
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasensitive recognition of AP sites in DNA at the single-cell level: one molecular rotor sequentially self-regulated to form multiple different stable conformations†
Beidou
Feng
a,
Kui
Wang
a,
Yonggang
Yang
a,
Ge
Wang
b,
Hua
Zhang
 *a,
Yufang
Liu
a and
Kai
Jiang
a
*a,
Yufang
Liu
a and
Kai
Jiang
a
aHenan Key Laboratory of Green Chemical Media and Reactions, Ministry of Education, Key Laboratory of Green Chemical Media and Reactions; Collaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, Henan Key Laboratory of Organic Functional Molecules and Drug Innovation, School of Chemistry and Chemical Engineering, School of Environment, College of Physics and Materials Science, Henan Normal University, Xinxiang 453007, China. E-mail: zhanghua1106@163.com
bXinxiang Medical University, Xinxiang 453000, P. R. China
First published on 20th September 2019
Abstract
The AP site is a primary form of DNA damage. Its presence alters the genetic structure and eventually causes malignant diseases. AP sites generally present a high-speed dynamic change in the DNA sequence. Thus, precisely recognizing AP sites is difficult, especially at the single-cell level. To address this issue, we provide a broad-spectrum strategy to design a group of molecular rotors, that is, a series of nonfluorescent 2-(4-vinylbenzylidene)malononitrile derivatives (BMN-Fluors), which constantly display molecular rotation in a free state. Interestingly, after activating the relevant specific-recognition reaction (i.e., hydrolysis reaction of benzylidenemalononitrile) only in the AP-site cavity within a short time (approximately 300 s), each of these molecules can be fixed into this cavity and can sequentially self-regulate to form different stable conformations in accordance with the cavity size. The different stable conformations possess various HOMO–LUMO energy gaps in their excited state. This condition enables the AP site to emit different fluorescence signals at various wavelengths. Given the different self-regulation abilities of the conformations, the series of molecules, BMN-Fluors, can emit different types of signals, including an “OFF–ON” single-channel signal, a “ratio” double-channel signal, and even a precise multichannel signal. Among the BMN-Fluors derivatives, d1-BMN can sequentially self-regulate to form five stable conformations, thereby resulting in the emission of a five-channel signal for different AP sites in situ. Thus, d1-BMN can be used as a probe to ultrasensitively recognize the AP site with precise fluorescent signals at the single-cell level. This design strategy can be generalized to develop additional single-channel to multichannel signal probes to recognize other specific sites in DNA sequences in living organisms.
Introduction
AP sites are specific sites with a hydrophobic cavity within a DNA sequence, having neither purine nor pyrimidine bases.1,2 The AP site is a classic damage marker for a DNA sequence. It is caused mainly by reactive oxygen species (ROS) and occurs within tens of seconds in living organisms.2 The presence of AP sites can indicate the degree of DNA damage. Increasingly more serious DNA damage is signified by a large AP-site cavity.1,2 In addition, when DNA damage forms, the fast-changing AP site in DNA can cause many severe hazards for a living organism. For example, the AP site stalls genetic transcription, thereby leading to the loss of genetic integrity and further accelerating the progression of some diseases, such as cancer, Alzheimer's disease and Parkinson's disease.1,2 AP sites in mitochondrial DNA sequences are passed on to subsequent generations through maternal inheritance to cause many extremely serious genetic diseases.3–8 Thus, a new research focus to recognize and investigate such specific sites in living organisms must be determined.AP sites possess a special structure and reactivities that can spontaneously induce some chemical reactions in living biological systems.9,10 Thus, developing active substrate molecules that can take part in the relevant chemical reactions induced by the AP site is immensely conducive for such site-specific recognition in a DNA sequence.11,12 To date, combined with nuclear magnetic resonance (NMR) spectrometry,13,14 mass spectrometry15,16 and optical techniques,17–20 some substrate molecules have been successfully implemented for quantifying AP sites in DNA.13–20 NMR spectrometry for detecting AP sites is characterized by highly selective signals. However, the sensitivity of signals is low.13,14 Other substrate molecules, under the condition of mass spectrometry, can emit a highly sensitive mass spectrometry signal but cannot in situ detect the AP sites in living cells.15,16 Still other substrate molecules with optical signals, such as UV-visible and fluorescence signals, only exhibit a single optical signal change at one wavelength.17–20 Thus, the stability of the recognition signal (especially fluorescence recognition signal) for the AP sites is relatively poor in living cells. In addition, inherent interference from complex living organisms (e.g., pH, hydrophilicity and hydrophobicity) for the stability of the recognition signal becomes difficult to eliminate. Furthermore, the response times are longer in these reported substrate molecules for AP sites than the formation time of the AP site. Thus, precisely detecting AP sites that appear with rapid and dynamic real-time change is extremely difficult in living cells, especially at the single-cell level. Moreover, these substrate molecules cannot recognize the differences in cavity size of the AP site and thus cannot provide feedback on the degree of DNA damage in real time. Therefore, the current major challenge is to enhance the sensitivity and fidelity of recognition for AP sites, especially for differentiating the cavity sizes of AP sites at the single-cell level.21–23
To enhance the recognition sensitivity and fidelity, inducing molecules to specifically generate a multichannel signal is an effective solution.24–26 Therefore, we constructed a series of 2-(4-vinylbenzylidene)malononitrile derivatives (BMN-Fluors), because BMN derivatives are typically chosen as the basic dye parent structure in the synthesis of chemosensors for nucleic acids. In addition, their spectra can be easily adjusted and are diverse.27,28 This series of BMN-Fluors, which are in constant rotation in a free state, can sequentially self-regulate their conformations to form different stable conformations when they encounter the AP site. BMN-Fluors can emit different types of signals, including an “OFF–ON” single-channel signal, a “ratio” double-channel signal, and even a precise multichannel signal. BMN-Fluors can ultrasensitively recognize AP sites with a high-fidelity multichannel signal at the single-cell level. This broad-spectrum molecular design strategy can be generalized to design additional probes, from single-channel probes to multichannel probes, to recognize specific sites in a DNA sequence in living organisms.
