
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScreening of metal ions and organocatalysts on solid support-coupled DNA oligonucleotides guides design of DNA-encoded reactions†
Marco
Potowski
,
Florian
Losch
,
Elena
Wünnemann
,
Janina K.
Dahmen
,
Silvia
Chines
and
Andreas
Brunschweiger
 *
*
Department of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn-Str. 6, 44227 Dortmund, Germany. E-mail: andreas.brunschweiger@tu-dortmund.de
First published on 1st November 2019
Abstract
DNA-encoded compound libraries are a widely used technology for target-based small molecule screening. Generally, these libraries are synthesized by solution phase combinatorial chemistry requiring aqueous solvent mixtures and reactions that are orthogonal to DNA reactivity. Initiating library synthesis with readily available controlled pore glass-coupled DNA barcodes benefits from enhanced DNA stability due to nucleobase protection and choice of dry organic solvents for encoded compound synthesis. We screened the compatibility of solid-phase coupled DNA sequences with 53 metal salts and organic reagents. This screening experiment suggests design of encoded library synthesis. Here, we show the reaction optimization and scope of three sp3-bond containing heterocyclic scaffolds synthesized on controlled pore glass-connected DNA sequences. A ZnCl2-promoted aza-Diels–Alder reaction with Danishefsky's diene furnished diverse substituted DNA-tagged pyridones, and a phosphoric acid organocatalyst allowed for synthesis of tetrahydroquinolines by the Povarov reaction and pyrimidinones by the Biginelli reaction, respectively. These three reactions caused low levels of DNA depurination and cover broad and only partially overlapping chemical space though using one set of DNA-coupled starting materials.
Introduction
DNA-encoded combinatorial chemistry is today a widely applied technology for target-based small molecule screening.1–7 It relies on the concept of phenotype–genotype coupling. The genotype enables combining large numbers of molecules with complex mixtures and screening them mostly on protein targets for identification of protein binders that can later be evolved in drug development projects (Fig. 1A). The most common approach to these DNA-encoded libraries (DELs) initiates encoded compound synthesis with a short hairpin-like DNA duplex and build up the barcoded molecule through combinatorial cycles of preparative organic compound synthesis and enzymatic barcode concatenation.8 Chemical methods for compound synthesis are restricted by the chemical reactivity of DNA.9,10 In particular low pH leads to DNA degradation by depurination.11 In addition, the high redox potential of many metal salts may lead to formation of reactive oxygen species which can result in degradation of DNA.12 A variety of metal-mediated DNA-damage instances such as nucleobase oxidation and double strand breaks have been observed upon incubation with several metal ions like copper13 or palladium ions.14 In particular, the purine base guanine is susceptible to formation of mutagenic 8-oxo-G due to its lower oxidation potential.15 Furthermore, the presence of water as a co-solvent in solution phase-based DEL synthesis precludes adaption of many reactions to the encoded format. The latter demand may be bypassed through immobilization of DNA on charged surfaces and exchange of the solvent.16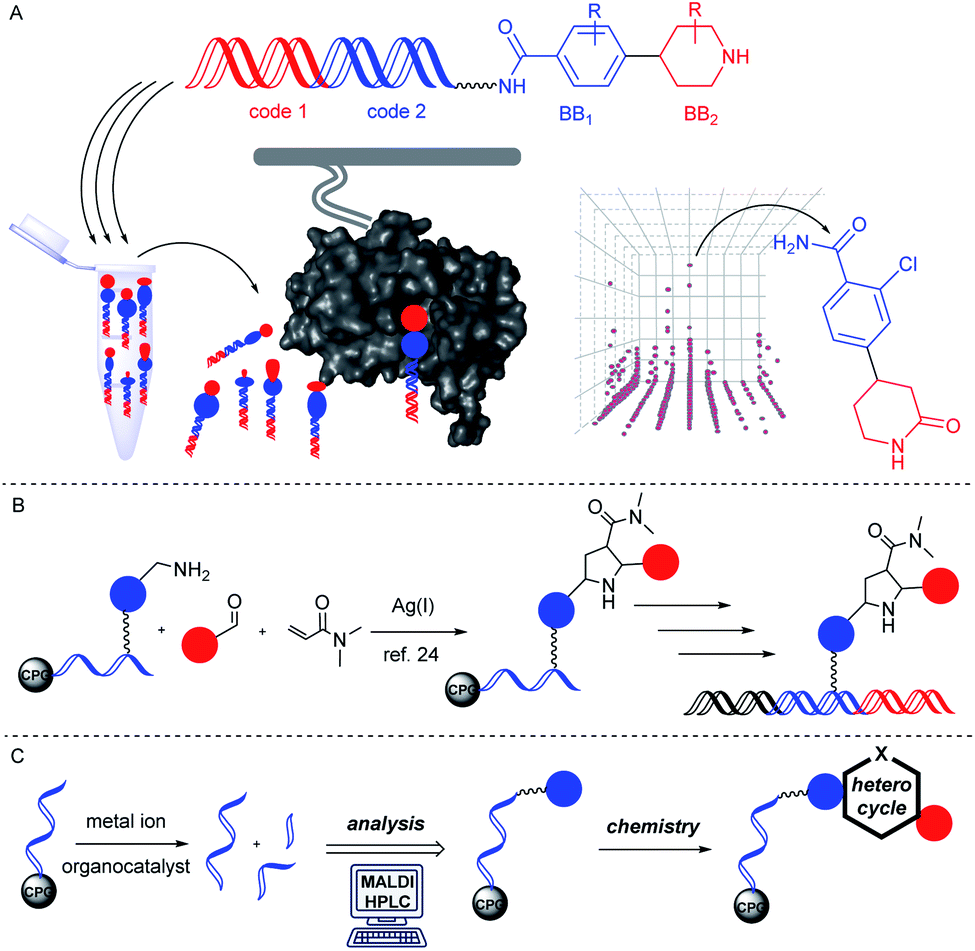 | ||
| Fig. 1 Solid phase-initiated access to DNA-encoded compound libraries. (A) Encoded chemistry provides DNA-barcode tagged small molecules that are concatenated to building blocks (BBs). These can be screened on protein targets by selection. (B) A solid phase-based encoding strategy allowed for synthesis of pyrrolidines by Ag(I)-mediated cycloaddition. (C) Screening of metal ions and organic reagents on DNA sequences suggests selection of preparative organic reactions for encoded compound synthesis. | ||
Alternatively, DNA-encoded compound synthesis can be initiated with the product of DNA oligonucleotide synthesis covalently coupled on a controlled pore glass (CPG) solid phase (Fig. 1B). This strategy follows the well-established concept of “post-synthetic” DNA modification, for instance by the Sonogashira reaction and others.17–19 It benefits from the choice of common (dry) organic solvents and the option of removing contaminants such as an excess of metal ions or reactants by washing steps.
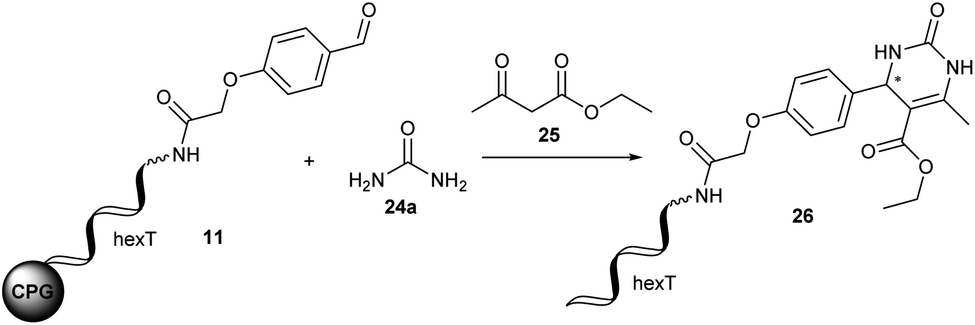
Furthermore, the CPG-coupled DNA carries nucleobase protecting groups. They safe-guard DNA against chemical introduction of mutations by nucleobase deamination and plausibly enhance its stability against depurination. Indeed, a few accounts demonstrated DEL synthesis initiated on a CPG solid phase and one of these showed an enlarged scope of amino acid starting materials enabled by facile removal of Boc protecting groups with TFA.20–23 We recently reported that CPG-coupled DNA barcodes tolerated Ag(I)-mediated (2 + 3) azomethine ylide cycloadditions, the Yb(III)-mediated Castagnoli–Cushman reaction,24 and isocyanide multicomponent reactions.25 Harsher reaction conditions such as the trifluoroacetic acid-mediated Pictet–Spengler reaction and Au(I)-mediated pyrazole synthesis were compatible with a chemically very stable, CPG-coupled hexathymidine adapter “hexT”.26 These reports prompted us to compare the stability of solid phase-coupled hexT, pyrimidine DNA and DNA barcodes to a larger number of reagents. They comprised various metal ions commonly used as catalysts in organic chemistry, organocatalysts, and oxidants. The findings of this study then served to guide encoded compound synthesis (Fig. 1C). To our surprise, the DNA barcode tolerated high concentrations of most metal ions at room temperature and a phosphoric acid-based organocatalyst well. Heterocyclic molecules are overrepresented in the chemical space of bioactive molecules, natural products, and drugs.27–32 In fact, 60% of all approved drugs contain a nitrogenous heterocycle.33 Thus, they are essential structures in drug development and indispensable in any screening library. In line with our aim to populate the chemical space of DNA-encoded libraries with diverse substituted heterocyclic scaffolds, we show here how catalyst screening on DNA guides the translation of three heterocycle-forming reactions to a solid phase-coupled DNA barcode. These were a ZnCl2-promoted aza-Diels–Alder reaction with Danishefsky's diene to pyridones and BINOL phosphoric acid-mediated Povarov and Biginelli reactions, respectively. All of them form heterocycles with sp3-connected substituents, a feature underrepresented in most encoded library synthesis schemes. Exemplary bioactive compounds 1–6 that were accessed through these reactions are given in Fig. 2.
