
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile triflic acid-catalyzed α-1,2-cis-thio glycosylations: scope and application to the synthesis of S-linked oligosaccharides, glycolipids, sublancin glycopeptides, and TN/TF antigens†
Sanyong
Zhu
a,
Ganesh
Samala
a,
Eric T.
Sletten
b,
Jennifer L.
Stockdill
 *a and
Hien M.
Nguyen
*a
*a and
Hien M.
Nguyen
*a
aDepartment of Chemistry, Wayne State University, Detroit, Michigan 48202, USA. E-mail: hmnguyen@wayne.edu; stockdill@wayne.edu
bDepartment of Chemistry, University of Iowa, Iowa City, Iowa 52242, USA
First published on 1st October 2019
Abstract
Studies of S-linked glycoconjugates have attracted growing interest because of their enhanced chemical stability and enzymatic resistance over O-glycoside counterparts. We here report a facile approach to access α-1,2-cis-S-linked glycosides using triflic acid as a catalyst to promote the glycosylation of a series of thiols with D-glucosamine, galactosamine, glucose, and galactose electrophiles. This method is broadly applicable for the stereoselective synthesis of S-linked glycopeptides, oligosaccharides and glycolipids in high yield and excellent α-selectivity. Many of the synthetic limitations associated with the preparation of these S-linked products are overcome by this catalytic method.
Introduction
Protein glycosylation, one of the most ubiquitous post-translational modifications, typically involves the attachment of carbohydrate chains to proteins through the hydroxyl group of serine or threonine (O-glycans) or the amido group of asparagine (N-glycans).1–5 The resulting glycoproteins exhibit a diverse array of biological functions such as cell adhesion, protein folding, signal transduction, and immune response.1–5 However, naturally occurring glycoproteins exist as mixtures of glycoforms, and their isolation as homogeneous species is a complicated process.6 As such, there is a great demand for methods to efficiently access structurally defined glycoproteins. Recently, replacement of the anomeric oxygen of O-linked glycosides with a sulfur atom to generate S-linked glycosides has attracted considerable attention because of the enhanced resistance of the latter to chemical and enzymatic hydrolysis.7,8 In addition, S-linked glycosides exhibit similar conformational preferences and equal or even improved biological activities compared to their native O-glycoside counterparts.9–11 In this context, S-linked glycan analogs could be utilized as structural mimetics and serve as powerful tools for the biological study of the natural O-linked substrates. The recent discovery of S-glycosylation, the addition of carbohydrate residues to the sulfur atom of cysteine on bacterial peptides,12–14 suggests that naturally existing S-linked glycoproteins may be more widespread than was previously thought and may lead to the development of new therapeutics.Given the significant importance of thiol-containing carbohydrate molecules, methods that enable access to the challenging α-1,2-cis thiol glycosidic linkages are of high synthetic value. Formation of β-1,2-trans glycosides can be readily accomplished by employing electrophiles with C(2)-participatory groups.15 The stereoselective formation of α-1,2-cis glycosides, however, has proven challenging, and a mixture of α- and β-glycosides is often obtained.15 A variety of strategies have been reported for the synthesis of mucin-related α-thiol-containing GalNAc glycopeptide mimetics.16–24 Representative methods include stereoselective preparation of α-GalNAc thiols followed by (1) an SN2 displacement with β-bromoalanine-containing peptides,16,17 (2) site-selective conjugation with aziridine-containing peptides,18,19 or (3) conjugate addition to dehydroalanine-containing peptides.20 SN2 reaction of α-glycosyl thiols with 4-axial triflate glycosides21,22 6-iodinated glycosides,23–25 enzymatic glycosylation,26–29 and metal-catalyzed cross coupling,30,31 have also been reported to generate α-1,2 cis S-linked oligosaccharides and glycopeptides. Despite these important advances over the past decades, numerous challenges remain, including the need for highly specialized coupling partners and the multistep preparation of bromoalanine-, aziridine-, and dehydroalanine-containing amino acid residues or peptides.32 In addition, most of the current methods are limited to specific classes of thiol nucleophiles. To date, there is only one reported method that is applicable for a variety of sulfur nucleophiles, and it uses glycosyl stannane to promote S-linked glycoside formation.31
We recently found that triflic acid, released from nickel triflate, can effectively promote the glycosylations of serine/threonine amino acids and hydroxyl groups of carbohydrates with the C(2)-N-ortho-(trifluoromethyl)benzylidenamino N-phenyl trifluoroacetimidates.33–36 This catalytic system features several advantages such as mild conditions, short reaction time, good yields and excellent levels of α-selectivity. However, the question remains whether it will be suited for S-glycosylations as Lewis acid-promoted reactions are known to be incompatible with thiol nucleophiles.37–40 Lewis acid-promoted S-glycosylations were underutilized with a few limited examples.37–40 The reactions generally proceed to provide the coupling products with low yields and require stoichiometric amount of promoter. An inherent issue with the S-nucleophiles is their tendency to undergo oxidation to form disulfide and other side products.38 They also react with the TMSOTf catalyst during the glycosylation process.40 All those factors make it more challenging to handle S-nucleophiles than their O-counterparts.
Herein we report a distinct approach to S-linked glycosylations: a triflic acid catalyst is shown to enable the facile α-1,2-cis synthesis of thiol-oligosaccharides, glycopeptide of antimicrobial sublancin, S-linked tumor-associated TN/TF antigens, and thiol-glycolipids. This strategy obviates the need for substrate prefunctionalization and can proceed by direct coupling of thiol-containing molecules or cysteine-containing peptides with N-phenyl trifluoroacetimidates.