Results and discussion
Design strategy of BMN-Fluors
This work aimed to develop a series of molecular rotors for the ultrasensitive recognition of AP sites in DNA, with a high-fidelity signal at the single-cell level. In terms of molecular design, activating a specific recognition reaction and regulating the recognition signal (i.e., the energy release pathway of a molecular excited state) were selected as the key points. Based on the new key points, we constructed a series of BMN-Fluors (Fig. 1 and Scheme S1†). The hydrolysis reaction of benzylidenemalononitrile derivatives was selected as the recognition reaction (Scheme S1†). Different numbers of the –ONH2 group were introduced as the basic binding sites for the AP site. Given that the –ONH2 group binds to the AP site, BMN-Fluors could be forced to steadily remain in the AP-site cavity. This process enables the AP site to activate the recognition reaction—specifically, the hydrolysis reaction of the BMN-Fluors (Scheme S1†).1,2,9,10,21–23,29 Then, to obtain a high-fidelity recognition signal, we introduced ethenyl as a linker of BMN-Fluors to self-regulate their conformations sequentially and thus form different stable states in the different cavity sizes of the AP sites. These different molecular stable conformations possess different HOMO–LUMO gaps, which enable the emission of different types of fluorescence signals. To extract the true and strong recognition signal further at the single-cell level, 3H-indole or 1H-benzo[d]imidazole derivative groups were introduced into molecules, considering that these derivative groups enable BMN-Fluors to target the AP site and effectively prevent additional damage to DNA.29 These groups can further enhance the hydrogen bonds between BMN-Fluors and the AP site. Thus, conformation stability in the same cavity of the AP site was ensured. Essentially, the presence of 3H-indole or 1H-benzo[d]imidazole derivative groups improved the intensity of recognition signal at the single-cell level by strengthening the molecular electronic push-and-pull system. The synthetic routes of BMN-Fluors and corresponding intermediate products are illustrated in Scheme S2.† By harnessing this design strategy, we aimed at BMN-Fluors to emit multichannel fluorescence signals in situ using the sequentially self-regulated molecule to form different stable conformations after specifically activating its hydrolysis reaction.21–23BMN-Fluors could be further used as a site-specific probe to monitor the AP site and evaluate the DNA damage with ultrasensitivity and high fidelity at the single-cell level. The design strategy for molecules possessing the different types of signals is finally described. Based on this design strategy, we supposed that many molecules possessing the precise signal can be designed to ultrasensitively recognize specific sites in DNA.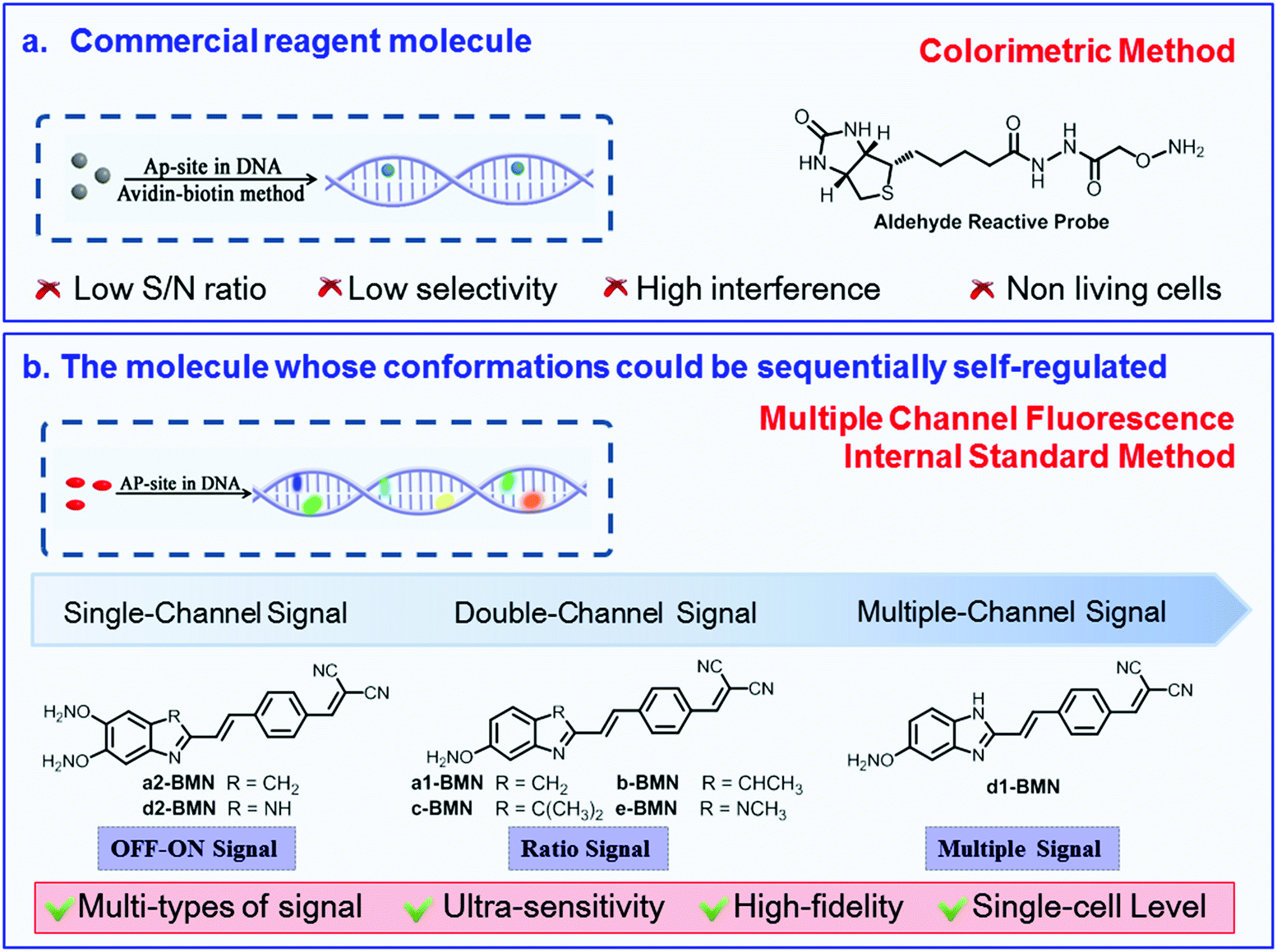 | ||
| Fig. 1 Rational design of the BMN-Fluors molecules with different types of signals for the AP-site. (a) Commercial reagent molecule based on the colorimetric method. (b) Molecule based on the multiple channel fluorescence internal standard method. BMN-Fluors could be sequentially self-regulated to form multiple different stable conformations in the cavity of the AP site and to emit different types of signals with ultra-sensitivity and high fidelity. | ||
Specific recognition of AP sites using different types of signals in PBS buffer
The specific recognition ability and recognition signal of BMN-Fluors for AP sites in DNA were investigated in PBS buffer (pH 7.4) at 37 °C. ROS was used to induce the generation of AP sites in DNA, in accordance with the previously reported method.23 Furthermore, the amount of AP sites in DNA was detected using an AP-site counting kit (Dojindo, Japan, ESI†).d1-BMN in all the molecules (BMN-Fluors) is a typical multichannel signal-emitting molecule used to explain the recognition behavior and signal for AP sites in PBS buffer. In the absence of AP sites, the experiment results showed that d1-BMN (3.0 μM, λex = 400 nm, ε = 1.5 × 105 M−1 cm−1) emits an extremely low signal (Fig. 2a, ΦFreeF = 0.021) at its maximum emission wavelength of 605 nm in PBS buffer. d1-BMN exhibited a unique spectral change when it encountered different AP sites in