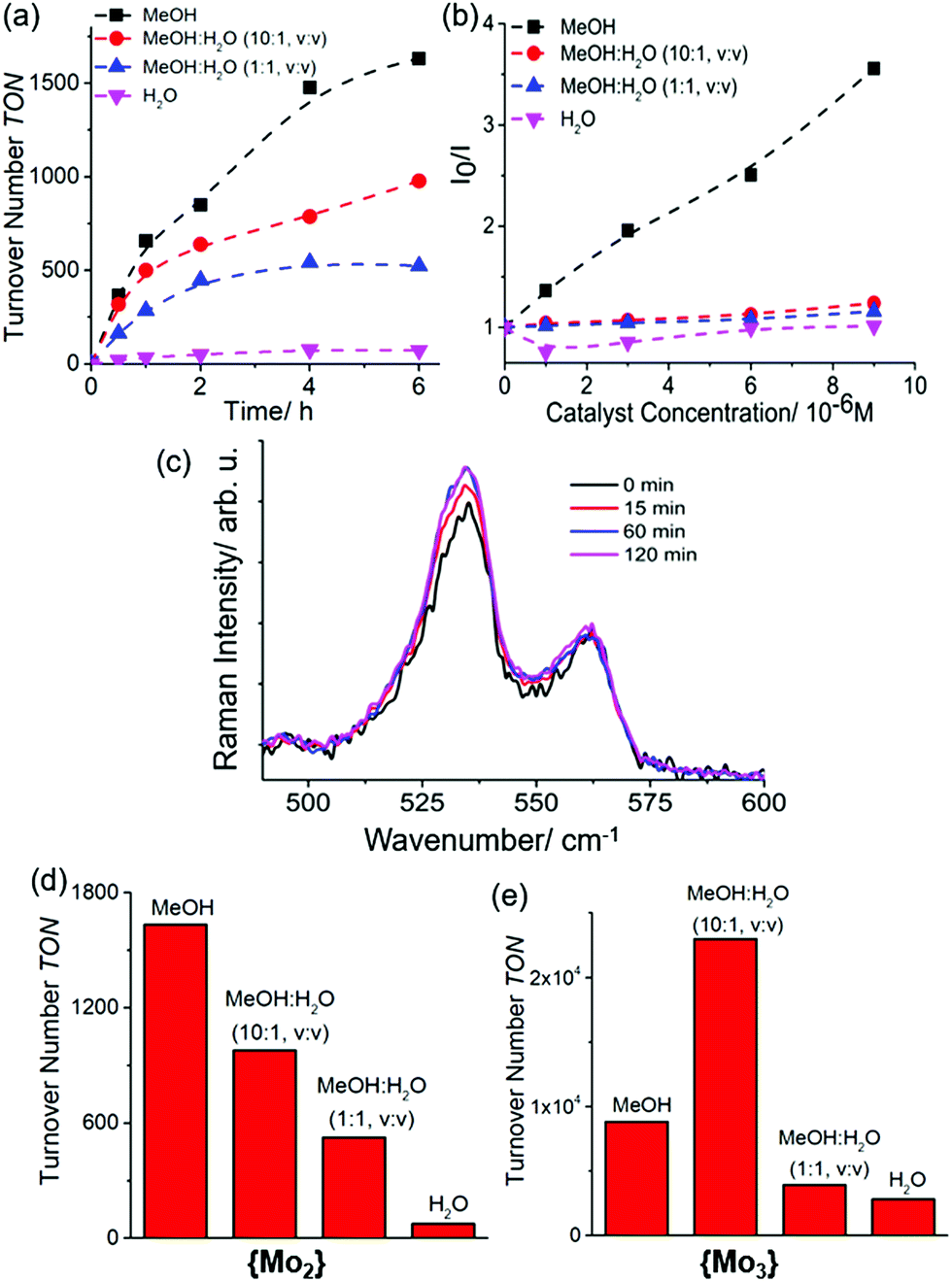 | ||
| Fig. 2 (a) Turnover number (TON) for the {Mo2}-catalyzed HER activity at different MeOH:H2O solvent ratios. Conditions: [catalyst] = 0.5 μM, [[Ru(bpy)3]2+] = 20 μM, [ascorbic acid] = 0.01 M. (b) Stern–Volmer plot (emission quenching) of the photosensitizer by {Mo2} in different MeOH:H2O mixtures. Conditions: [catalyst] = 0–10 μM, [[Ru(bpy)3]2+] = 20 μM. (c) Raman spectra of {Mo2} (excitation wavelength λexcitation = 785 nm, normalized to the solvent band at 650 cm−1, no significant changes of the disulfide Raman bands under irradiation over a period of 2 h. Conditions: solvent: DMF:H2O (1:1, v/v), [{Mo2}] = 2 mM; λirradiation = 470 nm (LED). (d) and (e) bar graphs showing the observed TONs for {Mo2} and {Mo3} at different MeOH:H2O ratios. | ||
Similar solvent-dependent reactivity has previously been observed for {Mo3} (Fig. 2e), however, while {Mo3} showed the highest TON-based reactivity in the presence of small amounts of water (to allow partial disulfide ligand exchange, see above and ESI, Fig. S4b,† we note that for {Mo2}, highest activity is observed in water-free MeOH. To further explore the solvent influence on the photosensitizer–catalyst interactions, we undertook emission quenching studies. As shown in the Stern–Volmer plot (Fig. 2b), different MeOH:H2O ratios significantly affect the [Ru(bpy)3]2+ quenching by {Mo2}. As a general trend, at higher H2O concentrations, quenching is less pronounced which is in line with the observed decrease in HER activity at increasing H2O concentrations. One reason for the decrease in quenching could be the formation of a hydration shell around the quenching partners when water is present. This could prevent efficient static or dynamic quenching processes.38 For {Mo3}, the quenching behaviour also correlates with the catalytic activity, i.e. the highest quenching is noted under the conditions where the highest TON-based reactivity is observed (ESI, Fig. S4b†). This initial data therefore suggests that photosensitizer–catalyst interactions play a crucial role in controlling homogenous light-driven HER activity for {Mo2} and {Mo3}.
To assess whether {Mo2} shows light-driven HER activity in combination with different PS, we used the literature-known PS [Ru(tbbpy)2(mmip)]3+ (see ESI Fig. S12† for structural details) as comparison. The compound features photophysical and electrochemical properties similar to [Ru(bpy)3]2+, undergoes reductive quenching by ascorbic acid and is sterically more shielded, so that different interactions with the catalyst can be explored.36,37 Stern–Volmer quenching studies showed significant interactions between this PS and {Mo2} (see ESI, Fig. S13†) under identical conditions as described for [Ru(bpy)3]2+ in Fig. 2b. Catalytic studies under the standard conditions described above (using 10:1, v/v MeOH:H2O as solvent) showed HER activity and gave lower overall TONs (∼550; −40%) compared with [Ru(bpy)3]2+ (see ESI, Fig. S14†).