Results and discussions
To test our hypothesis, we initiated our study by examining the glycosylation of N-Cbz cysteine methyl ester 2 with C(2)-N-ortho-(trifluoromethyl)benzylidene glucosamine N-phenyl trifluoroacetimidate 1 under previously established conditions (Table 1).36 To our delight, the coupling of 2 with 1 was successful using 15 mol% nickel triflate, Ni(OTf)2, (entry 1). As we expected based on the previously reported conditions,36 the reaction with 5 mol% triflic acid, TfOH, proceeded to completion within 1 h at 35 °C (entry 2) to afford the desired thiol-linked glycoconjugate 4 in 68% yield with excellent selectivity (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β > 20:1). This result is consistent with our recently reported mechanism of the triflic acid-catalyzed α-selective 1,2-cis glycosylation with N-phenyl trifluoroacetimidate electrophiles.36 Specifically, triflic acid engages in the activation of electrophile 1 to generate a glycosyl triflate intermediate, which then undergoes equilibration from the stable α-anomer to the more reactive β-anomer. Subsequent SN2-like displacement of the reactive β-anomer of glycosyl triflate by 2 results in the formation of 4 with exclusive α-configuration.
:β > 20:1). This result is consistent with our recently reported mechanism of the triflic acid-catalyzed α-selective 1,2-cis glycosylation with N-phenyl trifluoroacetimidate electrophiles.36 Specifically, triflic acid engages in the activation of electrophile 1 to generate a glycosyl triflate intermediate, which then undergoes equilibration from the stable α-anomer to the more reactive β-anomer. Subsequent SN2-like displacement of the reactive β-anomer of glycosyl triflate by 2 results in the formation of 4 with exclusive α-configuration.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 (equiv.) | 2 or 3 (equiv.) | Catalyst | Temp. (°C) | Time (h) |
4 or 5 yield (α:β) |
| a The reaction was conducted with 0.1–0.2 mmol of donor 1. Yields of the isolated product averaged two runs. The (α/β) ratios were determined by 1H NMR analysis. | ||||||
| 1 | 1 | 2 (1.5) | 15 mol% Ni(OTf)2 | 35 | 16 |
4: 66% (>20:1) |
| 2 | 1 | 2 (1.5) | 5 mol% TfOH | 35 | 1 |
4: 64% (>20:1) |
| 3 | 1 | 2 (1.5) | 5 mol% TfOH | 25 | 2 |
4: 68% (>20:1) |
| 4 | 1 | 2 (1.5) | 1 mol% TfOH | 25 | 20 |
4: 67% (>20:1) |
| 5 | 1.5 | 2 (1.0) | 3 mol% TfOH | 25 | 3 |
4: 76% (>20:1) |
| 6 | 2 | 2 (1.0) | 3 mol% TfOH | 25 | 3 |
4: 81% (>20:1) |
| 7 | 2 | 2 (1.0) | 5 mol% TfOH | 25 | 1 |
4: 80% (>20:1) |
| 8 | 2 | 3 (1.0) | 5 mol% TfOH | 25 | 1 |
5: 78% (>20:1) |
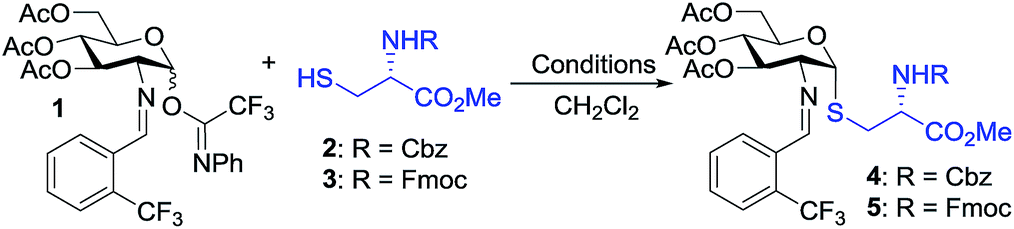
We observed that while the reaction proceeded to completion faster with use of triflic acid, the glycal elimination product derived from donor 1 was also detected (see Fig. S1† for details). Lowering the reaction temperature (entry 3) and catalyst loading (entry 4) provided a similar outcome (Table 1). To address the problem of 1 from undergoing elimination, the cysteine residue 2 was utilized as the limiting reagent in the presence of 1.5 equiv. of donor 1. This modification improved the yield to 76% (entry 5). Further increasing the donor 1 to two equivalents gave the desired coupling product 4 in an enhanced 81% yield while maintaining high levels of selectivity (entry 6). Most notably, the glycosylation reached completion within 1 h at 25 °C with use of 5 mol% TfOH to provide 4 with comparable yield and α-selectivity (entry 7). The cysteine residue 3 with N-Fmoc protection, commonly utilized in the solid-phase peptide synthesis (SPPS), also displayed good efficiency (entry 8) to provide the desired glycoconjugate 5 (Table 1) in good yield and α-selectivity.