the DNA. Within the first few seconds of this process, d1-BMN immediately emitted a strong signal at 605 nm, and its fluorescence quantum yield increased to 0.66 (31.4-fold, Table S1† and Fig. 2a). However, fluorescence signal intensity at 605 nm decreased after approximately 300 seconds (Fig. 2b). In addition, a new relevant signal was emitted at different wavelengths when d1-BMN encountered various amounts of AP sites. Fig. 2a shows that new signals appeared, in turn, at 414 nm (Φ = 0.93), 441 nm (Φ = 0.88), 468 nm (Φ = 0.84), 498 nm (Φ = 0.80), 524 nm (Φ = 0.78) and 552 nm (Φ = 0.71), excited at 354 nm (Table S1†), when the number of AP sites were 2, 4, 6, 8, 10 and 14 in double-stranded DNA segments (20 bp), respectively. Their signal intensities decreased in sequence. In other words, d1-BMN exhibited a “multichannel” recognition signal for the different amounts of AP sites. During the abovementioned processes, the recognition signal changes can be observed in the solution by the naked eye (Fig. 2c). However, other BMN-Fluors, such as a2-BMN and d2-BMN, only exhibited a single signal change at one wavelength, that is, an OFF–ON single-channel signal for the AP site (Fig. S1 and Table S1†). Moreover, a1-BMN, b-BMN, c-BMN, and e-BMN displayed a ratio double-channel signal for the AP site (Fig. S1 and Table S1†). Nevertheless, all BMN-Fluors derivatives only showed the relevant types of fluorescence signal changes for AP sites in DNA, even when they encounter other biomacromolecules with aldehyde groups (e.g., 5-formyluracil or 8-OHDG in DNA) and living cell substances in the testing system (Fig. S2†); BMN-Fluors derivatives cannot emit such ratio double-channel and multichannel recognition signal.
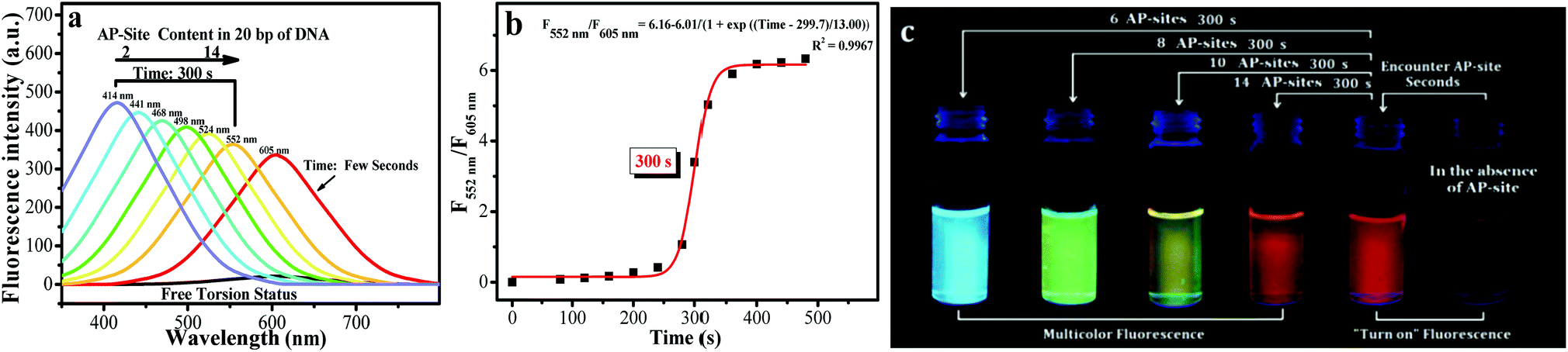 | ||
| Fig. 2 (a) Spectral data of d1-BMN (3.0 μM) for AP sites (2, 4, 6, 8, 10 and 14 AP sites/20 bp DNA, according to the given DNA sequence) in PBS buffer (pH = 7.4) at different reaction times (0, few seconds and 300 s). All DNA sequences with AP sites were synthesized by Thermo Fisher Scientific; the DNA sequence information is listed in the Procedures section of ESI.† Excitation wavelength = 400 nm and 354 nm. (b) The reaction time of d1-BMN (3.0 μM) for AP sites (14 AP sites/20 bp DNA) in PBS buffer (pH = 7.4). (c) d1-BMN emitting multicolor fluorescence, visible to the naked eye, for different contents of AP sites (6, 8, 10 and 14 AP sites/20 bp DNA) in PBS buffer (pH = 7.4) at different reaction times (0, few seconds and 300 s).23 The number of AP sites in the DNA sequence was quantitatively detected using an AP-site counting kit (Dojindo, Japan, see ESI†). | ||
Specific recognition mechanism
To explain this series of OFF–ON single-channel, ratio double-channel to multichannel signal change phenomena and illustrate the critical design factors for a molecule possessing the high-fidelity signals simultaneously to ultrasensitively recognize the AP site, 1H NMR spectra, HPLC-HRMS, Gaussian 16 molecular docking and molecular dynamics simulations were used to characterize this recognition process. AP sites result from the cleavage of the N-glycosidic bonds, leading to removal of the bases and formation of 2′-deoxyribose residues in DNA. That is, (3R,4R)-4-hydroxy-3,5-dimethoxypentanal (HDM, Scheme S1†) is the residue group in DNA after the bases are removed.30,31 To verify the binding effect of d1-BMN and AP sites and to characterize the molecular structure change in d1-BMN during the recognition process of AP sites, HDM was selected as the small-fragment molecule to replace macromolecular DNA containing AP sites in the NMR titration of this work.Gaussian 16 simulation (ESI-2.avi†) indicated that the molecular skeleton of the serial derivatives BMN-Fluors are constantly in rotation under unconstrained environments. In addition, their molecular excited-state energy is nearly lost by nonradiative transitions. This phenomenon resulted in an extremely low fluorescence (Fig. 2a, S1 and Table S1†) and is the basic reason for the OFF–ON single-channel signal.32,33