In addition to photosensitizer–catalyst interactions, structural changes (e.g. ligand exchange) of the catalyst have to be considered as a reason for decreasing catalytic activity. An initial analysis of the deactivation kinetics of {Mo2} (see Fig. 2a and ESI, Fig. S4a†) shows that TOF changes are on the timescale of hours. In contrast, for {Mo3}, the loss of catalytic reactivity (based on TOF changes) was observed on the minute timescale (see ESI, Fig. S4b†).18 For {Mo3}, exchange of the terminal disulfide ligands was proposed as catalyst deactivation mechanism.18 Based on these findings, we hypothesized that a similar process proceeding with slower kinetics could be active for {Mo2} also. To this end, we explored the stability of the native {Mo2} in solution using Raman spectroscopy. Under visible light irradiation in mixed DMF:H2O (1:1, v/v), we observed that the characteristic bridging and terminal disulfide vibrations remain virtually unchanged over a period of several hours (Fig. 2c). Only after prolonged irradiation (>12 h), changes of these characteristic signals are observed which could indicate structural changes of {Mo2} (isomerization has been proposed as one possibility in the literature).39
To substantiate the slow ligand exchange of {Mo2}, we performed catalyst deactivation studies in the presence of the coordinating ligands such as chloride and 2,2′-bipyridine (bpy), which are prototype competing ligands present under typical reaction conditions.18 When the standard {Mo2} HER catalysis (as described above) was carried out in the presence of small concentrations of tetra-n-butyl ammonium chloride (nBu4NCl) or bpy, (20 μM), no significant TON change is observed (see ESI, Fig. S8.† This is different compared to the identical experiments for {Mo3} where significantly reduced HER TONs were observed, see ESI, Fig. S8.† When increasing the concentration of chloride or bpy to 2 mM, we note a decrease in HER reactivity for {Mo2} see Fig. 3a. Notably, the HER reactivity decrease proceeds faster for the chloride-containing solutions, while for the bpy-containing solution, the deactivation process is only notable after several hours of irradiation (Fig. 3a), indicating slower deactivation kinetics in case of bpy. Initial Stern–Volmer analyses (Fig. 3b) show that in the presence of bpy or chloride, emission quenching of the photosensitizer by {Mo2} is slightly less efficient, which could be one contributing factor to the reduced HER activity. To provide further evidence for the disulfide/bpy/Cl− exchange in {Mo2} we performed time-dependent UV-vis spectroscopy under typical HER conditions. The data show that significant changes of the spectral features are observed upon irradiation in the presence of bpy and chloride. The changes occur on the timescale of hours, highlighting that ligand exchange in {Mo2} proceeds slower compared to {Mo3} where ligand exchange and deactivation was observed on the minute timescale (see ESI, Fig. S7a and b†).