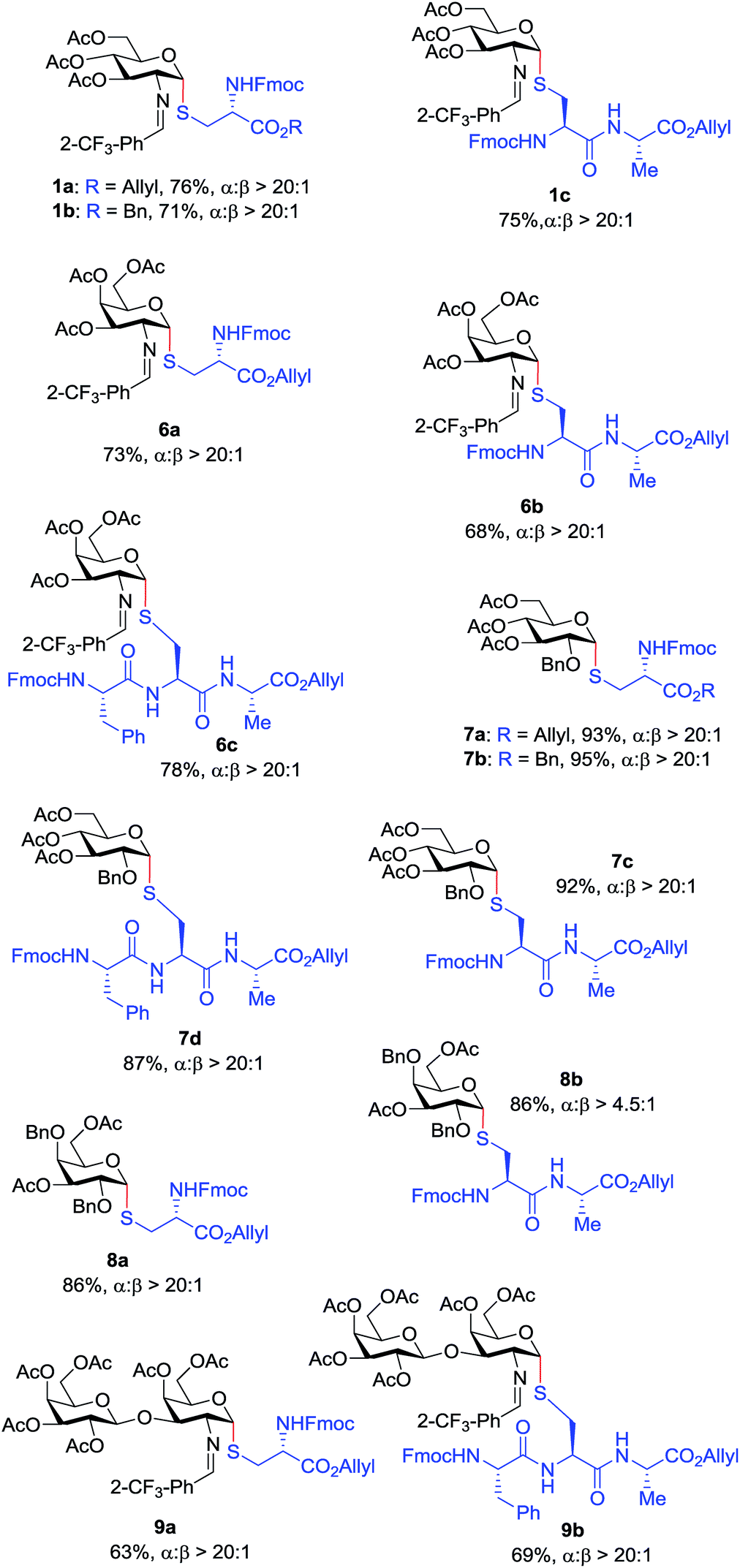
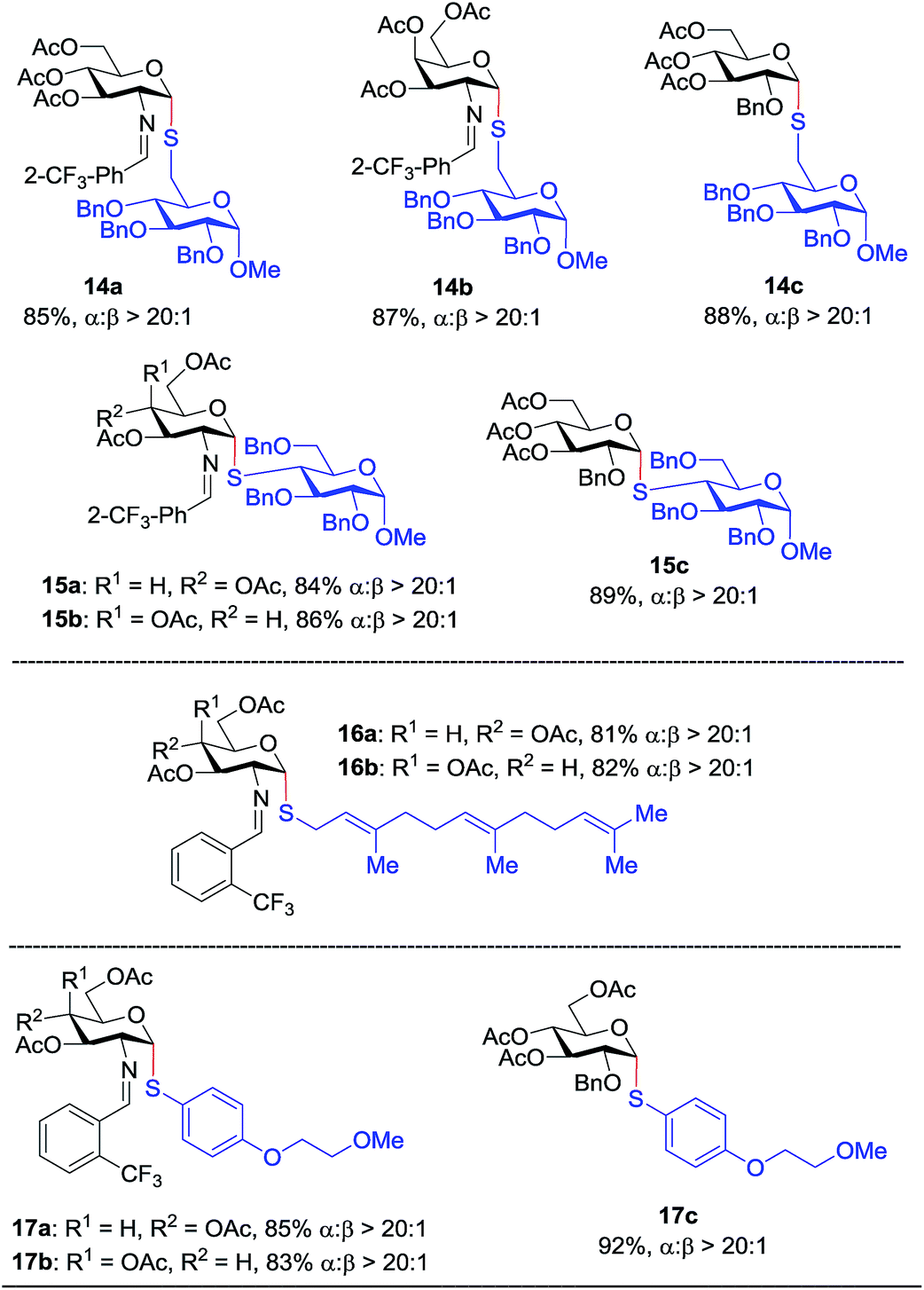
With the optimized conditions in hand, we evaluated the scope of the triflic acid-catalyzed S-glycosylations (Fig. 1) using glycosyl N-phenyl trifluoroacetimidate donors (1, 6–9) and thiol-containing molecules (10–17). Based on our previous study,36 we anticipated that N-phenyl trifluoroacetimidates 1, 6 and 7 would exhibit high α-selectivity because their C(2)-N-benzylidene and C(2)-benzyl ether can modulate the electronic properties of the anomeric carbon, facilitating isomerization of the glycosyl triflate intermediate. Indeed, both donors 1 and 6 were effectively glycosylated to cysteine amino acids 10 and 11 to provide 1a, 1b, and 6a, respectively, in good yield and with excellent α-selectivity (Table 2). There are several underlying factors that could influence the stereochemical outcome of the coupling event. To confirm that the α-selectivity arise from the directing ability of the C(2)-N-benzylidene group, we also coupled C(2)-azido donors 18 and 19 with cysteine residue 10 under standard conditions (Scheme 1). The desired products 18a and 19a were obtained with poor levels of α-selectivity (α:β = 1.6:1 to 2:1).
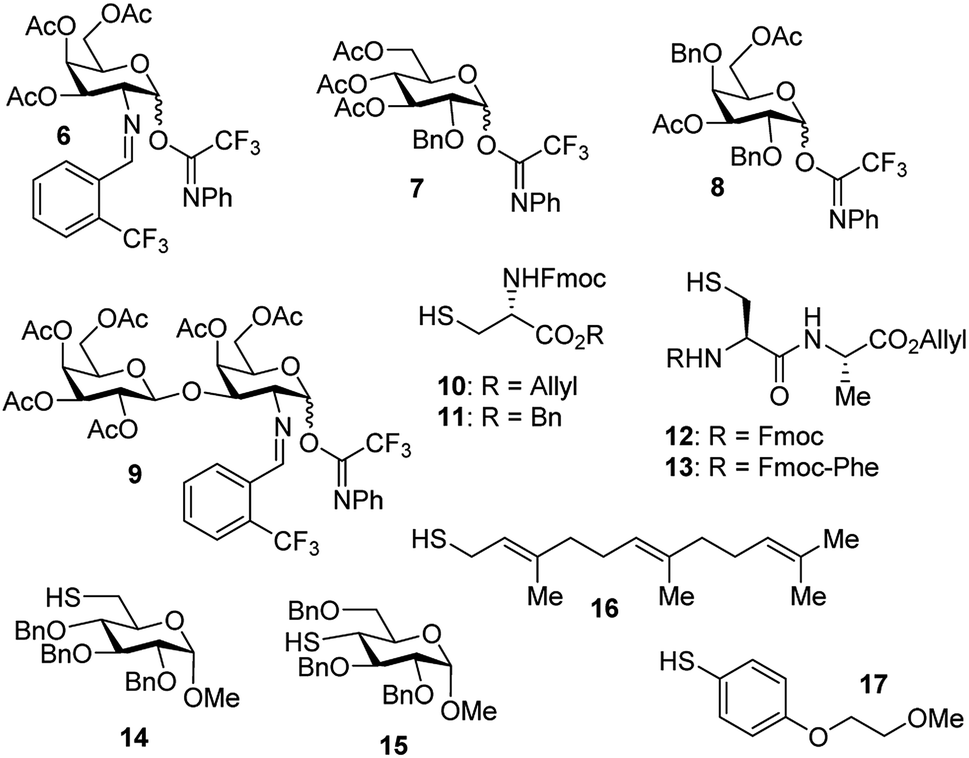 | ||
| Fig. 1 Carbohydrate donors and thiol-containing acceptors. | ||
| a All reactions were conducted with donor (2 equiv.), acceptor (1 equiv.) and 5 mol% TfOH in CH2Cl2 at 25 °C for 1 h. Yields of isolated S-linked glycopeptides averaged two runs. The (α/β) ratios were determined by 1H NMR analysis. |
|---|
|
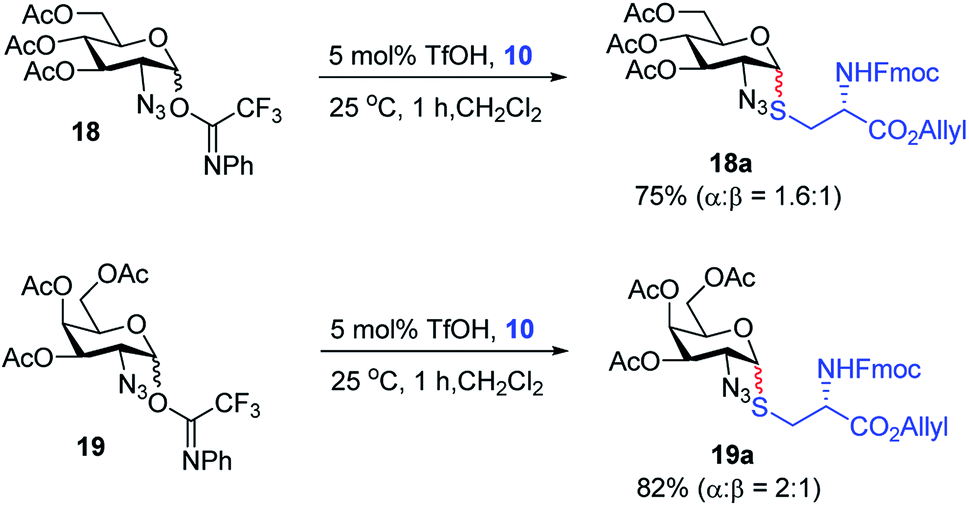 | ||
| Scheme 1 Glycosylation with C(2)-azido donors 18 and 19. | ||