Fig. 3a demonstrates the –ONH2 group of d1-BMN initially reacted with the –CHO group of HDM when it encountered an AP site, forming the new intermediate product d1-BMN-NO through aldehyde–ammonia condensation reaction. On the basis of this kind of structural transformation, d1-BMN can be bound to a biomolecule possessing the –CHO groups, such as an AP site and 5-formyluracil. This binding partly limits the molecular rotation, which is the main reason that all BMN-Fluors derivatives, including d1-BMN, emit enhanced red fluorescence for the AP site and 5-formyluracil (Fig. S2d†). Furthermore, 1H NMR spectra (Fig. 3b) showed that d1-BMN-NO subsequently generates the peculiar structure changes only in a simulation environment similar to the AP-site cavity, but not to 5-formyluracil. First, the –ph–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CN)2 group of d1-BMN-NO reacts to generate a –ph–CH(OH)–CH(CN)2 group and form another new intermediate product d1-BMN-ts (Fig. 3c). d1-BMN-ts generation is the key point that effectively accelerates the rate-limiting step of the entire recognition reaction, that is, the benzylidenemalononitrile hydrolysis reaction.21–23 With this kind of acceleration, the –ph–CH(OH)–CH(CN)2 group of d1-BMN-ts instantly changes to the –ph–CHO group to form the final product (d1-BMN-CHO, Fig. 3c). The molecular docking (Fig. 4a and b) results indicated that only d1-BMN-CHO can be forced to steadily stay in one DNA crystal structure that has many AP sites in a certain conformation by the hydrogen bonds (the classical hydrogen bonds, indicated by the green dotted line, and the non-classical hydrogen bonds, indicated by the white dotted line), the charge interaction (orange dotted line), and π–π stacking (purple dotted line). However, d1-BMN (Fig. 4b) cannot enter this cavity given the insufficient binding force, such as hydrogen bonds and the charge interaction. Furthermore, the molecular docking (Fig. 4a) and the molecular dynamics simulations (Fig. 4c and d) showed that d1-BMN-CHO exhibited six different representative conformations (Fig. 4c) with various intramolecular torsion angles in the different AP-site cavities of one DNA crystal structure (Fig. 4d and S3,† 17.9°, 41.8°, 53.8°, 65.7°, 71.8° and 77.6°). The aldehyde-group metabolites of all other BMN-Fluors derivatives also have different numbers of conformations. The molecular docking indicated that a2-BMN-CHO and d2-BMN-CHO only have one conformation, whereas a1-BMN-CHO, b-BMN-CHO, c-BMN-CHO and e-BMN-CHO have two conformations. This result was mainly due to the lack of sufficient –ONH2 and –NH– groups in the molecule to form hydrogen bonds and interact with the AP site.
C(CN)2 group of d1-BMN-NO reacts to generate a –ph–CH(OH)–CH(CN)2 group and form another new intermediate product d1-BMN-ts (Fig. 3c). d1-BMN-ts generation is the key point that effectively accelerates the rate-limiting step of the entire recognition reaction, that is, the benzylidenemalononitrile hydrolysis reaction.21–23 With this kind of acceleration, the –ph–CH(OH)–CH(CN)2 group of d1-BMN-ts instantly changes to the –ph–CHO group to form the final product (d1-BMN-CHO, Fig. 3c). The molecular docking (Fig. 4a and b) results indicated that only d1-BMN-CHO can be forced to steadily stay in one DNA crystal structure that has many AP sites in a certain conformation by the hydrogen bonds (the classical hydrogen bonds, indicated by the green dotted line, and the non-classical hydrogen bonds, indicated by the white dotted line), the charge interaction (orange dotted line), and π–π stacking (purple dotted line). However, d1-BMN (Fig. 4b) cannot enter this cavity given the insufficient binding force, such as hydrogen bonds and the charge interaction. Furthermore, the molecular docking (Fig. 4a) and the molecular dynamics simulations (Fig. 4c and d) showed that d1-BMN-CHO exhibited six different representative conformations (Fig. 4c) with various intramolecular torsion angles in the different AP-site cavities of one DNA crystal structure (Fig. 4d and S3,† 17.9°, 41.8°, 53.8°, 65.7°, 71.8° and 77.6°). The aldehyde-group metabolites of all other BMN-Fluors derivatives also have different numbers of conformations. The molecular docking indicated that a2-BMN-CHO and d2-BMN-CHO only have one conformation, whereas a1-BMN-CHO, b-BMN-CHO, c-BMN-CHO and e-BMN-CHO have two conformations. This result was mainly due to the lack of sufficient –ONH2 and –NH– groups in the molecule to form hydrogen bonds and interact with the AP site.