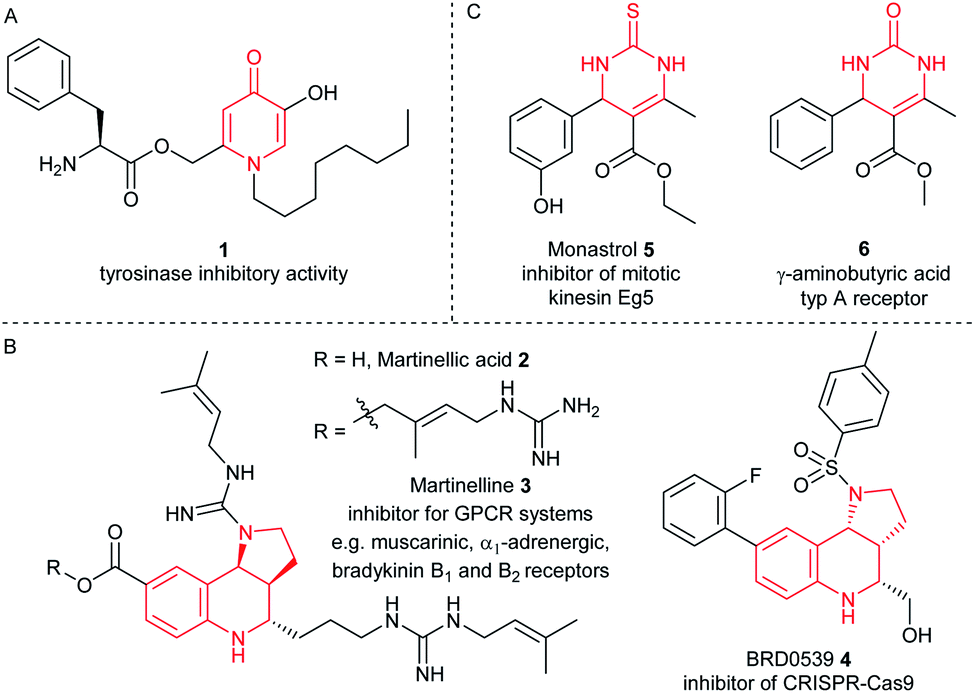 | ||
| Fig. 2 Representative bioactive compounds based on (A) pyridinone, (B) pyrrolotetrahydroquinoline, or (C) dihydropyrimidinone scaffolds. | ||
Results and discussion
Investigations into the stability of solid phase coupled oligonucleotides charged with metal ion salts and organocatalysts
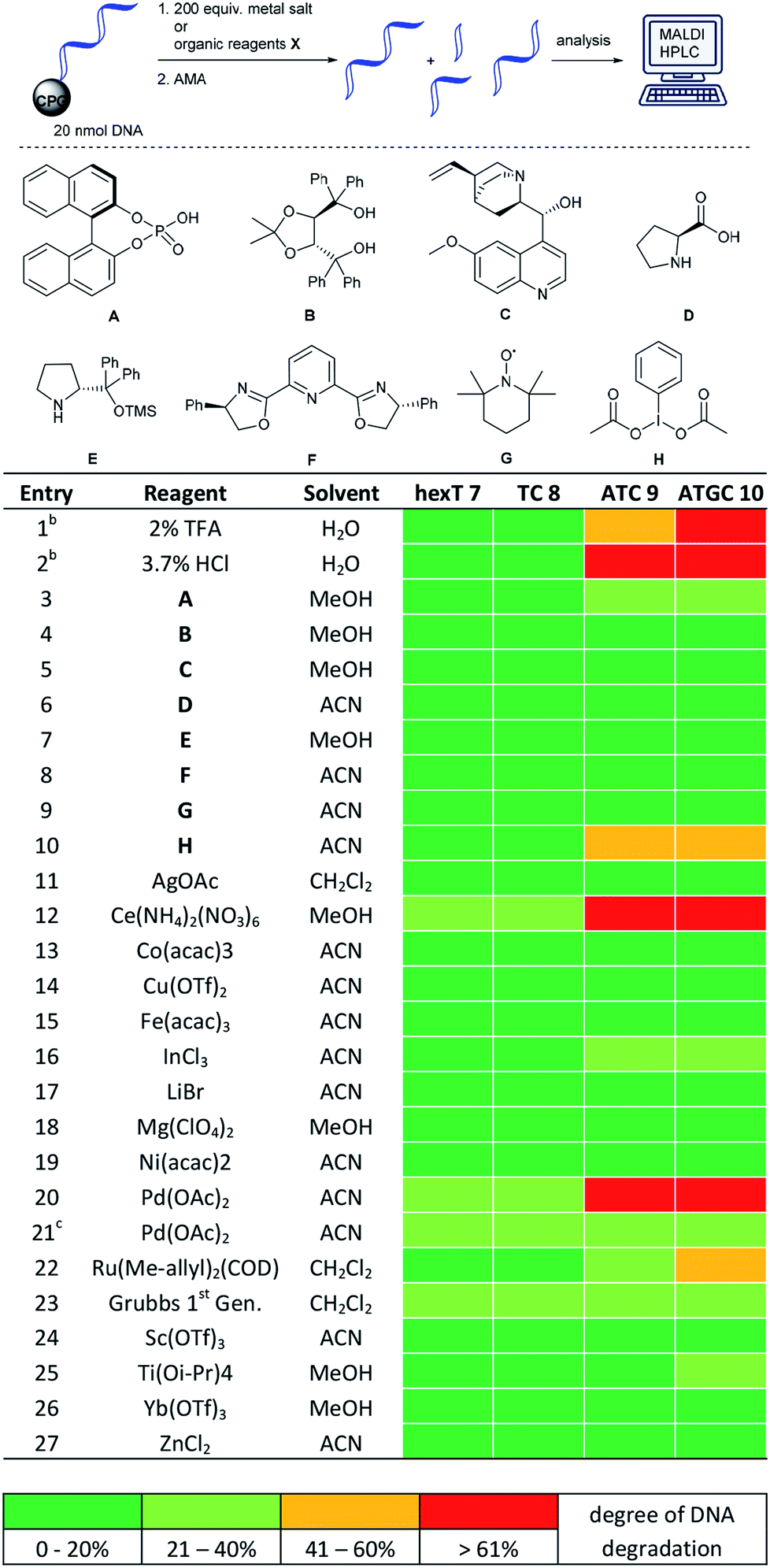
A prime concern in the selection of synthesis methods for encoded library synthesis is the stability of DNA to reagents and reaction conditions. An investigation into the stability of DNA in aqueous solutions of metal ions and organic reagents has been conducted earlier.34 DNA integrity was analysed by PCR which, as the authors mentioned, may not have assessed the extent of DNA degradation exactly. Taq polymerase reads through DNA lesions such as single depurination sites,35 8-oxopurines35 and deaminated nucleobases.36 These lesions introduce mutations into the compound barcode. We were interested in gaining a detailed picture of the stability of CPG-coupled DNA against commonly used catalysts to guide library synthesis design. Brief details of these experiments are given in Table 1, and a full overview of these experiments can be found in Tables S1–S4 in the ESI.† Our chemically very stable adapter oligonucleotide hexT 7 and three different decamer DNA oligonucleotides (TC 8, ATC 9, and ATGC 10) were incubated with three aqueous acids, as well as 33 metal salts and 20 organocatalysts dissolved in dry organic solvents. To reflect plausible reaction conditions,24,25 the catalysts were incubated with DNA strands at a 200-fold reagent excess for 22 hours at ambient temperature. After washing steps and standard DNA cleavage from solid support we quantitated and characterized DNA damage by RP-HPLC and MALDI-TOF mass spectrometric analysis. We expected higher degrees of DNA degradation for the purine-containing sequences since depurination is one of the most important causes of DNA degradation. In contrast, pyrimidine DNA should readily tolerate most if not all applied reagents.| a For each: 20 nmol DNA, 200 equiv. (transition) metal salt or organocatalyst A–H, 50 μL dry solvent, r.t., 22 h. b 50 μL of the indicated aqueous acid. c 5 equiv. of metal salt were used. hexT 7 = 5′-AcN–(CH2)6–TTT TTT-3′-CPG, TC 8 = 5′-TTC CTC TCC T-3′-CPG, ATC 9 = 5′-TTA CTA CCT A-3′-CPG, ATGC 10 = 5′-GTC ATG ATC T-3′-CPG, ACN = acetonitrile, MeOH = methanol. |
|---|
|
Indeed, in all experiments, including those with aqueous protic acids, we could observe almost no DNA damage for both hexT- and 10mer TC-sequences 7 and 8. Only Ce(III), Pd(II), Rh(II) and Ru(II) salts caused a still tolerable degradation between 20 and 40% (Table 1, entries 12, 20, and 22 and Table S1†).
The same experiments performed on ATC- and ATGC-sequences 9 and 10 yielded a different outcome. Most organocatalysts were well tolerated by both DNA strands. For instance, they were only degraded to 20–40% by 200 equiv. of the phosphoric acid organocatalyst A (Table 1, entry 3). However, we observed massive DNA degradation upon prolonged incubation even with low, 2% and 3.7%, concentrations of the strong protic acids TFA and HCl (Table 1, entries 1 and 2). Higher degrees of DNA degradation were caused by a hypervalent iodine oxidant H, too (Table 1, entry 10). In the case of metal ions we observed that both DNA sequences were hardly damaged by Lewis acids such as Bi(III), In(III), Li(I), Mg(II), Sb(III), Sc(III), Ti(IV), and Yb(III) salts (Table 1, entries 16–18 and 24–26 and Table S1†). The latter result is in line with an earlier observation from our group.24 The transition metal ions Ag(I),24 Co(II), Cu(II), Fe(III), Ni(II), and Zn(II) were well tolerated, too (Table 1, entries 11, 13–15, 19, and 27 and Table S1†).
However, both DNA strands suffered higher levels of DNA degradation upon incubation with Ce(III), an ion with a high redox potential. Rh(II), Ru(II), Pd(0) and Pd(II) led to higher degrees of degradation, too (Table 1, entries 12, 20, and 22 and Table S1†). Mass spectrometric analysis of these experiments shows extensive depurination and fragmentation of DNA at the depurination sites due to β- or δ-elimination during DNA deprotection and cleavage. A representative mass spectrometric analysis of DNA degradation products can be found in the ESI (Fig. S1†). For instance, Pd(0) and Pd(II) caused colouring of the solid support. (Fig. S2†). In particular, Pd(0) led to blackening of the CPG. Therefore, we cannot exclude the fact that DNA degradation occurred during DNA cleavage with an aqueous ammonia/methylamine solution. A reduced five-fold excess of Pd(II) avoided colouring of the solid support and caused much lower degrees of DNA degradation (Table 1, entry 21 and Table S1†).
We performed a number of metal screening experiments on ATC 9 and ATGC 10 at 40 °C in dry organic solvents to evaluate the impact of higher temperatures on DNA stability (Table S2†). Both DNA sequences were stable or only slightly damaged under these conditions by In(III), SeO2, Cu(II) and Yb(III) (Table S2,† entries 1–4). However, they were heavily damaged by Sc(III) (Table S2,† entry 5). In a third series of experiments we assessed the impact of non-dry benchtop solvents, i.e. trace water, on potential metal-mediated DNA degradation at ambient temperature (Table S3†). The presence of trace amounts of water did not impact DNA degradation levels.
With this catalyst tolerance profile of solid phase-coupled DNA in hand, we investigated three heterocycle-forming reactions for translation to the DNA barcoding format: a ZnCl2-promoted aza-Diels–Alder reaction, a (R)-(−)-BNDHP-mediated Povarov reaction, and a (R)-(−)-BNDHP-mediated Biginelli reaction.