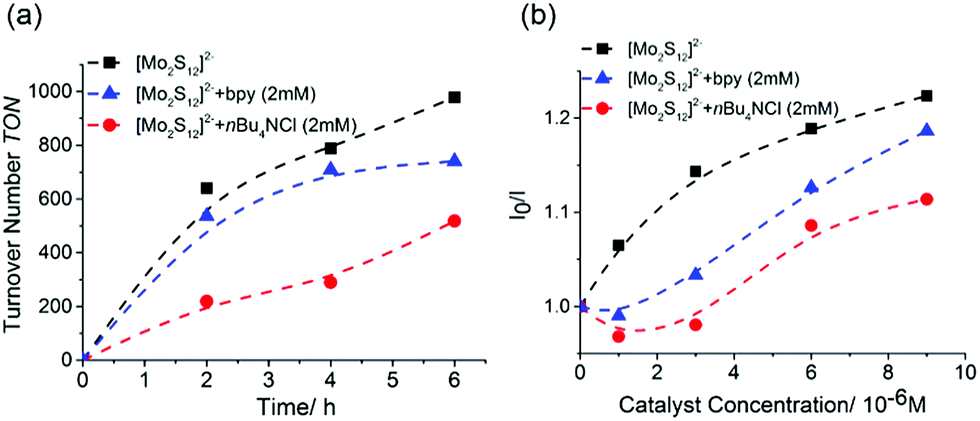 | ||
| Fig. 3 (a) Decreased HER activity of {Mo2} in the presence of nBu4NCl and 2,2′-bipyridine (bpy). Conditions: [bpy; Cl−] = 2 mM; MeOH:H2O (10:1, v/v), [{Mo2}] = 0.5 μM, [[(Ru(bpy)3]2+] = 20 μM, [ascorbic acid] = 0.01 M. (b) Stern–Volmer plot (emission quenching) of the photosensitizer by {Mo2} in the presence of nBu4NCl and 2,2′-bipyridine (bpy). Conditions: [bpy; Cl−] = 2 mM; solvent: MeOH:H2O (10:1, v/v), [catalyst] = 0–10 μM, [[(Ru(bpy)3]2+] = 20 μM. Samples were equilibrated for 2 h after mixing to allow for initial ligand exchange. | ||
Finally, we explored how degradation of the photosensitizer affects the HER activity of the system. To this end, the standard HER catalysis (in MeOH:H2O (10:1, v/v)) was performed (tirradiation = 6 h; TON ∼ 900, see ESI Fig. S9b†). After this period, UV-Vis spectroscopy of the reaction solution indicated significant degradation of the photosensitizer (see ESI, Fig. S9a†). An additional aliquot of [Ru(bpy)3]2+ (20 μM) was then added under inert atmosphere and the sample was irradiated again for 6 h. This procedure led to sustained hydrogen evolution, giving a total TON of ∼1750 at tirradiation = 12 h. This initial experiment therefore shows that degradation of the photosensitizer is a significant factor affecting the loss of catalytic reactivity of the system, so that the coupling of more stable light-absorbers with {Mo2} could lead to significantly longer operating times.40
In summary, we provide the first example of visible light driven, homogeneous hydrogen evolution reactivity of the smallest thiomolybdate cluster, {Mo2}. We report high reactivity and notable reactivity differences when compared with the recently studied model system {Mo3}. Exchange of the terminal disulfide ligands, which is a dominant and fast deactivation pathway for {Mo3} (deactivation on the minute timescale) is less pronounced for {Mo2}, where deactivation occurs on the timescale of several hours. This study therefore opens new pathways to explore reactivity control and deactivation mechanisms in molecular molybdenum sulfides as a means of rationalizing and tuning the reactivity of molecular and solid-state molybdenum sulfide HER catalysts. Future works will use more detailed in operando mechanistic studies and theoretical analyses to rationalize thiomolybdate reactivity under homogeneous conditions and when heterogenized on solid substrates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ulm University and the Helmholtz Gemeinschaft (HGF) are gratefully acknowledged for financial support. The Deutsche Forschungsgemeinschaft is gratefully acknowledged for financial support (TRR234 “CataLight”, Projects A4 and A5 (C. S., T. J., S. R.); ST330/3-1 (S. T.)).Notes and references