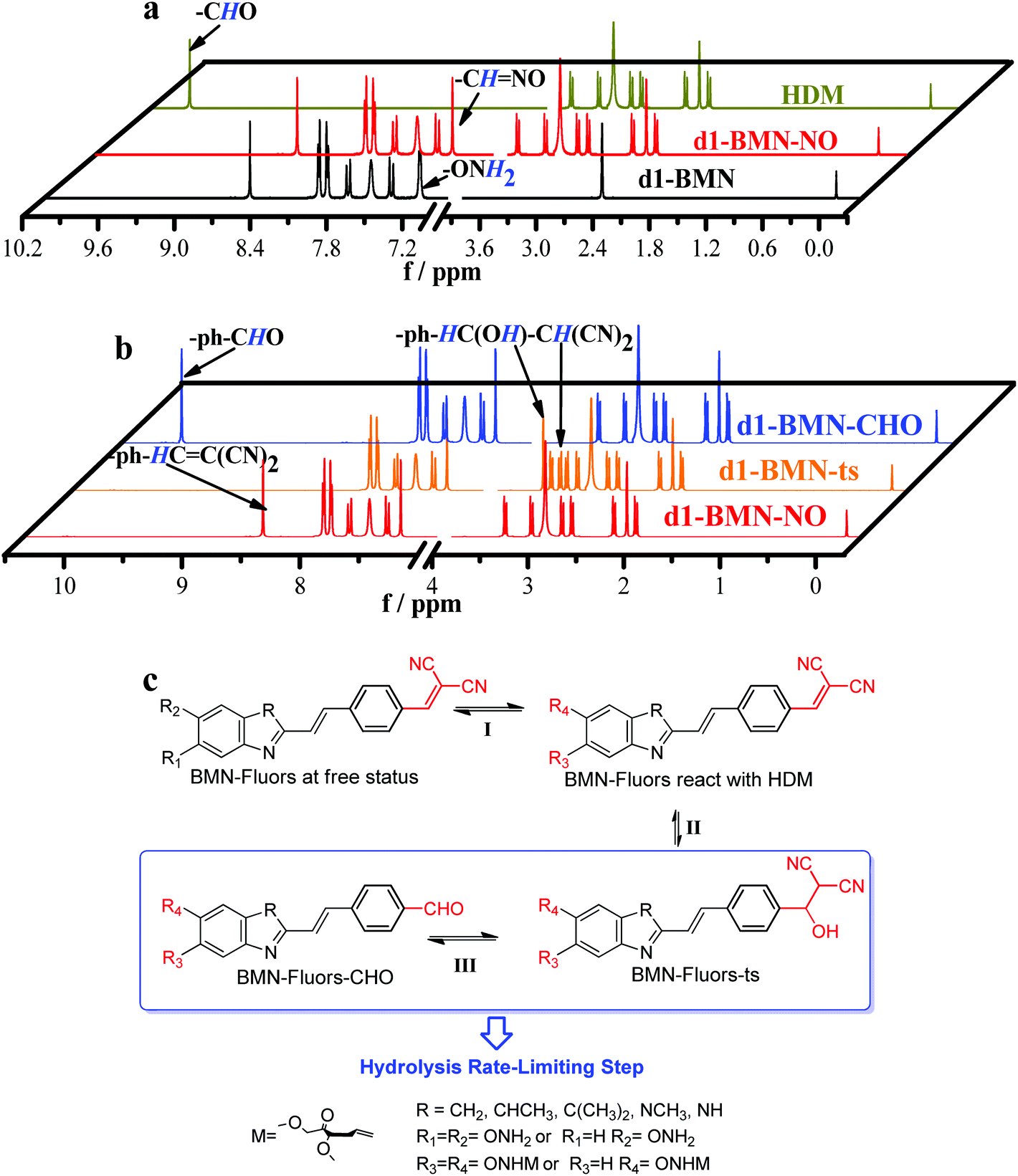 | ||
| Fig. 3 (a) NMR titration between d1-BMN (3.0 mM) and HDM (3.0 mM) in D2O. Black line: d1-BMN; dark yellow line: HDM; red line: d1-BMN-NO. (b) NMR spectra monitoring the molecular structure changes during the recognition process in Fig. 1a and c. Red line: d1-BMN-NO; orange line: d1-BMN-ts; blue line: d1-BMN-CHO. (c) The inferred molecular structures of BMN-Fluors change during the recognition process. | ||
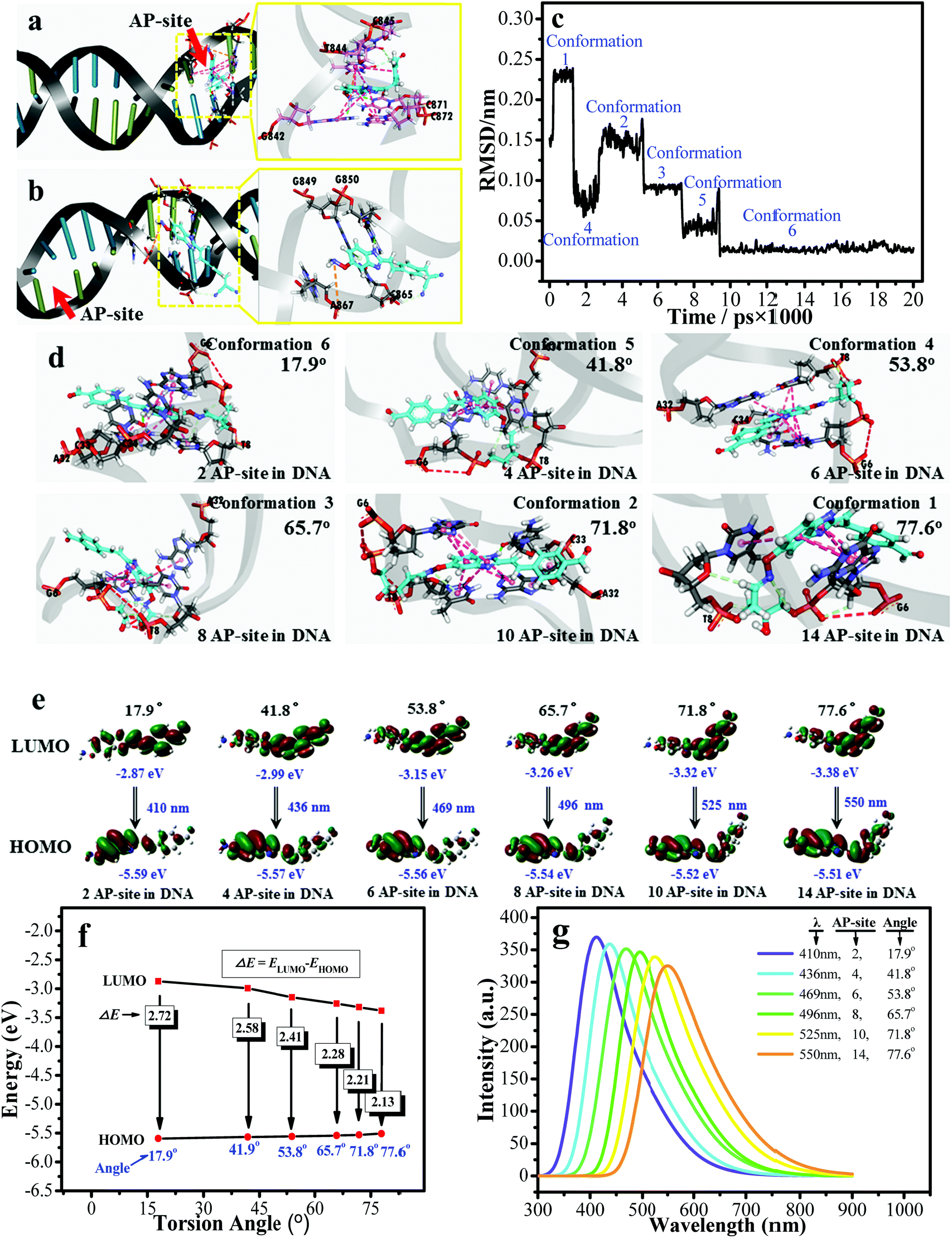 | ||
| Fig. 4 The molecular docking results of (a) d1-BMN-CHO and (b) d1-BMN. (c) The RMSD analysis between the different molecular conformations obtained by the molecular dynamics simulations. The PDB code is 4Q45. (d) The six preferential conformations during the RMSD analysis. (e) and (f) The orbital energy of HOMO and LUMO using Gaussian 16 at different intermolecular torsion angles (obtained by the molecular docking) when the number of AP sites are 2, 4, 6, 8, 10 and 14. (g) The simulated fluorescence emission spectra using Gaussian 16 at different intermolecular torsion angles when the number of AP sites are 2, 4, 6, 8, 10 and 14. | ||
Gaussian 16 was used to calculate the ground and excited states of these different representative conformations collected by the molecular docking (Fig. 4a) and the molecular dynamics simulations (Fig. 4c and d). The energy gap (ΔE) between HOMO and LUMO of d1-BMN-CHO in the excited state gradually decreased (Fig. 4e and f). The simulated fluorescence emission spectra (Fig. 4g) were consistent with those of Fig. 2a, that is, a multichannel signal. Furthermore, through the same method, the simulated fluorescence emission spectra of other aldehyde-group metabolites (i.e.a2-BMN-CHO, d2-BMN-CHO, a1-BMN-CHO, b-BMN-CHO, c-BMN-CHO, and e-BMN-CHO) were basically consistent with their experimental data (Fig. S4†). In other words, they emitted OFF–ON single-channel signal and ratio double-channel signal.
The results mentioned above indicated that the different types of signal (i.e. OFF–ON single-channel signal, ratio double-channel signal, and multichannel signal) are mainly attributed to the sequential self-regulation of one molecule between the unstable and stable states of molecular conformations. In the free state, the aldehyde-group metabolites (BMN-Fluors-CHO, Fig. 3c) of BMN-Fluors derivatives were in a real-time rotation state, that is, molecular conformations are in an unstable state. Furthermore, these molecules are bound in an AP site cavity by different binding effects (i.e., hydrogen bonds, charge interaction, and π–π stacking). In accordance with the strength of binding effect, the degree of molecular torsion, and the cavity size of AP sites, one molecule could sequentially self-regulate from one unstable state to form different stable-state conformations. The molecules in different stable-state conformations possessed different energy gaps (ΔE) between their HOMO and LUMO orbits in the excited state. Thus, different types of signals were emitted.
Ultrasensitive recognition of AP sites in DNA at the single-cell level