Development of a ZnCl2-mediated aza-Diels–Alder reaction on CPG-coupled DNA oligonucleotides
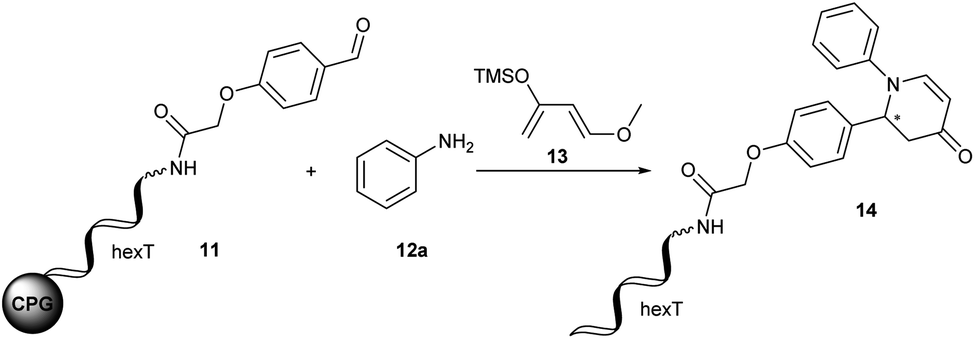
The Diels–Alder reaction is one of the most frequently used methods for construction of six-membered, partially saturated ring structures in preparative organic chemistry.37–39 It has previously been employed in the synthesis of encoded libraries.40 A heterocycle-forming variant is the aza-Diels–Alder reaction of imines with Danishefsky's diene41,42 which has been used as a key step in the synthesis of e.g. natural product-inspired libraries and in diversity-oriented synthesis.43,44 It gives rise to diverse substituted pyridinones from a broad scope of aromatic aldehydes and amines. The core heterocycle projects one substituent out of the plane, thus providing three-dimensionality. This can be found in several bioactive compounds.43–45 One example is the tyrosinase inhibitor 1 described by Li et al. (Fig. 2A).45 Therefore, the aza-Diels–Alder reaction is a highly attractive chemistry for encoded library synthesis.Based on the original report of Danishefsky et al. using ZnCl2 (ref. 41) as the catalyst we started our investigation of the aza-Diels–Alder reaction on CPG-coupled DNA. We initiated the imine formation of hexT–aldehyde conjugate 11 with 500 equivalents of aniline 12a in tetrahydrofuran/triethyl orthoformate (TEOF) for four hours followed by the addition of 500 equivalents of Danishefsky's diene 13 and 50 equivalents of ZnCl2. To our delight we obtained the target product 14 with 62% conversion (Table 2, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | 12a [equiv.] | 13 [equiv.] | Solvent | Conversionb [%] |
a Condensation of CPG-coupled hexT aldehyde conjugate 11 (20 nmol) with aniline 12a (X equiv.) in 36 μL of indicated solvent/triethyl orthoformate (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at ambient temperature for 4 h, followed by addition of ZnCl2 (50 equiv.) suspended in 30 μL of the indicated solvent and Danishefsky's diene 13 (Y equiv.) and shaking of the reaction mixture at ambient temperature for 1 h. Afterwards AMA (30% aqueous ammonia/40% aqueous methylamine, 1:1 (vol/vol)) was added at ambient temperature for 0.5 h.
b Determined by analytical RP-HPLC analysis.
c Incomplete solubility of ZnCl2 in the indicated solvent.
d 100 equiv. of ZnCl2 were used.
e Reaction was performed in the absence of ZnCl2. DCE = dichloroethane, THF = tetrahydrofuran, ACN = acetonitrile. :1) at ambient temperature for 4 h, followed by addition of ZnCl2 (50 equiv.) suspended in 30 μL of the indicated solvent and Danishefsky's diene 13 (Y equiv.) and shaking of the reaction mixture at ambient temperature for 1 h. Afterwards AMA (30% aqueous ammonia/40% aqueous methylamine, 1:1 (vol/vol)) was added at ambient temperature for 0.5 h.
b Determined by analytical RP-HPLC analysis.
c Incomplete solubility of ZnCl2 in the indicated solvent.
d 100 equiv. of ZnCl2 were used.
e Reaction was performed in the absence of ZnCl2. DCE = dichloroethane, THF = tetrahydrofuran, ACN = acetonitrile.
|
||||
| 1 | 500 | 500 | THF | 62 |
| 2 | 500 | 500 | CH2Cl2 | 61c |
| 3 | 500 | 500 | DCE | 28c |
| 4 | 500 | 500 | ACN | 82 |
| 5 | 500 | 500 | MeOH | 80 |
| 6 | 500 | 500 | EtOH | 80 |
| 7 | 500 | 500 | Toluene | 65 |
| 8 | 1000 | 1000 | THF | 49 |
| 9 | 2000 | 2000 | THF | 43 |
| 10 | 4000 | 4000 | THF | 34 |
| 11 | 4000 | 500 | THF | 33 |
| 12 | 500 | 4000 | THF | 73 |
| 13d | 500 | 500 | THF | 82 |
| 14e | 500 | 500 | THF | <5 |
In order to improve product yields we conducted multi-parametric reaction optimization. Evaluation of different solvents (Table 2, entries 2–7) in combination with TEOF revealed the low solubility of ZnCl2 in dichloromethane and dichloroethane. While the Diels–Alder product 14 was formed with 61% conversion in dichloromethane (Table 2, entry 2), the conversion dropped to 28% in dichloroethane (Table 2, entry 3). The highest conversions were obtained in polar solvents such as MeOH, EtOH and acetonitrile with up to 82% product formation (Table 2, entries 4–6). Increasing both aniline 12a and Danishefsky's diene 13 from 500 to 4000 equivalents resulted in a continuous decrease of product formation (Table 2, entries 1 and 8–10). Keeping the amount of Danishefsky's diene 13 constant at 500 equivalents and increasing the excess of aniline 12a to 4000-fold led to the same result with 33% conversion (Table 2, entry 11). Reversing the reactant excess revealed 73% conversion, a better result than that of the initial experiment (Table 2, entries 1 and 12). Finally, we investigated the impact of ZnCl2 concentration on the reaction. Employing 100 instead of 50 equivalents resulted in a 20% higher conversion (Table 2, entries 1 and 13). In the absence of ZnCl2 no product formation was observed (Table 2, entry 14).
We concluded from these experiments that the optimized reaction conditions for the aza-Diels–Alder reaction on DNA were imine formation of the DNA–aldehyde conjugate and 500 equivalents of aniline 12a in acetonitrile/triethyl orthoformate for four hours at ambient temperature, followed by the addition of 1000 equivalents of Danishefsky's diene 13 and 100 equivalents of ZnCl2 dissolved in acetonitrile for one hour at ambient temperature for heterocycle formation.
With these optimized reaction conditions in hand we translated the aza-Diels–Alder reaction to both a 10mer DNA oligonucleotide–aldehyde conjugate 15a as well as to an aniline conjugate 17.
To our delight, DNA oligonucleotide–aldehyde conjugate 15a readily reacted with aniline 12a and Danishefsky's diene 13 to form the desired product 16a with 80% conversion and no observable degree of DNA degradation (Fig. 3). However, reaction of ATGC–aniline conjugate 17 with benzaldehyde 18a and Danishefsky's diene 13 led only to traces of product 19a under these conditions (Table S7†). Changing the reaction conditions slightly revealed that dry tetrahydrofuran as the solvent together with 1500 equivalents of benzaldehyde 18a was required to obtain the desired aza-Diels–Alder product 19a with 62% conversion. Formation of a side product with an additional mass of 32 Da was observed in MALDI-TOF analysis.46 Since we never observed this side product during the reaction optimization campaign on hexT, we assumed a conjugate addition of methylamine during DNA deprotection and cleavage. While hexT is routinely cleaved by a short 30 minutes treatment with AMA (30% aqueous ammonia/40% aqueous methylamine, 1:1 (vol/vol)), the CPG-coupled ATGC conjugates are suspended for 4 hours in AMA solution, explaining the addition of methyl amine to the double bond. Therefore, we resorted to cleaving the DNA from CPG with 30% aqueous ammonia at 50 °C. Indeed after six hours the desired ATGC conjugate 19a was cleaved from CPG cleanly without side products due to conjugate addition (Fig. 4).