- R. Schlögl, Angew. Chem., Int. Ed., 2011, 50, 6424–6426 CrossRef PubMed.
- C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671–2681 CrossRef CAS PubMed.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878 RSC.
- T. Wang, H. Xie, M. Chen, A. D'Aloia, J. Cho, G. Wu and Q. Li, Nano Energy, 2017, 42, 69–89 CrossRef CAS.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- B. Seo and S. H. Joo, Nano Convergence, 2017, 4, 19 CrossRef PubMed.
- M.-L. Grutza, A. Rajagopal, C. Streb and P. Kurz, Sustainable Energy Fuels, 2018, 2, 1893–1904 RSC.
- Z. Huang, W. Luo, L. Ma, M. Yu, X. Ren, M. He, S. Polen, K. Click, B. Garrett, J. Lu, K. Amine, C. Hadad, W. Chen, A. Asthagiri and Y. Wu, Angew. Chem., Int. Ed., 2015, 54, 15181–15185 CrossRef CAS PubMed.
- L. R. L. Ting, Y. Deng, L. Ma, Y.-J. Zhang, A. A. Peterson and B. S. Yeo, ACS Catal., 2016, 6, 861–867 CrossRef CAS.
- B. Lassalle-Kaiser, D. Merki, H. Vrubel, S. Gul, V. K. Yachandra, X. Hu and J. Yano, J. Am. Chem. Soc., 2015, 137, 314–321 CrossRef CAS PubMed.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS PubMed.
- B. R. Garrett, K. A. Click, C. B. Durr, C. M. Hadad and Y. Wu, J. Am. Chem. Soc., 2016, 138, 13726–13731 CrossRef CAS PubMed.
- B. R. Garrett, S. M. Polen, K. A. Click, M. He, Z. Huang, C. M. Hadad and Y. Wu, Inorg. Chem., 2016, 55, 3960–3966 CrossRef CAS PubMed.
- P. D. Tran, T. V. Tran, M. Orio, S. Torelli, Q. D. Truong, K. Nayuki, Y. Sasaki, S. Y. Chiam, R. Yi, I. Honma, J. Barber and V. Artero, Nat. Mater., 2016, 15, 640–646 CrossRef CAS PubMed.
- Y. Huang, R. J. Nielsen, W. A. Goddard and M. P. Soriaga, J. Am. Chem. Soc., 2015, 137, 6692–6698 CrossRef CAS PubMed.
- A. Hijazi, J. C. Kemmegne-Mbouguen, S. Floquet, J. Marrot, J. Fize, V. Artero, O. David, E. Magnier, B. Pégot and E. Cadot, Dalton Trans., 2013, 42, 4848–4858 RSC.
- A. Müller, E. Diemann, R. Jostes and H. Bögge, Angew. Chem., Int. Ed. Engl., 1981, 20, 934–955 CrossRef.
- M. Dave, A. Rajagopal, M. Damm-Ruttensperger, B. Schwarz, F. Naegele, L. Daccache, D. Fantauzzi, T. Jacob and C. Streb, Sustainable Energy Fuels, 2018, 2, 1020–1026 RSC.