The AP site, as a specific site of DNA damage, can accelerate the progression of certain diseases, such as genetic diseases and cancers.1–8 An AP site that can be precisely monitored at the single-cell level is useful for studying related diseases at the molecular level.34,35 Given that they produce multichannel fluorescence signals, if combined with the single-cell gel electrophoresis assay (also known as comet assay), d1-BMN could potentially be used for the precise monitoring of AP sites at the single-cell level.36,37 To offer accurate evaluation data, we should initially study the biocompatibility of d1-BMN, including photostability (Fig. S5†), biological stability (Fig. S6†), toxicity (Fig. S7†), and site damage (Fig. S8–S10†). Results showed that d1-BMN presents excellent biocompatibility and negligible damage and cytotoxicity to DNA in HepG 2 cells (Fig. S5–S10†). The HepG 2 cells were incubated with nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) to establish the AP site in a DNA sequence. In Fig. 5a, a circular fluorescence signal appeared in a nuclear matrix. In addition, no trailing phenomenon was observed when a DNA sequence has no AP site. When an AP site appeared in DNA sequences after NNK induction, the original circular red fluorescence area gradually diffused in one direction and displayed a trailing phenomenon resembling a comet tail (Fig. 5b and c). This event was mainly due to the decrease in DNA molecular weight with the increase in the amount of AP sites, thus gradually diffusing to the positive pole. The signals were emitted at different wavelengths (e.g., 414 nm, 441 nm, 468 nm, 498 nm, 524 nm and 552 nm) from the nuclear matrix to comet tail. These wavelengths were given different pseudo colors (red-orange-yellow-green-blue). Such a trailing phenomenon worsened with the increase in AP sites (Fig. 5b and c). This multichannel fluorescence signal phenomenon was directly proportional to the amount of AP sites in the DNA and the degree of DNA damage (Fig. 5d and Table S2†). The amount of AP sites can be quantified on the basis of the multichannel fluorescence integration of the tail/head. All the results are similar to those obtained from the control group, which was stained by ethidium bromide (EB, Fig. 5d and Table S2†). Cells with different degrees of damage could be promptly and precisely sorted by flow cytometry in accordance with the multichannel signal (Fig. 5e and S11†). Therefore, by using d1-BMN, we could accurately monitor the changes in AP sites in the DNA sequences at the single-cell level and evaluate the degree of DNA damage through this kind of multichannel fluorescence signal.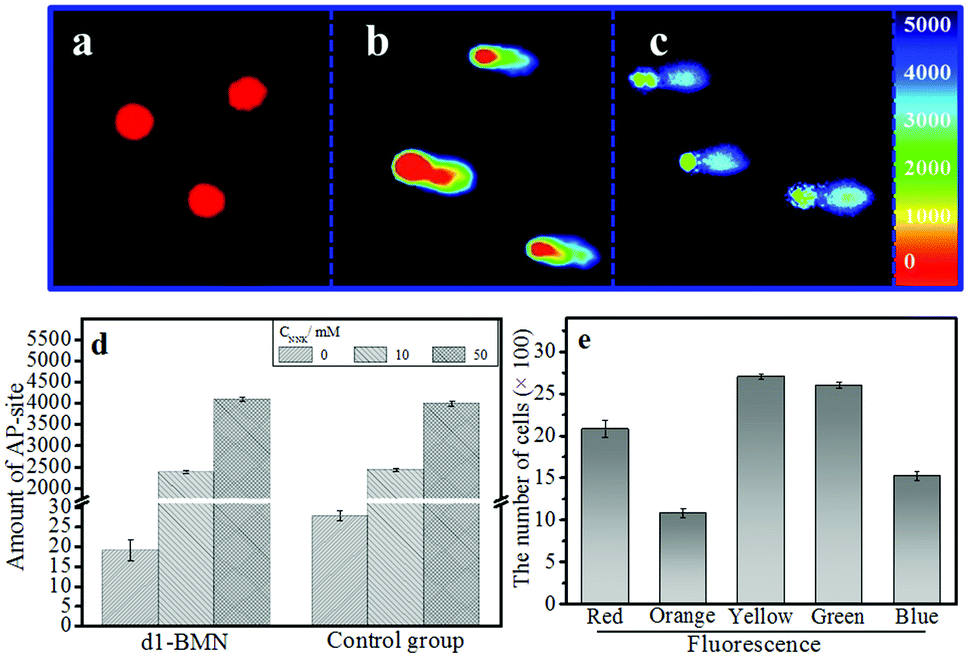 | ||
| Fig. 5 (a–c) The single-cell gel electrophoresis assay of d1-BMN (5.0 μM) in HepG 2 cell. The concentration of NNK in (a, b and c) are 0, 10, and 50 mM. (d) The amount of AP sites. (e) The cells damaged at different degrees was sorted by flow cytometry through multicolor fluorescence. | ||
Conclusions