These results in Scheme 1 support the importance of the C(2)-N-benzylidene group in the triflic acid-catalyzed stereoselective α-1,2-cis S-linked glycosylation. Next, we expanded nucleophilic scope from single cysteine amino acid to more complex peptides. Both dipeptide 12 and tripeptide 13 (Fig. 1) performed well under the standard conditions with 1 and 6 to afford glycopeptides 1c, 6b, and 6c (Table 2), presaging the potential utility of this transformation for access S-linked glycopeptides of tumor-associated mucin TN and TF antigens (vide infra).11,44
In addition to C(2)-N-benzylidene electrophiles 1 and 6, the C(2)-O-benzyl protecting group is tolerated. Glucose donor 7 (Table 2) was an effective electrophilic partner, providing the coupling products 7a–7d in excellent yields (87–95%) with exclusive α-configuration. More importantly, donor 7 does not generate a glycal elimination product. Employing the axial 4-O-benzyl protected donor 8 in place of the equatorial 4-O-acetyl group (7) slightly diminished the α-selectivity (8a: α:β > 20:1, 8b: α:β = 4.5:1). Nevertheless, this result illustrates the ability of this catalytic system to overturn the inherent bias of D-galactose donors whose axial C(4)-O-benzyl protecting group has been reported to favor β-products.41 The difference in α-selectivity between the coupling products 8a and 8b could be explained by the relative rate for anomerization of the α-to the β-glycosyl triflate intermediate generated from the reaction of glycoyl electrophile 8 with triflic acid. Since a dipeptide is a more reactive nucleophile than a cysteine amino acid residue, less time is allowed for anomerization of the α-triflate to the more reactive β-triflate, resulting in decreasing the α-selectivity observed in the coupling product 8b. Finally, disaccharide donor 9, a carbohydrate motif of TF antigen,42 was readily coupled to provide the corresponding 1,2-cis S-glycoconjugates 9a and 9b (63–69%, α:β > 20:1).