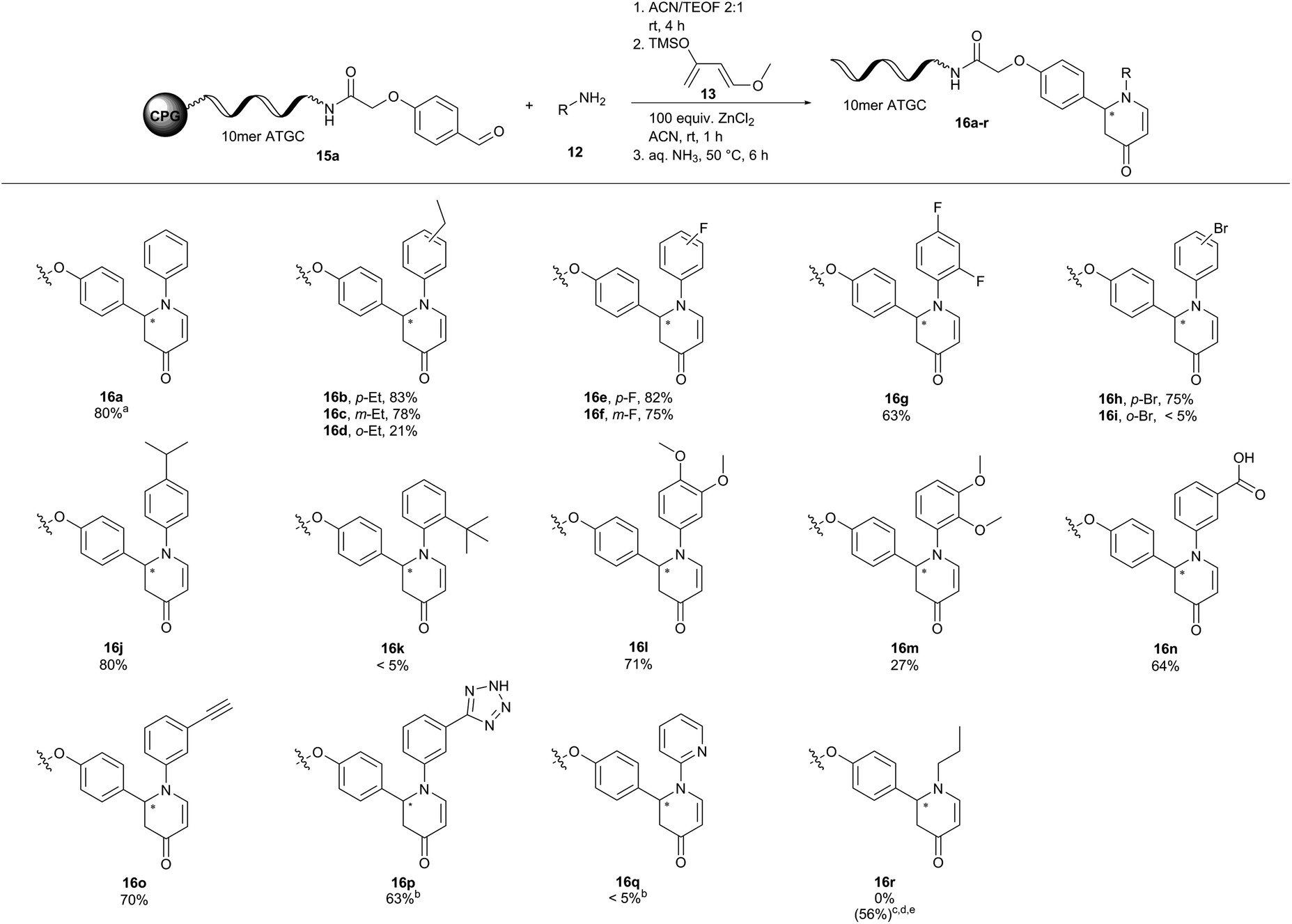 | ||
| Fig. 3 Scope of a ZnCl2-promoted aza-Diels–Alder reaction of CPG-coupled 10mer ATGC oligonucleotide–aldehyde conjugate 15a with Danishefsky's diene 13 and amines 12. Reaction conditions: Condensation of CPG-coupled ATGC aldehyde conjugate 15a (20 nmol) with amine 12 (500 equiv., 10 μmol) in 36 μL acetonitrile/triethyl orthoformate (2:1) at ambient temperature for 4 h, followed by addition of ZnCl2 (100 equiv., 2 μmol) dissolved in 30 μL acetonitrile and Danishefsky's diene 13 (1000 equiv., 20 μmol) at ambient temperature for 1 h. DNA cleavage with 30% aqueous ammonia at 50 °C for 6 h. aDetermined by analytical RP-HPLC analysis. bDimethyl sulfoxide was used instead of acetonitrile. c1000 equiv. of amine 12 were used. dYb(OTf)3 was used instead of ZnCl2. eThe 2nd step of the reaction was performed overnight at 35 °C. 10mer ATGC = 5′-GTC ATG ATC T-3′, ACN = acetonitrile. | ||
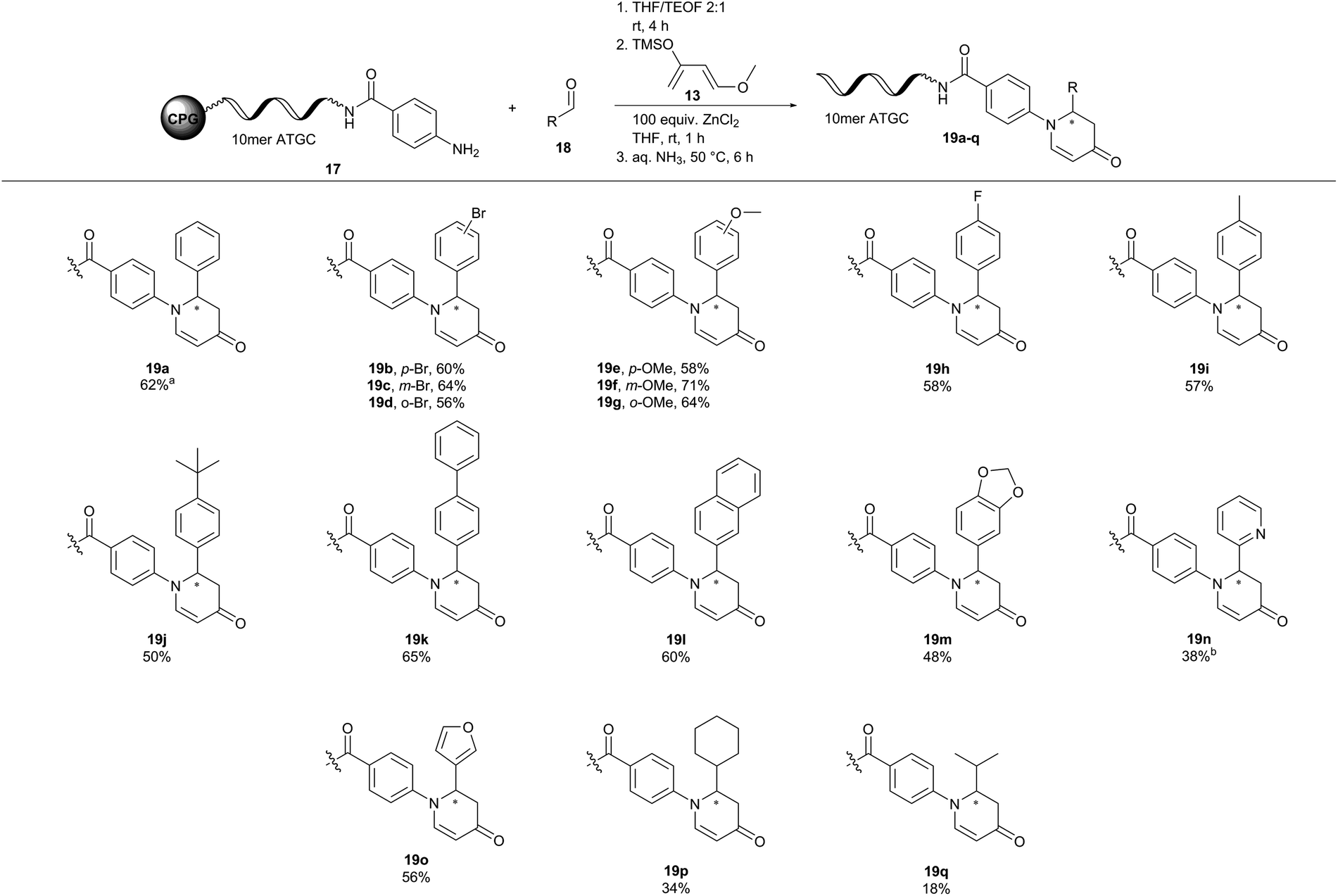 | ||
| Fig. 4 Scope of a ZnCl2-mediated aza-Diels–Alder reaction of CPG-coupled 10mer ATGC oligonucleotide–aniline conjugate 17 with Danishefsky's diene 13 and aldehydes 18. Reaction conditions: Condensation of CPG-coupled ATGC aniline conjugate 17 (20 nmol) with aldehyde 18 (1500 equiv., 30 μmol) in 36 μL tetrahydrofuran/triethyl orthoformate (2:1) at ambient temperature for 4 h, followed by addition of ZnCl2 (100 equiv., 2 μmol) dissolved in 30 μL tetrahydrofuran and Danishefsky's diene 13 (1000 equiv., 20 μmol) at ambient temperature for 1 h. DNA cleavage with 30% aqueous ammonia at 50 °C for 6 h. aDetermined by analytical RP-HPLC analysis. bDimethyl sulfoxide was used instead of acetonitrile. 10mer ATGC = 5′-GTC ATG ATC T-3′, THF = tetrahydrofuran. | ||
Next, we explored the substrate scope for both variants of the aza-Diels–Alder reaction (Fig. 3 and 4). In the first set of reactions we reacted the 10mer DNA oligonucleotide–aldehyde conjugate 15a with a variety of anilines 12 and Danishefsky's diene 13. Electron withdrawing and electron donating substituents were well tolerated in the reaction yielding the products 16b–j and l–p with conversions of up to 83%. Product conversions were sensitive to both substituent positioning and size. Substituents at the para-position such as ethyl or fluoro gave slightly better conversions of 83% (16b) and 82% (16e) as compared to the same substituents positioned at meta with 78% (16c) and 75% (16f), respectively. Substituents at the ortho-position mostly caused a steep drop in product conversions. A fluoro-substituent at this position was still tolerated with a conversion of 63% (16g). The slightly larger ethyl- and methoxy-substituents decreased the conversion to 21% (16d) and 27% (16m). Finally, bulky groups like bromine (16i) or tert-butyl (16k) were completely unreactive. Methyl 3-aminobenzoate (16n) and 3-ethynylaniline (16o) containing functional groups that can be further modified in a later step of library synthesis were viable reactants with reasonable conversions of 64% and 70%, respectively. The methyl ester of product 16n was hydrolysed either during the reaction or during DNA cleavage. Heterocyclic substituents at the aniline (16p) were tolerated, too. Due to solubility issues dimethyl sulfoxide had to be used as the solvent in this case. Unfortunately, no conversion towards the desired products was observed with 2-aminopyridine (16q) and propylamine (16r). The metal ion screening data indicated that alternative metal salts could be tested to extend the reaction scope. Indeed, an aliphatic amine could be reacted with moderate conversion of 56% to the target pyridone 16r by exchanging ZnCl2 with Yb(OTf)3 and running the reactions at 35 °C overnight.48
Next, we investigated the substrate scope of the inverse aza-Diels–Alder reaction with CPG-coupled DNA-aniline conjugate 17, a diverse set of aldehydes 18, and Danishefsky's diene 13 (Fig. 4). Electron donating substituents such as a methoxy group and electron withdrawing functionalities such as fluorine were well tolerated in this aza-Diels–Alder approach on DNA. The desired products (19b–m) were formed with conversions between 48 and 71%. Here, the position of the substituent on the benzaldehyde 18 did not affect the reaction outcome. Para-, meta- and even ortho-substituted products alike were formed with comparable conversions (19b, p-Br = 60%, 19c, m-Br = 64%, 19d, o-Br = 56%). To our delight, heterocyclic aldehydes 2-pyridinecarboxaldehyde (19n) and 3-furaldehyde (19o) were viable reactants showing conversions of up to 56%. Aliphatic aldehydes such as cyclohexanecarboxaldehyde (19p) and the enolizable isobutyraldehyde (19q) gave the desired products, too, albeit at lower conversions of 20% and 34%, respectively. In conclusion, the aza-Diels–Alder reaction showed a broad scope and mostly good-to-high conversions for both DNA-aldehyde and DNA-aniline conjugates 15a and 17.