In summary, to realize the accurate recognition of AP sites at the single-cell level, we provided a rational strategy to design a series of molecular rotors, BMN-Fluors. The conformations of this series could sequentially self-regulate between the unstable and stable states. Fluorescence recognition signal was quenched in a free torsion state given the constant molecular rotation in the free state. The recognition rate-limiting reaction—hydrolysis reaction of BMN-Fluors—was strategically activated to enhance the recognition specificity for the AP sites. The ensuing recognition products were specifically forced to remain in the AP site cavity steadily through hydrogen bonds, charge interaction, and π–π stacking. Depending on the number of hydrogen bonds and bumps, different cavity sizes limited the unstable-state molecule to form different stable-state molecular conformations, thereby emitting in situ and highly sensitive OFF–ON single-channel signals, ratio double-channel signals and multichannel signals for the different AP sites. Among them, d1-BMN could emit a five-channel signal through the conformation self-regulation in different cavity sizes of the AP site. Thus, d1-BMN enabled us to detect AP sites that presented a high-speed dynamic change at the single-cell level in real time. DNA damage was precisely evaluated using AP sites as marker, and precise multichannel signal was obtained at the single-cell level. This property allowed us to sort cells with different degrees of damage through flow cytometry. Overall, the key points to designing molecules possessing different types of signals to recognize a specific site are as follows: (1) specific activation of recognition reaction, (2) easy-to-form multiple different conformations by self-regulation in the cavity of a specific site, (3) the conformations with different HOMO–LUMO energy gaps. Based on this design strategy, we supposed that several molecules possessing the precise signal can be designed to ultrasensitively recognize other kinds of specific sites in DNA.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21722501), Program for Science Technology Innovation Talents in Universities of Henan Province (18HASTIT001). Key Scientific Research Project of Higher Education of Henan Province (18A150046).Notes and references
- B. E. Tropp and D. Freifelder, Molecular Biology, Sudbury, 2012, p. 455 Search PubMed.
- E. C. Friedberg, Nature, 2003, 421, 436–440 CrossRef PubMed.
- M. Neiman and D. R. Taylor, Proc. R. Soc. B, 2009, 276, 1201–1209 CrossRef CAS PubMed.
- A. M. Furda, A. M. Marrangoni, A. Lokshin and B. Van Houten, DNA Repair, 2012, 11, 684–692 CrossRef CAS.
- E. A. Schon, S. DiMauro and M. Hirano, Nat. Rev. Genet., 2012, 13, 878–890 CrossRef CAS.
- D. Schuermann, S. P. Scheidegger, A. R. Weber, M. Bjoras, C. J. Leumann and P. Schar, Nucleic Acids Res., 2016, 44, 2187–2198 CrossRef CAS.
- A. Bender, K. J. Krishnan and C. M. Morris, Nat. Genet., 2006, 38, 515–517 CrossRef CAS PubMed.
- C. A. Rebbeck, A. M. Leroi and A. Burt, Science, 2011, 331, 303 CrossRef CAS PubMed.
- Y. Yuan, Y. Y. Zhao, L. Q. Chen, J. S. Wu, G. Y. Chen, S. Li, J. W. Zou, R. Chen, J. Wang, F. Jiang and Z. Tang, Nucleic Acids Res., 2017, 45, 8676–8683 CrossRef CAS.
- O. A. Kladova, M. Bazlekowa-Karaban, S. Baconnais, O. Pietrement, A. A. Ishchenko, B. T. Matkarimov, D. A. Iakovlev, A. Vasenko, O. S. Fedorova, E. Le Cam, B. Tudek, N. A. Kuznetsov and M. Saparbaev, DNA Repair, 2018, 64, 10–25 CrossRef CAS PubMed.
- H. W. Liu, K. Li, X. X. Hu, L. Zhu, Q. Rong, Y. Liu, X. B. Zhang, J. Hasserodt, F. L. Qu and W. H. Tan, Angew. Chem., Int. Ed., 2017, 39, 11788–11792 CrossRef PubMed.