Inspired by the above discovery, we next examined the scope with respect to nucleophilic coupling partners 14–17. As illustrated in Table 3, the glycosylations of primary (14) and secondary (15) thiol acceptors with donors 1, 6, and 7 proceeded smoothly to produce the desired S-linked disaccharides 14a–c and 15a–c in good yields (84–89%) with exclusive α-selectivity. The trans,trans-farnesyl mercaptan 16 was also a suitable nucleophile (16a, b), revealing the potential utility of this method for preparation of S-linked glycolipids. Recently, Messaoudi and co-workers reported the Pd-mediated cross coupling of aryl iodides with β-glycosyl thiols to generate S-linked glycosides with exclusive β-configuration.30 For comparison, we examined the coupling efficiency of our method with aryl thiol 17. To our delight, the reaction compared favorably with excellent α-stereoselectivity and yield (17a–c).
We expect that the triflic acid-catalyzed α-1,2-cis S-linked glycosylation method will be particularly useful when applied to the synthesis of bioactive glycopeptides. To establish the potential of a late-stage glycosylation approach, we prepared the tetrapeptide sequence surrounding the β-linked D-glucose unit in sublancin 20 (Scheme 2). This S-linked glycosyl unit is essential for antimicrobial activity.12 SunS, the recently discovered S-glycosyl transferase enzyme, is responsible for the unusual glycosylation of cysteine residues with carbohydrates.12 It has a relaxed substrate specificity and is able to glycosylate other hexose sugars, which has allowed its use in the preparation of sublancin analogs bearing other β-linked glycans.12,13 However, to the best of our knowledge, the activity of α-1,2-cis S-linked sublancin analogs has not been investigated,10 presumably because of the difficulty of α-1,2-cis S-glycosylations. Our current catalytic method would provide an ideal approach for the assembly of such analogs. Accordingly, we investigated the coupling of tetrapeptide 21 with glucose donor 7 in the presence of 5 mol% triflic acid. The glycosylation proceeded to completion within an hour to provide glycopeptide 22 in 65% yield with exclusive α-selectivity (Scheme 2).