Development of a (R)-(−)-BNDHP-mediated Povarov reaction on CPG-coupled DNA oligonucleotides
Another highly attractive three-component reaction for synthesis of screening collections is the Povarov reaction. It yields tetrahydroquinolines (THQs) from readily available anilines, aldehydes and electron-rich olefins.47–52 The Povarov reaction of aromatic imines with 2,3-dihydropyrrole allowed for preparation of the pyrroloquinoline scaffold present in diverse natural products like martinellic acid 2 and martinelline 3 (ref. 53) and in several bioactive compounds, for instance in the CRISPR inhibitor BRD0539 4 (ref. 54) (Fig. 2B). Very recently, we described a Brønsted acid promoted Povarov reaction on DNA-tagged aldehydes in an aqueous micellar dispersion.55 Here, we explored this reaction for our solid-phase based encoding strategy.Initial experiments were performed on the hexT-adapter oligonucleotide. From the many catalysts that are reported for the Povarov reaction, we selected Yb(OTf)3 and the phosphoric acid (R)-(−)-BNDHP A for our initial experiments on DNA. In analogy to the aza-Diels–Alder reaction we first condensed the CPG-coupled hexT-aldehyde conjugate 11 with 500 equivalents of aniline 12a in dichloromethane/triethyl orthoformate for four hours at ambient temperature. Afterwards 500 equivalents of N-Boc-2,3-dihydro-1H-pyrrole 20 and 50 equivalents of Yb(OTf)3 dissolved in dichloromethane were added and the reaction was shaken for 16 hours at ambient temperature. The desired THQ 21 was formed with 42% conversion in the initial experiment (Table 3, entry 1). Starting from this encouraging result we performed a multi-parametric reaction optimization campaign, using (R)-(−)-BNDHP A as a second potential reaction promoter. First, we investigated the impact of the reaction temperature and solvent on heterocycle formation. The Yb(III)-mediated heterocycle formation step gave a higher conversion of 63% towards THQ 21 in THF at 50 °C (Table 3, entry 2). Substituting the Lewis acid with the phosphoric acid organocatalyst (R)-(−)-BNDHP A resulted in even higher product conversions of 82% (50 equiv. of A) and 94% (100 equiv. of A), respectively (Table 3, entries 3 and 4). Interestingly, in the absence of any mediator the product 21 was still formed with 34% conversion (Table 3, entry 5). This observation could plausibly be explained by deprotection of the phosphate backbone of the DNA under the basic conditions of amide bond formation, generating a slightly acidic solid phase matrix. In the next series of experiments, we investigated the impact of reagent excess. Keeping the amount of N-Boc-2,3-dihydro-1H-pyrrole 20 constant at 500 equivalents and using 50 equivalents of (R)-(−)-BNDHP A we increased the excess of the aniline 12a stepwise to 4000 equivalents (Table 3, entries 3 and 6–8). With an increasing excess of aniline 12a, the product conversion dropped to 7% indicating that the reaction was sensitive to the aniline to acid ratio.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 12a [equiv.] | 20 [equiv.] | Mediator | Solvent | Conversionb [%] |
|
a Condensation of CPG-coupled hexT aldehyde conjugate 11 (20 nmol) with aniline 12a (X equiv.) in 36 μL of indicated solvent/triethyl orthoformate (2:1) at ambient temperature for 4 h, followed by addition of a mediator (50 equiv.) suspended in 30 μL of the indicated solvent and N-Boc-2,3-dihydro-1H-pyrrole 20 (Y equiv.). The reaction mixture was shaking at 50 °C for 16 h. Afterwards AMA (30% aqueous ammonia/40% aqueous methylamine, 1:1 (vol/vol)) was added at ambient temperature for 0.5 h.
b Determined by analytical RP-HPLC analysis.
c reaction was performed at ambient temperature.
d 100 equiv. of (R)-(−)-BNDHP were used.
e Boc cleavage with 10% TFA for 4 h at ambient temperature prior to DNA deprotection.
|
|||||
| 1c | 500 | 500 | Yb(OTf)3 | CH2Cl2 | 42 |
| 2 | 500 | 500 | Yb(OTf)3 | THF | 63 |
| 3 | 500 | 500 | (R)-(−)-BNDHP A | THF | 82 |
| 4d | 500 | 500 | (R)-(−)-BNDHP A | THF | 94 |
| 5 | 500 | 500 | — | THF | 34 |
| 6 | 1000 | 500 | (R)-(−)-BNDHP A | THF | 55 |
| 7 | 2000 | 500 | (R)-(−)-BNDHP A | THF | 11 |
| 8 | 4000 | 500 | (R)-(−)-BNDHP A | THF | 7 |
| 9 | 500 | 1000 | (R)-(−)-BNDHP A | THF | 63 |
| 10 | 500 | 2000 | (R)-(−)-BNDHP A | THF | 67 |
| 11 | 500 | 4000 | (R)-(−)-BNDHP A | THF | 86 |
| 12 | 500 | 500 | (R)-(−)-BNDHP A | EtOH | >95 |
| 13 | 500 | 500 | (R)-(−)-BNDHP A | DMF | 50 |
| 14e | 500 | 500 | (R)-(−)-BNDHP A | THF | 82 |
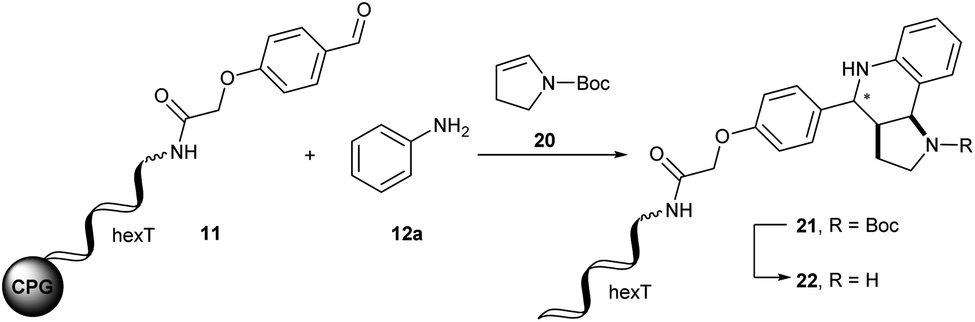
The same experiments with aniline 12a set to 500 equivalents and increasing amounts of N-Boc-2,3-dihydro-1H-pyrrole 20 revealed a slightly decrease of conversion for 1000 and 2000 equivalents (Table 3, entries 9 and 10). However, with 4000 equivalents of N-Boc-2,3-dihydro-1H-pyrrole 20 we observed a conversion of 86% comparable to that of the initial experiments with 500 equivalents (Table 3, entries 3 and 11). In the last set of experiments we investigated different solvents for the Povarov reaction (Table 3, entries 3, 12 and 13). Full product conversions were achieved using polar protic ethanol (Table 3, entry 12). Towards further library synthesis, we removed the Boc-protecting group from the THQ 21. CPG-coupled hexT-THQ conjugate 21 was treated with 10% trifluoroacetic acid for four hours at ambient temperature yielding the desired deprotected product 22 (Table 3, entry 14).
Optimized reaction conditions called for condensation of the DNA-aldehyde conjugate with 500 equivalents of aniline 12a in ethanol/triethyl orthoformate for four hours at ambient temperature, followed by the addition of 500 equivalents of N-Boc-2,3-dihydro-1H-pyrrole 20 and 100 equivalents of (R)-(−)-BNDHP A dissolved in ethanol and heterocycle formation for 16 hours at 50 °C.
According to our catalyst screening, these reaction conditions should be compatible with DNA. Indeed, we obtained similar conversions to the desired THQ conjugate 23a from DNA aldehyde conjugate 15a with no concomitant DNA degradation. Yet, the protocol for removal of the Boc protecting group had to be adapted to account for the susceptibility of DNA to depurination.23 The protecting group could be removed from the THQs by repeated incubation of the CPG-coupled DNA with 75% TFA for 30 seconds. Next, we investigated the scope of the reaction with a set of diverse substituted anilines (Fig. 5). In most cases, we observed excellent conversions of more than 80%. Notable exceptions were the ortho-ethyl-substituted aniline yielding 39% THQ conjugate 23l and ortho-tert-butyl-aniline which yielded the product 23m only in trace amounts. This contrasted e.g. with ortho-bromo aniline which gave the product 23b with a good conversion of 57%. Furthermore, aminopyridines did not yield THQs 23t and 23u. Finally, we tested two further