- X. Bai, Y. Huang, M. Lu and D. Yang, Angew. Chem., Int. Ed., 2017, 56, 12873–12877 CrossRef CAS.
- B. Martin, F. Radovan, K. Iva, B. Klara, M. Radek, S. Janos, S. Vladimir and V. Michaela, Nucleic Acids Res., 2014, 42, 14031–14041 CrossRef.
- N. Kojima, T. Takebayashi, A. Mikami, E. Ohtsuka and Y. Komatsu, J. Am. Chem. Soc., 2009, 131, 13208–13209 CrossRef CAS PubMed.
- C. X. Zhao, Q. Dai and T. Seino, Chem. Commun., 2006, 11, 1185–1187 RSC.
- H. Chen, L. Yao, C. Brown, C. J. Rizzo and R. J. Turesky, Anal. Chem., 2019, 91, 7403–7410 CrossRef CAS PubMed.
- Y. Wang, Q. Q. Sun, L. L. Zhu, J. Y. Zhang, F. Y. Wang, L. L. Lu, H. J. Yu, Z. A. Xu and W. Zhang, Chem. Commun., 2015, 51, 7958–7961 RSC.
- R. Feng, G. Liang, L. H. Guo and Y. P. Wu, Sens. Actuators, B, 2017, 248, 512–518 CrossRef CAS.
- L. J. Wang, M. Ren, Q. Y. Zhang, B. Tang and C. Y. Zhang, Anal. Chem., 2017, 89, 4488–4494 CrossRef CAS PubMed.
- Y. H. Hu, F. Lin, T. Wu, Y. F. Zhou, Q. S. Li, Y. Shao and Z. A. Xu, Anal. Chem., 2017, 89, 2181–2185 CrossRef CAS.
- X. Y. Zhang, Z. Xu, Z. Y. Ge, X. H. Ouyang and W. J. Ji, J. Photochem. Photobiol., A, 2014, 290, 22–30 CrossRef CAS.
- Y. L. Song, T. Tian, Y. Z. Shi, W. L. Liu, Y. Zou, T. Khajvand, S. L. Wang, Z. Zhu and C. Y. Yang, Chem. Sci., 2017, 8, 1736–1751 RSC.
- C. H. leung, H. J. Zhong, H. Z. He, L. H. Lu, D. S. Chan and D. L. Ma, Chem. Sci., 2013, 4, 3781–3795 RSC.
- M. W. Li, Y. Yuan and Y. L. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 1237–1243 CrossRef CAS PubMed.
- B. A. Smith, B. W. Xie, E. R. Beek, I. Que, V. Blankevoort, S. Z. Xiao, E. L. Cole, M. Hoehn, E. L. Kaijzel, C. W. G. M. Löwik and B. D. Smith, ACS Chem. Neurosci., 2012, 3, 530–537 CrossRef CAS PubMed.
- H. B. Xiao, P. Li, X. F. Hu, X. H. Shi, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 6 Search PubMed.
- N. P. EPionnier, H. Sjoberg, V. C. Chunda, F. F. Fombad, P. W. Chounna, A. J. Njouendou, H. M. Metuge, B. L. Ndzeshang, N. V. Gandjui, D. N. Akumtoh, D. B. Tayong, M. J. Taylor, S. Wanji and J. D. Turner, Nat. Commun., 2019, 10, 1–11 Search PubMed.
- J. M. Ramanjulu, G. S. Pesiridis, J. S. Yang, N. Concha, R. Singhaus, S. Y. Zhang, J. L. Tran, P. Moore, S. Lehmann, H. C. Eberl, M. Muelbaier, J. L. Schneck, J. Clemens, M. Adam, J. Mehlmann, J. Romano, A. Morales, J. Kang, L. Leister, T. L. Graybill, A. K. Charnley, G. Ye, N. Nevins, K. Behnia, A. I. Wolf, V. Kasparcova, K. Nurse, L. P. Wang, Y. Li, M. Klein, C. B. Hopson, J. Guss, M. Bantscheff, G. Bergamini, M. A. Reilly, Y. Q. Lian, K. J. Duffy, J. Adams, K. P. Foley, P. J. Gough, R. W. Marquis, J. Smothers, A. Hoos and J. Bertin, Nature, 2018, 564, 439–443 CrossRef CAS PubMed.
- X. Peng, T. Wu, J. Fan, J. Wang, S. Zhang, F. Song and S. Sun, Angew. Chem., Int. Ed., 2011, 50, 4180–4183 CrossRef CAS PubMed.
- C. X. Liu, Y. F. Wang, W. Yang, F. Wu, W. W. Zeng, Z. G. Chen, J. G. Huang, G. G. Zou, X. Zhang, S. R. Wang, Z. G. Wu, Y. Zhou and X. Zhou, Chem. Sci., 2017, 8, 7443–7447 RSC.
- O. D. Schaerer, J. Y. Ortholand, A. Ganesan, K. Ezaz-Nikpay and G. L. Verdine, J. Am. Chem. Soc., 1995, 117, 6623–6624 CrossRef CAS.
- C. X. Liu, Y. F. Wang, X. Zhang, F. Wu, W. Yang, G. G. Zou, Q. Yao, J. Q. Wang, Y. Q. Chen, S. R. Wang and X. Zhou, Chem. Sci., 2017, 8, 4505–4510 RSC.
- E. E. Visvardis, Mutat. Res., 1997, 383, 71–80 CAS.
- R. R. Tice, Environ. Mol. Mutagen., 2000, 35, 206–221 CrossRef CAS.
- S. Nandhakumar, S. Parasuraman, M. M. Shanmugam, K. R. Rao, P. Chand and B. V. Bhat, J. Pharmacol. Pharmacother., 2011, 2, 107–111 CrossRef CAS.
- R. Rahimoff, O. Kosmatchev, A. Kirchner, F. Pfaffeneder, T. Spada, V. Brantl, M. Müller and T. Carell, J. Am. Chem. Soc., 2017, 139, 10359–10364 CrossRef CAS.
- K. Hata, A. Urushibara, S. Yamashita, N. Shikazono, A. Yokoya and Y. Katsumura, Biochem. Biophys. Res. Commun., 2013, 434, 341–345 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04140k |
| This journal is © The Royal Society of Chemistry 2019 |