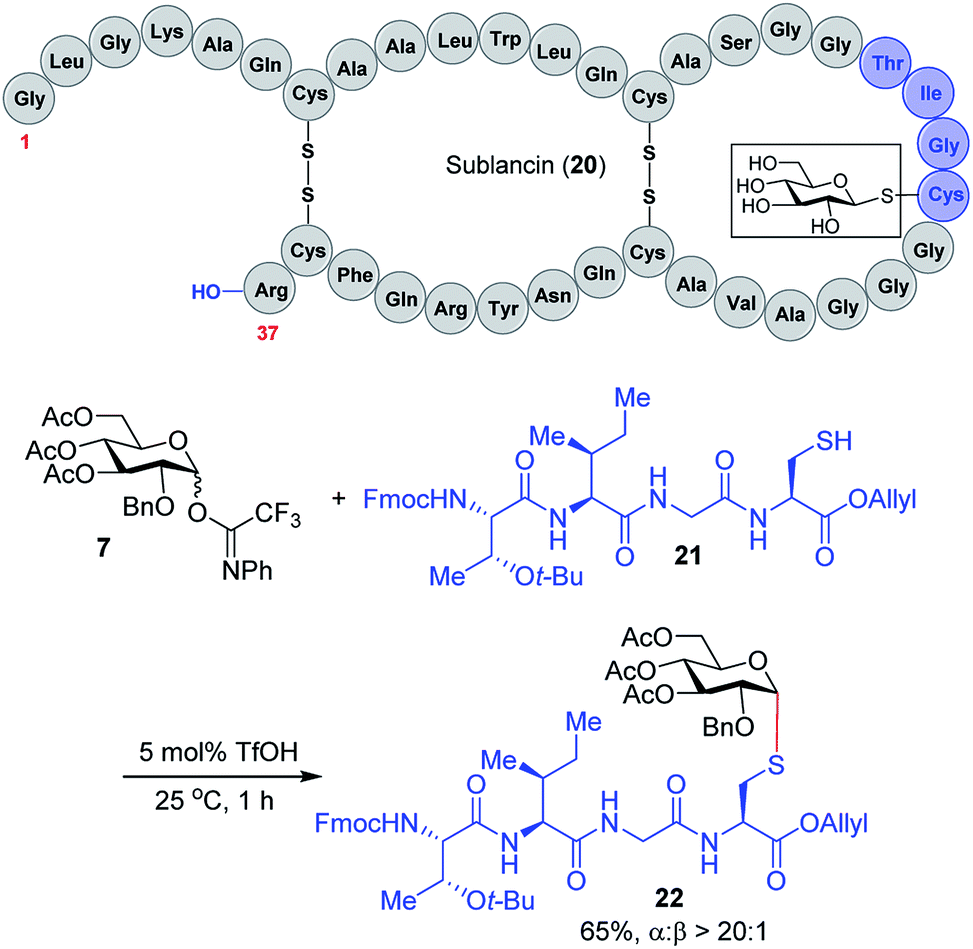 | ||
| Scheme 2 Synthesis of the sublancin glycopeptide fragment. | ||
Another important application of this method is the preparation of S-linked glycopeptides as analogs of MUC1 type tumor-associated TN/TF antigens. MUC1 is a glycoprotein that consists of a tandem 20 amino acids repeating unit, with five possible O-glycosylation sites (serine or threonine) (Scheme 3).43 In normal cells, the protein backbone is decorated with complex oligosaccharides, while the glycosylation is incomplete in cancer cells. As such, multiple epitopes such as TN/TF antigens are exposed to the immune system and can be targeted for the development of antitumor vaccines. A therapeutic vaccine employing multivalent TN-antigen clusters and CS4+ T-cell epitopes (MAG-Tn3) has entered to clinical trials.44 It has been reported that anti-TN monoclonal antibodies (mAbs) have a binding preference for TN-Ser antigen, and the short MUC1 pentapeptide, GVTSA, is a suitable binding motif for mAbs.43 Since the S-linked TN antigen has been proved to enhance the immunogenicity,11 we sought to prepare S-linked TN/TF antigen mimetics by exchanging the  sequence (I) for the
sequence (I) for the  sequence (II) (Scheme 3). Accordingly, we investigated the coupling of GVTCA pentapeptide 23 with both monosaccharide donor 6 and disaccharide donor 9 in the presence of 5 mol% TfOH. The coupling proceeded smoothly at 35 °C for 1 h to provide the corresponding 1,2-cis S-linked glycopeptides 24 (TN) and 25 (TF) in 76% and 73% yield, respectively, with exclusive α-selectivity (Scheme 3).
sequence (II) (Scheme 3). Accordingly, we investigated the coupling of GVTCA pentapeptide 23 with both monosaccharide donor 6 and disaccharide donor 9 in the presence of 5 mol% TfOH. The coupling proceeded smoothly at 35 °C for 1 h to provide the corresponding 1,2-cis S-linked glycopeptides 24 (TN) and 25 (TF) in 76% and 73% yield, respectively, with exclusive α-selectivity (Scheme 3).