DNA aldehyde conjugates, namely meta-substituted 15b and ortho-substituted 15c. The former starting material 15b gave THQ 23v with full conversion, whereas the latter aldehyde 15c proved unreactive. The Povarov reaction can give rise to diastereomeric mixtures of compounds. Unlike enantiomers, these could be readily observed by HPLC analysis in all cases. In one case (23d), we were even able to separate the two diastereomers and prove their identical masses by MALDI-TOF analysis. Treatment of the isolated second diastereomer 23d-dia2 with AMA solution for four hours led to 10% isomerisation towards the first diastereomer 23d-dia1 as analysed by analytical HPLC (ESI†). This finding indicates that product isomerisation might occur during the cleavage of DNA from the solid support. The four meta-substituted anilines gave complex product mixtures that are plausibly explained by formation of diastereo- and regioisomers. Unfortunately, the Povarov reaction failed with a DNA aniline conjugate under the evaluated conditions. In summary, the Povarov reaction on solid support-coupled DNA showed a broad reactant scope and mostly high conversions. Facile removal of the Boc protecting group provided compounds that could be readily used for further library synthesis, e.g. by amide bond formation, as demonstrated previously.55
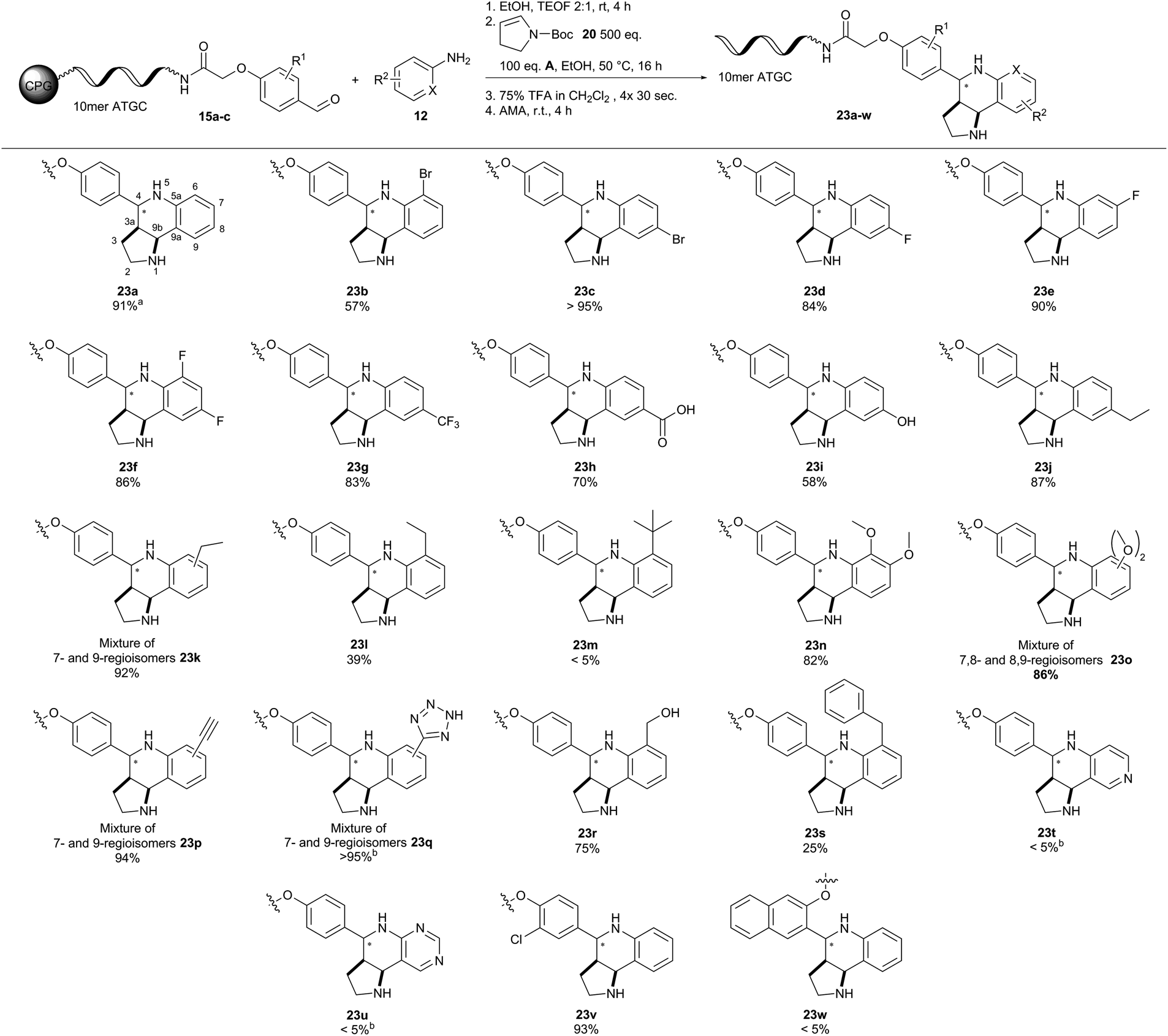 | ||
| Fig. 5 Scope of the (R)-(−)-BNDHP A-mediated Povarov reaction of CPG-coupled 10mer ATGC oligonucleotide–aldehyde conjugates 15 with N-Boc-2,3-dihydro-1H-pyrrole 20 and anilines 12. Reaction conditions: Condensation of CPG-coupled ATGC aldehyde conjugate 15 (20 nmol) with aniline 12 (500 equiv., 10 μmol) in 36 μL ethanol/triethyl orthoformate (2:1) at ambient temperature for 4 h, followed by addition of (R)-(−)-BNDHP A (100 equiv., 2 μmol) dissolved in 30 μL ethanol and N-Boc-2,3-dihydro-1H-pyrrole 20 (500 equiv., 10 μmol) at 50 °C for 16 h. Boc removal by repeated incubation with 75% TFA for 30 seconds. Afterwards AMA (30% aqueous ammonia/40% aqueous methylamine, 1:1 (vol/vol)) was added at ambient temperature for 4 h. aDetermined by analytical RP-HPLC analysis. bDimethyl sulfoxide was used instead of ethanol. 10mer ATGC = 5′-GTC ATG ATC T-3′. | ||
Development of a (R)-(−)-BNDHP-mediated Biginelli reaction on CPG-coupled DNA oligonucleotides
The Biginelli reaction gives rise to dihydropyrimidinones from aldehydes, N-substituted ureas, and acetoacetates.56–61 A prominent example of this class of heterocycles is the mitotic kinesin Eg5 inhibitor monastrol 5 (Fig. 2C).62 A second example is γ-aminobutyric acid type A receptor ligand 6 (Fig. 2C).63 In order to translate this reaction to the barcoded format disclosed in ref. 24 we investigated the DNA-compatible reagents Yb(OTf)3, Mg(ClO4)2 and (R)-(−)-BNDHP A that were all reported in the literature for the Biginelli reaction. CPG-coupled hexT-aldehyde conjugate 11 was reacted with 500 equivalents of urea 24a dissolved in 30 μL tetrahydrofuran and 500 equivalents of ethyl acetoacetate 25 (Table 4).|
|
|||||
|---|---|---|---|---|---|
| Entry | Mediator | Solvent | T [°C] | t [h] | Conversionb [%] |
|
a CPG-coupled hexT aldehyde conjugate 11 (20 nmol) was suspended in indicated solvent with urea 24a (500 equiv.) and mediator (50 equiv., 1 μmol) each dissolved or suspended in 30 μL of indicated solvent and ethyl acetoacetate 25 (500 equiv.), reaction mixture was shaken at indicated temperature for 20 h. Afterwards AMA (30% aqueous ammonia/40% aqueous methylamine, 1:1 (vol/vol)) at ambient temperature for 0.5 h.
b Determined by RP-HPLC analysis.
|
|||||
| 1 | Yb(OTf)3 | THF | 25 | 20 | <5 |
| 2 | (R)-(−)-BNDHP A | THF | 25 | 20 | <5 |
| 3 | Yb(OTf)3 | THF | 50 | 20 | 24 |
| 4 | (R)-(−)-BNDHP A | THF | 50 | 20 | 28 |
| 5 | Yb(OTf)3 | EtOH | 25 | 20 | 47 |
| 6 | Mg(ClO4)2 | EtOH | 25 | 20 | <5 |
| 7 | (R)-(−)-BNDHP A | EtOH | 25 | 20 | 13 |
| 8 | Yb(OTf)3 | EtOH | 50 | 20 | 65 |
| 9 | Mg(ClO4)2 | EtOH | 50 | 20 | 65 |
| 10 | (R)-(−)-BNDHP A | EtOH | 50 | 20 | 97 |
| 11 | (R)-(−)-BNDHP A | EtOH | 50 | 4 | 14 |
| 12 | (R)-(−)-BNDHP A | EtOH | 50 | 8 | 40 |
In the first series of experiments 50 equivalents of Yb(OTf)3 or (R)-(−)-BNDHP A, respectively, dissolved in 30 μL tetrahydrofuran were added and the reaction mixtures were shaken for 20 hours at ambient temperature (Table 4, entries 1 and 2). However, product formation was not observed in either case.
Increasing the temperature to 50 °C led to the target dihydropyrimidinone 26 with moderate conversions of 24% and 28% (Table 4, entries 3 and 4). Since urea was not well dissolved in tetrahydrofuran we tested polar protic ethanol and Yb(OTf)3, (R)-(−)-BNDHP A, and Mg(ClO4)2 to improve product conversions. At ambient temperature Yb(OTf)3 was superior to (R)-(−)-BNDHP A and Mg(ClO4)2 giving 26 with a conversion of 47% (Table 4, entries 5–7). In all cases increasing the temperature to 50 °C improved the reaction outcome. Both Yb(OTf)3 and Mg(ClO4)2 gave the desired product 26 with 65% conversion (Table 4, entries 8 and 9), and (R)-(−)-BNDHP A even mediated full conversion of the DNA-coupled aldehyde to the target dihydropyrimidinone 26 (Table 4, entry 10). Decreased reaction times of eight and four hours led to a sharp drop in product formation (Table 4, entries 11 and 12). Therefore, the optimized conditions for the Biginelli reaction on DNA were incubation of the CPG-coupled oligonucleotide–aldehyde conjugate with 500 equivalents of urea 24 and 50 equivalents of (R)-(−)-BNDHP A each dissolved in 30 μL ethanol and 500 equivalents of ethyl acetoacetate for 20 hours at 50 °C.
Fortunately, translation of the reaction conditions to a DNA-barcode worked smoothly with 90% conversion and importantly without detectable DNA degradation (27a, Fig. 6). Next, we investigated the substrate scope of the Biginelli reaction (Fig. 6). Both N-ethyl- and N-benzyl-urea led to the desired products 27b and 27d with good conversions of 59 and 85%, respectively. However, N-phenylurea gave 27c merely in traces with 10% conversion. Increasing the amount of (R)-(−)-BNDHP A to 200 equivalents and reaction time to 44 hours improved product conversion to 75%. However, approximately 30% DNA-degradation occurred under these reaction conditions, revealing a limit of DNA stability. Further CPG-coupled DNA-aldehyde conjugates 15b and 15c were investigated as well. The meta-substituted aldehyde conjugate 15b led to 92% conversion of product 27e. For ortho-substituted aldehyde conjugate 15c no product formation (27f) could be detected. Surprisingly, we did not observe ethyl ester aminolysis upon cleavage of DNA from CPG. We would like to remark that standard HPLC-based analysis of DNA conjugates does not resolve racemic compound mixtures, which are likely formed in the Biginelli reaction on DNA.