- Y. Lei, M. Yang, J. Hou, F. Wang, E. Cui, C. Kong and S. Min, Chem. Commun., 2018, 54, 603–606 RSC.
- C.-H. Lee, S. Lee, Y.-K. Lee, Y. C. Jung, Y.-I. Ko, D. C. Lee and H.-I. Joh, ACS Catal., 2018, 8, 5221–5227 CrossRef CAS.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed.
- M. Kokko, F. Bayerköhler, J. Erben, R. Zengerle, P. Kurz and S. Kerzenmacher, Appl. Energy, 2017, 190, 1221–1233 CrossRef CAS.
- T. Weber, J. C. Muijsers and J. W. Niemantsverdriet, J. Biophys. Chem., 1995, 99, 9194–9200 CAS.
- Y. Deng, L. Rui, L. Ting, P. Hui, L. Neo, Y. Zhang, A. A. Peterson and B. S. Yeo, ACS Catal., 2016, 6, 7790 CrossRef CAS.
- Y. Shang, X. Xu, B. Gao and Z. Ren, ACS Sustainable Chem. Eng., 2017, 5(10), 8908–8917 CrossRef CAS.
- V. Artero and J.-M. Saveant, Energy Environ. Sci., 2014, 7, 3808–3814 RSC.
- D. Recatalá, R. Llusar, A. L. Gushchin, E. A. Kozlova, Y. A. Laricheva, P. A. Abramov, M. N. Sokolov, R. Gómez and T. Lana-Villarreal, ChemSusChem, 2015, 8, 148–157 CrossRef PubMed.
- M. Kan, J. Jia and Y. Zhao, RSC Adv., 2016, 6, 15610–15614 RSC.
- K. Du, L. Zheng, T. Wang, J. Zhuo, Z. Zhu, Y. Shao and M. Li, ACS Appl. Mater. Interfaces, 2017, 9, 18675–18681 CrossRef CAS PubMed.
- Y. Lei, M. Yang, J. Hou, F. Wang, E. Cui, C. Kong and S. Min, Chem. Commun., 2018, 54, 603–606 RSC.
- M. Dave, A. Rajagopal, M. Damm-Ruttensperger, B. Schwarz, F. Naegele, L. Daccache, D. Fantauzzi, T. Jacob and C. Streb, Sustainable Energy Fuels, 2018, 2, 1020–1026 RSC.
- A. Müller, W.-O. Nolte and B. Krebs, Angew. Chem., 1978, 90, 286–287 CrossRef.
- B. Kirchhoff, S. Rau and C. Streb, Eur. J. Inorg. Chem., 2016, 2016, 1425–1429 CrossRef CAS.
- S. Schönweiz, S. A. Rommel, J. Kübel, M. Micheel, B. Dietzek, S. Rau and C. Streb, Chem. Eur. J., 2016, 22, 12002–12005 CrossRef PubMed.
- S. Schönweiz, M. Heiland, M. Anjass, T. Jacob, S. Rau and C. Streb, Chem. Eur. J., 2017, 23, 15370–15376 CrossRef PubMed.
- R. Staehle, C. Reichardt, J. Popp, D. Sorsche, L. Petermann, K. Kastner, C. Streb, B. Dietzek and S. Rau, Eur. J. Inorg. Chem., 2015, 2015, 3932–3939 CrossRef CAS.
- L. Petermann, R. Staehle, T. D. Pilz, D. Sorsche, H. Görls and S. Rau, Eur. J. Inorg. Chem., 2015, 2015, 750–762 CrossRef CAS.
- D. Krenske, S. Abdo, H. Van Damme, M. Cruz and J. J. Fripiat, J. Phys. Chem., 1980, 84, 2447–2457 CrossRef CAS.
- A. Müller and E. Krickemeyer, in Inorganic Syntheses, ed. A. P. Ginsberg, New York, 1990, pp. 47–51 Search PubMed.
- L. Petermann, R. Staehle, M. Pfeifer, C. Reichardt, D. Sorsche, M. Wächtler, J. Popp, B. Dietzek and S. Rau, Chem.–Eur. J., 2016, 22, 8240–8253 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical and catalytic details are provided. See DOI: 10.1039/c8se00346g |
| This journal is © The Royal Society of Chemistry 2019 |