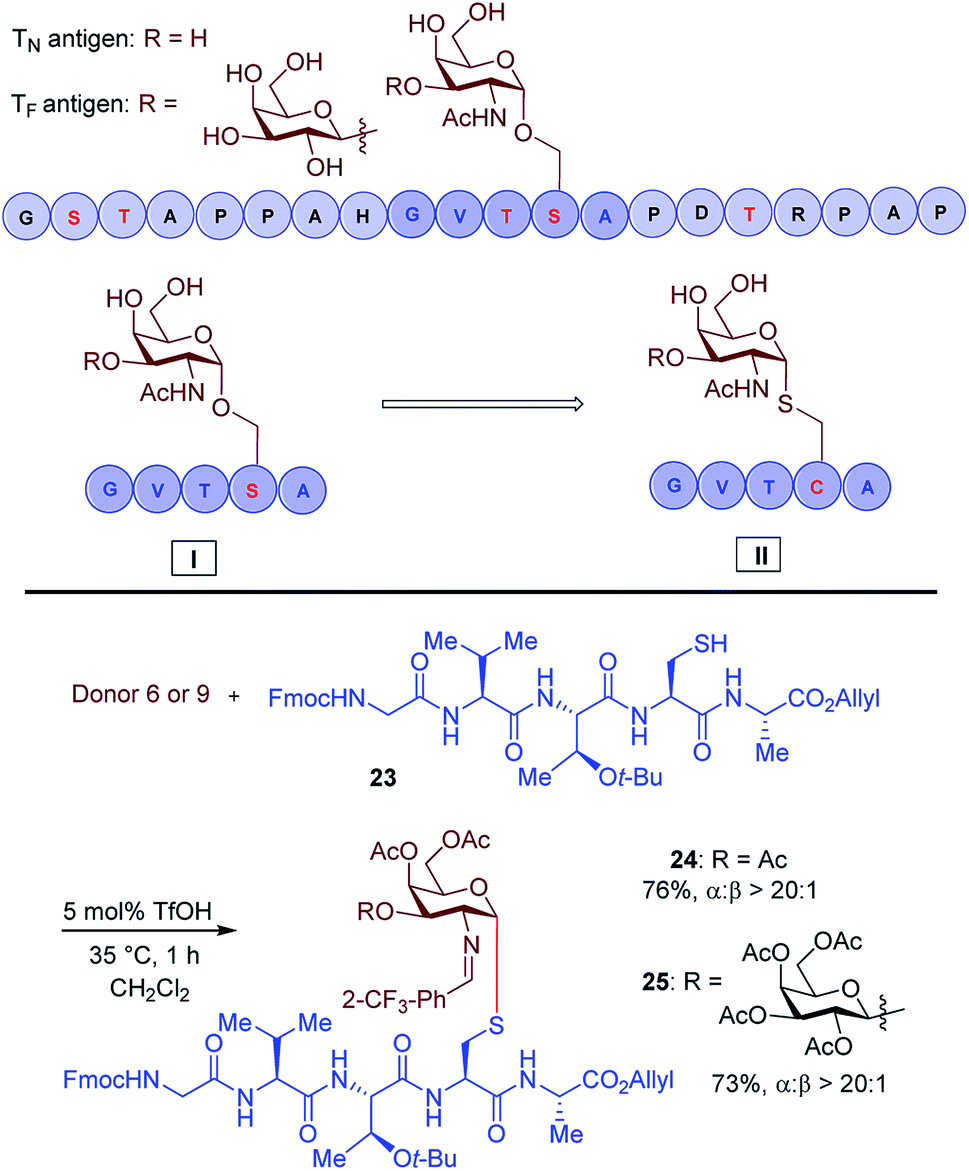 | ||
| Scheme 3 Substrate synthesis of S-linked TN and TF glycopeptide fragments. | ||
Conclusions
In conclusion, we have illustrated the utility of the triflic acid-catalyzed α-1,2-cis thiol glycosylation reaction using stable glycosyl N-phenyl trifluoroacetimidates and thiol-containing molecules. This catalytic system obviates the need for substrate prefunctionalization and furnishes a diverse collection of synthetically valuable S-linked glycopeptides, oligosaccharides, and glycolipids with excellent α-selectivity under mild and operationally simple conditions. Notably, the facile nature of this reaction have also successfully applied to the synthesis of S-linked glycopeptides of antimicrobial sublancin and tumor-associated mucin TN/TF antigens. Our future investigations will focus on transforming the current method to automated synthesis for the synthesis of the α-1,2-cis S-linked glycosides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the National Institutes of Health (R01 GM129475 for J. L. S. and R01 GM098285 for H. M. N.) and the National Science Foundation (CAREER Award CHE-1554752 for J. L. S.). We also thank the Lumigen Center at Wayne State University for instrumental assistance.Notes and references
- J. J. Beintema, J. Mol. Evol., 1986, 24, 118 CrossRef CAS PubMed.
- M. Kanagawa and T. Toda, J. Biochem., 2018, 163, 359 CrossRef CAS PubMed.
- N. Kaur and P. Jauhari, Mol. Cancer Ther., 2013, 12, A27 Search PubMed.
- J. Z. Wang, I. GrundkeIqbal and K. Iqbal, Nat. Med., 1996, 2, 871 CrossRef CAS PubMed.
- H. Watarai, R. Nozawa, A. Tokunaga, N. Yuyama, M. Tomas, A. Hinohara, K. Ishizaka and Y. P. Ishii, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13251 CrossRef CAS PubMed.
- C. Li and L.-X. Wang, Chem. Rev., 2018, 118, 8359 CrossRef CAS PubMed.
- C. Aydillo, I. Companon, A. Avenoza, J. H. Busto, F. Corzana, J. M. Peregrina and M. M. Zurbano, J. Am. Chem. Soc., 2014, 136, 789 CrossRef CAS PubMed.
- C. A. De Leon, P. M. Levine, T. W. Craven and M. R. Pratt, Biochemistry, 2017, 56, 3507 CrossRef CAS PubMed.
- A. M. Melo, L. Zhang, E. F. Dockry, A. Petrasca, Y. G. Ghnewa, E. P. Breen, M. E. Morrissey, C. O'Reilly, R. Bruen, A. O'Meara, J. Lysaght, X. M. Zhu and D. G. Doherty, Glycobiology, 2018, 28, 512 CrossRef CAS PubMed.
- Z. Amso, S. W. Bisset, S. H. Yang, P. W. R. Harris, T. H. Wright, C. D. Navo, M. L. Patchett, G. E. Norris and M. A. Brimble, Chem. Sci., 2018, 9, 1686 RSC.
- I. Companon, A. Guerreiro, V. Mangini, J. Castro-Lopez, M. Escudero-Casao, A. Avenoza, J. H. Busto, S. Castillon, J. Jimenez-Barbero, J. L. Asensio, G. Jimenez-Oses, O. Boutureira, J. M. Peregrina, R. Hurtado-Guerrero, R. Fiammengo, G. J. L. Bernardes and F. Corzana, J. Am. Chem. Soc., 2019, 141, 4063 CrossRef CAS PubMed.
- T. J. Oman, J. M. Boettcher, H. Wang, X. N. Okalibe and W. A. van der Donk, Nat. Chem. Biol., 2011, 7, 78 CrossRef CAS PubMed.
- S. Biswas, C. V. Garcia De Gonzalo, L. M. Repka and W. A. van der Donk, ACS Chem. Biol., 2017, 12, 2965 CrossRef CAS PubMed.