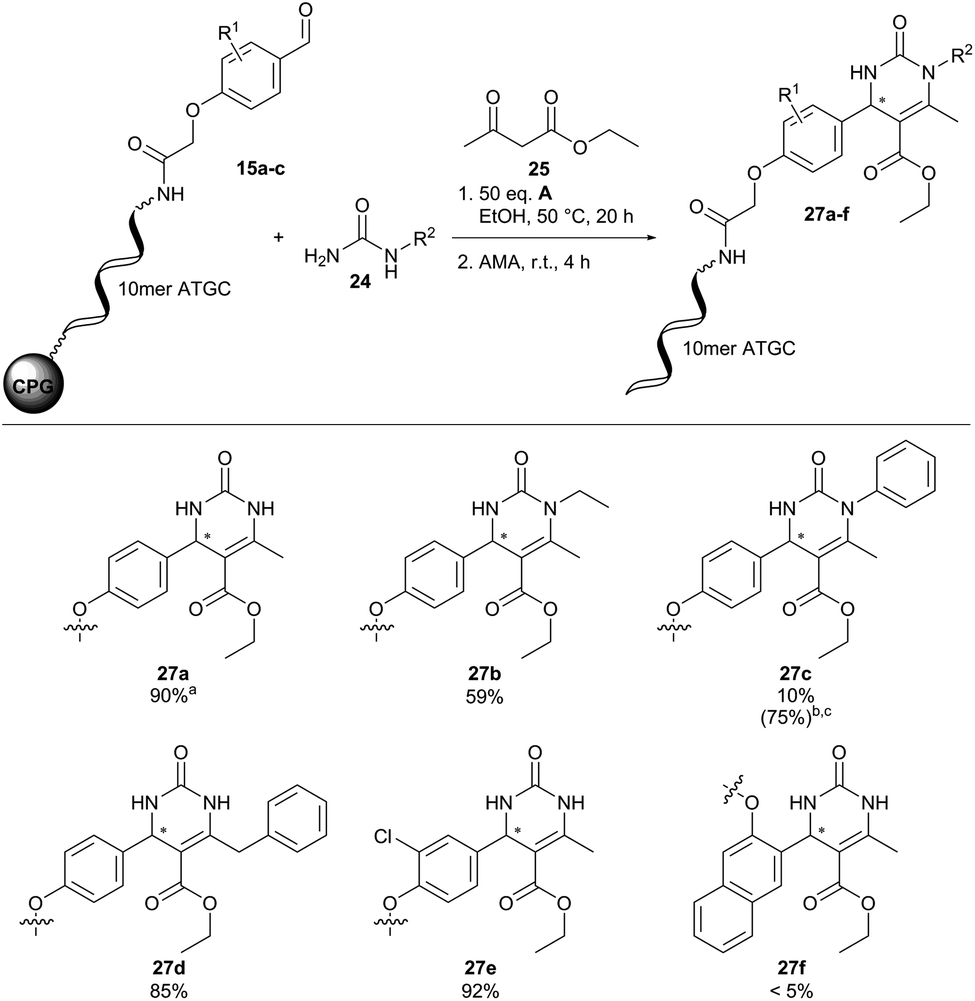 | ||
| Fig. 6 Scope of the (R)-(−)-BNDHP A-mediated Biginelli reaction on CPG-coupled 10mer ATGC oligonucleotide–aldehyde conjugates 15 with ureas 24 and ethyl acetoacetate 25. Reaction conditions: CPG-coupled ATGC aldehyde conjugate 15 (20 nmol) was suspended with urea 24 (500 equiv.) and (R)-(−)-BNDHP A (50 equiv., 1 μmol) each dissolved in 30 μL of ethanol and ethyl acetoacetate 25 (500 equiv.), and the reaction mixture was shaken at 50 °C for 20 h. Afterwards AMA (30% aqueous ammonia/40% aqueous methylamine, 1:1 (vol/vol)) was added at ambient temperature for 4 h. aDetermined by analytical RP-HPLC analysis. b200 equiv. of (R)-(−)-BNDHP A were used. cThe reaction was performed at 50 °C for 44 h. 10mer ATGC = 5′-GTC ATG ATC T-3′. | ||
Cheminformatics analysis of simulated encoded heterocycle libraries
We analysed four in silico libraries to gain insight into chemical space coverage: these were generated by simulated Diels–Alder reactions from DNA-aldehyde conjugates (DA-1) yielding 301.303 compounds, “reverse” Diels–Alder reactions from DNA-amine conjugates (DA-2, 389.388 compounds), Povarov (P, 318.175 compounds) and Biginelli reactions (B, 332.702 compounds).Prior to library design, the building blocks were filtered according to their molecular weight; the threshold was set to 200 Da for each starting material class to ensure a reasonable molecular weight of the final product. Building blocks that contained bulky ortho-substituents or the linker site to DNA at the ortho-position were excluded, too, as they proved unreactive. All the starting materials were separately filtered to exclude substructures that might show undesired reactivity.64 Furthermore, specific filters were applied to each compound class per library to account for reaction scope (ESI†). For comparison to commercially available screened molecules we used the Enamine REAL database:65 this dataset contains 24 million drug-like compounds. Each library was characterized by normalized molecular quantum numbers (MQNs 1–42).66 This analysis was visualized using principal component analysis (PCA) plots for chemical diversity and principal moments of inertia (PMI) plots for compound shape analysis (Fig. 7, S3 and S4†).67 From the PCA plots, we learnt that a higher chemical diversity was achieved by the simulated DA-1 library, which was almost comparable to that of a commercial compound database (Fig. S3†). As expected, the “reverse Diels–Alder” library (DA-2) showed a very similar outcome; slight differences in chemical space coverage were explained by the more restricted availability of acid functionalized anilines (Fig. S5†). Because of this high similarity the DA-2 was excluded from the PMI calculation. The Povarov and Biginelli libraries cover a more narrow chemical space that only partially overlaps with the DA library and the commercially available compounds (Fig. S6†). This can be plausibly explained by the more restricted reactant scope of these two reactions. Heteroaromatic anilines had to be excluded from the Povarov reaction and substituted β-ketoesters and ureas for the Biginelli reaction are only scarcely available. In agreement with an earlier library analysis,25 we observed that application of different synthesis methods (here Diels–Alder, Povarov and Biginelli reactions) led to only partially overlapping chemical space from common DNA-coupled starting materials.
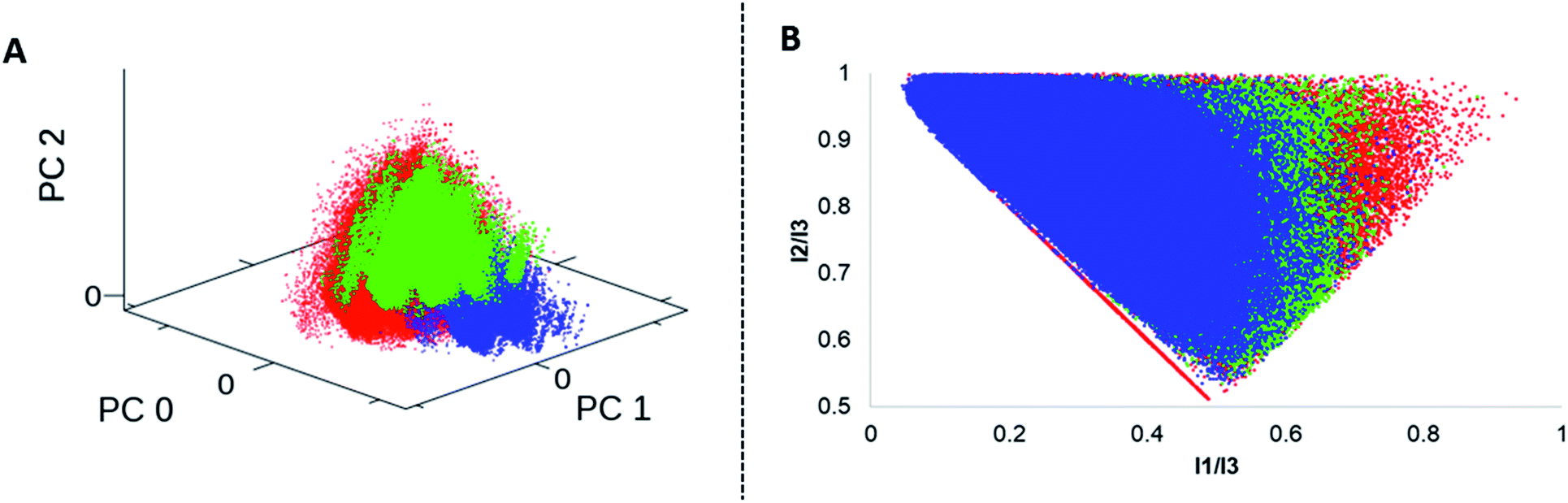 | ||
| Fig. 7 Cheminformatics analysis: A = PCA plot of the DA-1(red), P (blue) and B (green) libraries. B = PMI plot of the DA-1(red), P (blue) and B (green) libraries. | ||
The PMI compound shape analysis mostly confirmed the PCA analysis, with the Diels–Alder library covering the broadest shape space, which exceeded that of even the commercially available compounds (Fig. S4†). In comparison, the Povarov library was more biased towards linear and disc-shaped compounds plausibly due to its aromatic sub-structure in the heterocyclic scaffold. The Biginelli library occupied a central space in the plot, denoting rather rotatable structures, with a slight preference towards the disc shape.
Conclusions
Initiating DNA-encoded library synthesis on controlled pore glass (CPG)-coupled DNA barcodes21–24 benefits from free solvent choice and enhanced chemical stability of nucleobase-protected DNA. Here, we report a systematic analysis of the stability of pyrimidine and purine DNA sequences to 53 metal ions and organic reagents commonly used in preparative organic chemistry. All oligonucleotides tolerated the majority of reagents even at high equivalents and for prolonged reaction times. However, purine DNA was degraded by strong protic acids, oxidants and higher concentrations of transition metal catalysts such as Pd(II). We exploited this insight into chemical DNA stability to translate three heterocycle-forming reactions to a barcoded format: the ZnCl2-promoted aza-Diels–Alder reaction and the (R)-(−)-BNDHP A-promoted Povarov and Biginelli reactions, respectively. All three reactions give rise to attractive scaffolds that project diverse substituents out of plane. While both the Povarov and Biginelli reactions yield scaffolds that can be readily substituted with e.g. carbonyl chemistry, the aza-Diels–Alder reaction with Danishefsky's diene requires bifunctional or trifunctional building blocks for further library synthesis. Alternatives to Danishefsky's diene that provide a functional group for further library synthesis have been demonstrated.68 Cheminformatics analysis revealed that the three heterocycle-forming reactions lead to only partially overlapping chemical space. Notably, the aza-Diels–Alder reaction provided the most comprehensive representation of molecular shapes. We are currently exploring translation of further reactions to a DNA-barcoded format and we are synthesizing encoded libraries based on the three heterocyclic scaffolds. In future studies, it is conceivable to explore enantioselective on-DNA compound synthesis, too. The DNA stability profile disclosed in this study will surely aid chemists in designing further reactions for encoded library synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the generous support of this work through a Boehringer Ingelheim Foundation Exploration Grant and DFG grant No. BR 5049/3-1. We thank Christiane Ehrt for her support in the cheminformatics analysis. We would like to thank the group of Dr Andrey P. Antonchick (TU Dortmund University) for donation of several chemicals.References
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5381–5383 CrossRef CAS PubMed.
- R. Franzini, D. Neri and J. Scheuermann, Acc. Chem. Res., 2014, 47, 1247–1255 CrossRef CAS PubMed.
- H. Salamon, M. Klika Škopić, K. Jung, O. Bugain and A. Brunschweiger, ACS Chem. Biol., 2016, 11, 296–307 CrossRef CAS PubMed.
- R. A. Goodnow Jr, C. E. Dumelin and A. D. Keefe, Nat. Rev. Drug Discovery, 2017, 16, 131–147 CrossRef.
- B. Shi, Y. Zhou, Y. Huang, J. Zhang and X. Li, Bioorg. Med. Chem. Lett., 2017, 27, 361–369 CrossRef CAS.
- J. Ottl, L. Leder, J. V. Schaefer and C. E. Dumelin, Molecules, 2019, 24, E1629 CrossRef PubMed.
- L. Mannocci, Y. Zhang, J. Scheuermann, M. Leimbacher, G. DeBellis, E. Rizzi, C. Dumelin, S. Melkko and D. Neri, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17670–17675 CrossRef CAS.
- M. A. Clark, R. A. Acharya, C. C. Arico-Muendel, A. L. Belyanskaya, D. R. Benjamin, N. R. Carlson, P. A. Centrella, C. H. Chiu, S. P. Creaser, J. W. Cuozzo, C. P. Davie, Y. Ding, G. J. Franklin, K. D. Franzen, M. L. Gefter, S. P. Hale, N. J. Hansen, D. I. Israel, J. Jiang, M. J. Kavarana, M. S. Kelley, C. S. Kollmann, F. Li, K. Lind, S. Mataruse, P. F. Medeiros, J. A. Messer, P. Myers, H. O'Keefe, M. C. Oliff, C. E. Rise, A. L. Satz, S. R. Skinner, J. L. Svendsen, L. Tang, K. van Vloten, R. W. Wagner, G. Yao, B. Zhao and B. A. Morgan, Nat. Chem. Biol., 2009, 5, 647–654 CrossRef CAS PubMed.