- C. Wu, S. Biswas, C. V. Garcia De Gonzalo and W. A. van der Donk, ACS Infect. Dis., 2019, 5, 454 CrossRef CAS PubMed.
- M. J. McKay and H. M. Nguyen, ACS Catal., 2012, 2, 1563 CrossRef CAS PubMed.
- C. F. Liang, M. C. Yan, T. C. Chang and C. C. Lin, J. Am. Chem. Soc., 2009, 131, 3138 CrossRef CAS PubMed.
- X. M. Zhu and R. R. Schmidt, Chem.–Eur. J., 2004, 10, 875 CrossRef CAS PubMed.
- D. P. Galonic, N. D. Ide, W. A. van der Donk and D. Y. Gin, J. Am. Chem. Soc., 2005, 127, 7359 CrossRef CAS PubMed.
- D. P. Galonic, W. A. van der Donk and D. Y. Gin, J. Am. Chem. Soc., 2004, 126, 12712 CrossRef CAS PubMed.
- M. I. Gutierrez-Jimenez, C. Aydillo, C. D. Navo, A. Avenoza, F. Corzana, G. Jimenez-Oses, M. M. Zurbano, J. H. Busto and J. M. Peregrina, Org. Lett., 2016, 18, 2796 CrossRef CAS PubMed.
- L. Lazar, M. Csavas, M. Herczeg, P. Herczegh and A. Borbas, Org. Lett., 2012, 14, 4650 CrossRef CAS PubMed.
- S. Mandal and U. J. Nilsson, Org. Biomol. Chem., 2014, 12, 4816 RSC.
- G. J. L. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. El Oualid, T. D. W. Claridge and B. G. F. Davis, Angew. Chem., Int. Ed., 2008, 47, 2244–2247 CrossRef CAS PubMed.
- X. M. Zhu, T. Haag and R. R. Schmidt, Org. Biomol. Chem., 2004, 2, 31 RSC.
- E. Calce, G. Digilio, V. Menchise, M. Saviano and S. De Luca, Chem.–Eur. J., 2018, 24, 6231 CrossRef CAS PubMed.
- J. R. Rich, A. Szpacenko, M. M. Palcic and D. R. Bundle, Angew. Chem., Int. Ed., 2004, 43, 613 CrossRef CAS PubMed.
- J. R. Rich, W. W. Wakarchuk and D. R. Bundle, Chem.–Eur. J., 2006, 12, 845 CrossRef CAS PubMed.
- G. Tegl, J. Hanson, H. M. Chen, D. H. Kwan, A. G. Santana and S. G. Withers, Angew. Chem., Int. Ed., 2019, 58, 1632 CrossRef CAS PubMed.
- H. Wang, T. J. Oman, R. Zhang, C. V. G. De Gonzalo, Q. Zhang and W. A. van der Donk, J. Am. Chem. Soc., 2014, 136, 84 CrossRef CAS PubMed.
- D. Montoir, M. Amoura, Z. E. Ababsa, T. M. Vishwanatha, E. Yen-Pon, V. Robert, M. Beltramo, V. Piller, M. Alami, V. Aucagne and S. Messaoudi, Chem. Sci., 2018, 9, 8753 RSC.
- F. Zhu, E. Miller, S. Q. Zhang, D. Yi, S. O'Neill, X. Hong and M. A. Walczak, J. Am. Chem. Soc., 2018, 140, 18140 CrossRef CAS PubMed.
- K. Pachamuthu and R. R. Schmidt, Chem. Rev., 2006, 106, 160 CrossRef CAS PubMed.
- E. A. Mensah and H. M. Nguyen, J. Am. Chem. Soc., 2009, 131, 8778 CrossRef CAS PubMed.
- E. A. Mensah, F. Yu and H. M. Nguyen, J. Am. Chem. Soc., 2010, 132, 14288 CrossRef CAS PubMed.
- F. Yu, M. S. McConnell and H. M. Nguyen, Org. Lett., 2015, 17, 2018 CrossRef CAS PubMed.
- E. T. Sletten, Y.-J. Tu, H. B. Schlegel and H. M. Nguyen, ACS Catal., 2019, 9, 2110 CrossRef CAS.
- C. A. De Leon, G. Lang, M. I. Saavedra and M. R. Pratt, Org. Lett., 2018, 20, 5032 CrossRef CAS PubMed.
- E. Repetto, V. E. Manzano, M. L. Uhrig and O. Varela, J. Org. Chem., 2012, 77, 253 CrossRef CAS PubMed.
- A. Noel, B. Delpech and D. Crich, Org. Lett., 2012, 14, 4138 CrossRef CAS PubMed.
- G. H. Xin and X. M. Zhu, Tetrahedron Lett., 2012, 53, 4309 CrossRef CAS.
- S. Chatterjee, S. Moon, F. Hentschel, K. Gilmore and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 11942 CrossRef CAS PubMed.
- S. Nath and P. Mukherjee, Trends Mol. Med., 2014, 20, 332 CrossRef CAS PubMed.
- H. Coelho, T. Matsushita, G. Artigas, H. Hinou, F. J. Canada, R. Lo-Man, C. Leclerc, E. J. Cabrita, J. Jimenez-Barbero, S. Nishimura, F. Garcia-Martin and F. Marcelo, J. Am. Chem. Soc., 2015, 137, 12438 CrossRef CAS PubMed.
- R. Lo-Man, S. Vichier-Guerre, R. Perraut, E. Deriaud, V. Huteau, L. BenMohamed, O. M. Diop, P. O. Livingston, S. Bay and C. Leclerc, Cancer Res., 2004, 64, 4987 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, analytical data, and copties of 1H and 13C NMR spectra. See DOI: 10.1039/c9sc04079j |
| This journal is © The Royal Society of Chemistry 2019 |