- M. L. Malone and B. M. Paegel, ACS Comb. Sci., 2016, 18, 182–187 CrossRef CAS.
- A. S. Ratnayake, M. E. Flanagan, T. L. Foley, J. D. Smith, J. G. Johnson, J. Bellenger, J. I. Montgomery and B. M. Paegel, ACS Comb. Sci., 2019, 21, 650–655 CrossRef CAS PubMed.
- A. L. Satz, J. Cai, Y. Chen, R. Goodnow, F. Gruber, A. Kowalczyk, A. Petersen, G. Naderi-Oboodi, L. Orzechowski and Q. Strebel, Bioconjugate Chem., 2015, 26, 1623 CrossRef CAS PubMed.
- C. Angelé-Martínez, C. Goodman and J. Brumaghim, Metallomics, 2014, 6, 1358 RSC.
- A. M. Fleming, J. G. Muller, I. Ji and C. J. Burrows, Org. Biomol. Chem., 2011, 9, 3338–3348 RSC.
- T. Z. Liu, T. F. Lin, D. T. Y. Chiu, K.-J. Tsai and A. Stern, Free Radical Biol. Med., 1997, 23, 155–161 CrossRef CAS.
- C. E. Crespo-Hernandez, D. M. Close, L. Gorb and J. Leszczynski, J. Phys. Chem. B, 2007, 111, 5386 CrossRef CAS.
- D. T. Flood, S. Asai, X. Zhang, J. Wang, L. Yoon, Z. C. Adams, B. C. Dillingham, B. B. Sanchez, J. C. Vantourout, M. E. Flanagan, D. W. Piotrowski, P. Richardson, S. A. Green, R. A. Shenvi, J. S. Chen, P. S. Baran and P. E. Dawson, J. Am. Chem. Soc., 2019, 141(25), 9998–10006 CrossRef CAS.
- M. W. Grinstaff and S. I. Khan, J. Am. Chem. Soc., 1999, 121, 4704–4705 CrossRef.
- M. Rist, N. Amann and H. A. Wagenknecht, Eur. J. Org. Chem., 2003, 13, 2498 CrossRef.
- A. Krause, A. Hertl, F. Muttach and A. Jäschke, Chem.–Eur. J., 2014, 20, 16613–16619 CrossRef CAS PubMed.
- B. N. Tse, T. M. Snyder, Y. Shen and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 15611–15626 CrossRef CAS.
- R. M. Franzini, T. Ekblad, N. Zhong, M. Wichert, W. Decurtins, A. Nauer, M. Zimmermann, F. Samain, J. Scheuermann, P. J. Brown, J. Hall, S. Gräslund, H. Schüler and D. Neri, Angew. Chem., Int. Ed., 2015, 54, 3927–3931 CrossRef CAS.
- L. H. Yuen, S. Dana, Y. Liu, S. I. Bloom, A. Thorsell, D. Neri, A. J. Donato, D. B. Kireev, H. Schüler and R. M. Franzini, J. Am. Chem. Soc., 2019, 141, 5169–5181 CrossRef CAS PubMed.
- D. L. Usanov, A. I. Chan, J. P. Maianti and D. R. Liu, Nat. Chem., 2018, 10, 704–714 CrossRef CAS PubMed.
- M. Potowski, V. B. K. Kunig, F. Losch and A. Brunschweiger, MedChemComm, 2019, 10, 1082–1093 RSC.
- V. B. K. Kunig, C. Ehrt, A. Dömling and A. Brunschweiger, Org. Lett., 2019, 21, 7238–7243 CrossRef CAS PubMed.
- M. Klika Škopić, H. Salamon, O. Bugain, K. Jung, A. Gohla, L. J. Doetsch, D. dos Santos, A. Bhat, B. Wagner and A. Brunschweiger, Chem. Sci., 2017, 8, 3356–3361 RSC.
- M. Feher and J. M. Schmidt, J. Chem. Inf. Comput. Sci., 2003, 43, 218–227 CrossRef CAS PubMed.
- A. A. Shelat and R. K. Guy, Nat. Chem. Biol., 2007, 3, 442–446 CrossRef CAS PubMed.
- S. Wetzel, R. S. Bon, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 10800–10826 CrossRef CAS PubMed.
- Y. Hu, D. Stumpfe and J. Bajorath, J. Med. Chem., 2016, 59, 4062–4076 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2017, 56, 7971–7974 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- Y. Chen, A. S. Kamlet, B. J. Steinman and D. R. Liu, Nat. Chem., 2011, 3, 146–153 CrossRef CAS PubMed.
- J. A. Sikorsky, D. A. Primerano, T. W. Fenger and J. Denvir, Biochem. Biophys. Res. Commun., 2007, 355, 431–437 CrossRef CAS PubMed.
- M. Hofreiter, V. Jaenicke, D. Serre, A. von Haeseler and S. Pääboa, Nucleic Acids Res., 2001, 29, 4793–4799 CrossRef CAS PubMed.
- B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926–7953 RSC.
- X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515–5546 CrossRef CAS PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- F. Buller, L. Mannocci, Y. Zhang, C. E. Dumelin, J. Scheuermann and D. Neri, Bioorg. Med. Chem. Lett., 2008, 18, 5926–5931 CrossRef CAS PubMed.
- S. Danishefsky and J. F. Kerwin Jr, J. Org. Chem., 1982, 47, 3183–3184 CrossRef CAS.
- P. R. Girling, T. Kiyoi and A. Whiting, Org. Biomol. Chem., 2011, 9, 3105–3121 RSC.
- A. Nören-Müller, I. Reis-Corrêa Jr, H. Prinz, C. Rosenbaum, K. Saxena, H. J. Schwalbe, D. Vestweber, G. Cagna, S. Schunk, O. Schwarz, H. Schiewe and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(28), 10606–10611 CrossRef PubMed.
- H. S. Beckmann, F. Nie, C. E. Hagerman, H. Johansson, Y. S. Tan, D. Wilcke and D. R. Spring, Nat. Chem., 2013, 5, 861–867 CrossRef CAS PubMed.
- D.-F. Li, P.-P. Hu, M.-S. Liu, X.-L. Kong, J.-C. Zhang, R. C. Hider and T. Zhou, J. Agric. Food Chem., 2013, 61, 6597–6603 CrossRef CAS PubMed.
- L. H. Yuen and R. M. Franzini, Bioconjugate Chem., 2017, 28, 1076–1083 CrossRef CAS PubMed.
- S. Kobayashi, H. Ishitani and S. Nagayama, Synthesis, 1995, 1195–1202 CrossRef CAS.
- M. Fochi, L. Caruana and L. Bernardi, Synthesis, 2014, 46, 135–157 CrossRef.
- B. Gerard, M. Welzel O'Shea, E. Donckele, S. Kesavan, L. B. Akella, H. Xu, E. N. Jacobsen and L. A. Marcaurelle, ACS Comb. Sci., 2012, 14, 621–630 CrossRef CAS PubMed.
- G. Dagousset, J. Zhu and G. Masson, J. Am. Chem. Soc., 2011, 133, 14804–14813 CrossRef CAS PubMed.
- M. Hadden, M. Nieuwenhuyzen, D. Potts, P. J. Stevenson and N. Thompson, Tetrahedron, 2001, 57, 5615–5624 CrossRef CAS.
- H. Xu, S. J. Zuend, M. G. Woll, Y. Tao and E. N. Jacobsen, Science, 2010, 327, 986–990 CrossRef CAS PubMed.
- K. M. Witherup, R. W. Ransom, A. C. Graham, A. M. Bernard, M. J. Salvatore, W. C. Lumma, P. S. Anderson, S. M. Pitzenberger and S. L. Varga, J. Am. Chem. Soc., 1995, 117, 6682–6685 CrossRef CAS.
- B. Maji, S. A. Gangopadhyay, M. Lee, M. Shi, P. Wu, R. Heler, B. Mok, D. Lim, S. U. Siriwardena, B. Paul, V. Dančík, A. Vetere, M. F. Mesleh, L. A. Marraffini, D. R. Liu, P. A. Clemons, B. K. Wagner and A. Choudhary, Cell, 2019, 177, 1067–1079 CrossRef CAS PubMed.
- M. Klika Škopić, K. Götte, C. Gramse, M. Dieter, S. Pospich, S. Raunser, R. Weberskirch and A. Brunschweiger, J. Am. Chem. Soc., 2019, 141(26), 10546–10555 CrossRef PubMed.
- H. Nagarajaiah, A. Mukhopadhyay and J. N. Moorthy, Tetrahedron Lett., 2016, 57, 5135–5149 CrossRef CAS.
- M. K. Raj, H. S. P. Rao, S. G. Manjunatha, R. Sridharan, S. Nambiar, J. Keshwan, J. Rappai, S. Bhagat, B. S. Shwetha, D. Hegde and U. Santhosh, Tetrahedron Lett., 2011, 52, 3605–3609 CrossRef.
- L.-Z. Gong, X.-H. Chen and X.-Y. Xu, Chem.–Eur. J., 2007, 13, 8920–8926 CrossRef CAS PubMed.
- M. Stucchi, G. Lesma, F. Meneghetti, G. Rainoldi, A. Sacchetti and A. Silvani, J. Org. Chem., 2016, 81, 1877–1884 CrossRef CAS PubMed.
- Y. Ma, C. Qian, L. Wang and M. Yang, J. Org. Chem., 2000, 65, 3864–3868 CrossRef CAS PubMed.
- B. C. Ranu, A. Hajra and U. Jana, J. Org. Chem., 2000, 65, 6270–6272 CrossRef CAS PubMed.
- J. C. Cochran, J. E. Gatial III, T. M. Kapoor and S. P. Gilbert, J. Biol. Chem., 2005, 280(13), 12658–12667 CrossRef CAS PubMed.
- R. W. Lewis, J. Mabry, J. G. Polisar, K. P. Eagen, B. Ganem and G. P. Hess, Biochem. J., 2010, 49, 4841–4851 CrossRef CAS PubMed.
- J. B. Baell and J. W. M. Nissink, ACS Chem. Biol., 2018, 13, 36–44 CrossRef CAS PubMed.
- https://enamine.net/library-synthesis/real-compounds/real-compound-libraries .
- K. T. Nguyen, L. C. Blum, R. van Deursen and J.-L. Reymond, ChemMedChem, 2009, 4, 1803–1805 CrossRef CAS PubMed.
- W. H. B. Sauer and M. K. Schwarz, J. Chem. Inf. Comput. Sci., 2003, 43, 987–1003 CrossRef CAS PubMed.
- J. Barluenga, C. Mateos, F. Aznar and C. Valde's, Org. Lett., 2002, 4(11), 1971–1974 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and additional tables for screening experiments, reaction optimization experiments and reaction scope. Copies of HPLC traces and MALDI-MS spectra are shown for all discussed DNA conjugates. See DOI: 10.1039/c9sc04708e |
| This journal is © The Royal Society of Chemistry 2019 |