
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScale-up of microdroplet reactions by heated ultrasonic nebulization†
Chengyuan
Liu‡
a,
Jia
Li‡
a,
Hao
Chen
 b and
Richard N.
Zare
*a
b and
Richard N.
Zare
*a
aDepartment of Chemistry, Fudan University, Shanghai 200438, China. E-mail: rnz@fudan.edu.cn
bDepartment of Chemistry & Environmental Science, New Jersey Institute of Technology, Newark, NJ 07102, USA
First published on 19th August 2019
Abstract
Dramatically higher rates for a variety of chemical reactions have been reported in microdroplets compared with those in the liquid bulk phase. However, the scale-up of microdroplet chemical synthesis has remained a major challenge to the practical application of microdroplet chemistry. Heated ultrasonic nebulization (HUN) was found as a new way for scaling up chemical synthesis in microdroplets. Four reactions were examined, a base-catalyzed Claisen–Schmidt condensation, an oximation reaction from a ketone, a two-phase oxidation reaction without the use of a phase-transfer-catalyst, and an Eschenmoser coupling reaction. These reactions show acceleration of one to three orders of magnitude (122, 23, 6536, and 62) in HUN microdroplets compared to the same reactions in bulk solution. Then, using the present method, the scale-up of the reactions was achieved at an isolated rate of 19 mg min−1 for the product of the Claisen–Schmidt condensation, 21 mg min−1 for the synthesis of benzophenone oxime from benzophenone, 31 mg min−1 for the synthesis of 4-methoxybenzaldehyde from 4-methoxybenzyl alcohol, and 40 mg min−1 for the enaminone product of the Eschenmoser coupling reaction.
Introduction
A number of recent studies,1–11 including work of our own,12–16 have reported the acceleration of reaction rates in microdroplets formed in the course of spray-based ionization4,13,17 and microfluidics.18,19 Microdroplets show a strikingly different reactive environment from that of the corresponding bulk phase.5,18,20 The alteration of pH,21 accumulation of reagents5 on the microdroplet surface, solvent evaporation, confinement of reagents, and high surface-to-volume ratio of microdroplets are thought to contribute to the reaction acceleration.1 The acceleration phenomenon in confined volumes has also been reported in Leidenfrost levitated droplets22 and thin films.23,24 The applicability of microdroplets in organic chemical synthesis is attracting increasing attention because in addition to reaction rate acceleration, sometimes microdroplet reactions can also proceed well without the use of a catalyst.12,16The droplets in microfluidic channels can be used as chemical microreactors for performing many reactions on a small scale.25–28 Thus droplet-based microfluidics has been widely used for the high-throughput screening of diverse chemical and biochemical reactions.29–32 Preparative organic synthesis in microemulsions can be facilitated by continuous-flow microfluidic devices.33–36 Recently, many organic reactions1,3,4,6,13,14,37–40 have been demonstrated to occur on the millisecond timescale in microdroplet reactions generated by electrospray ionization mass spectrometry (ESI-MS). However, the flow rate (ca. 10 μL min−1) for the microdroplet reaction in ESI-MS is not high enough for preparative organic synthesis. Although multiplexed ESI sprayer tips3,16 have even been used in the preparative method, the scale-up of microdroplet reactions in air is still challenging.
Ultrasonic nebulization (UN), i.e., the emission of a fog of microdroplets from an acoustically excited liquid–air interface, has been well-known since the landmark work reported by Wood and Loomis in 1927.41 Since then, UN has been used commercially in air humidifiers, medical nebulizers, and other such devices. UN has also been used as a method of sample atomization for subsequent ionization such as inductively coupled plasma,42,43 electrospray ionization (ESI),44,45 extractive electrospray ionization,46 and photoionization47,48 in MS. Ultrasonically generated microdroplets have also been used as microreactors for the synthesis of semiconductor nanoparticles,49 porous carbon powder,50 and other nanostructured materials.51 Ambient ionization MS was even applied for the real-time monitoring of microreactions in acoustically levitated droplets for which at least an order of magnitude faster reaction rate was observed.52 Herein, we demonstrate the use of heated ultrasonic nebulization (HUN)-generated microdroplets to perform the scale-up of four different chemical syntheses.
Fig. 1 presents the experimental design for HUN microdroplet synthesis. When the ultrasonic nebulizer (Model 402AI, Yuwell Medical Equipment & Supply Corp., Suzhou, China) is switched on, the reactant solution in the nebulization cell can be continuously atomized into large amounts of microdroplets due to the so-called “ultrasonic fountain”. Compared with the traditional ESI sprayer, the HUN setup enabled the reuse of reactant solution for the microdroplet reaction. The working frequency of the transducer of the ultrasonic nebulizer is 1.7 MHz ± 10%. The glass nebulization cell consists of a cylindrical upper part (I.D. 35 mm, height 90 mm) and a lower part (I.D. 22 mm, height 15 mm). A thin polyethylene membrane transparent to ultrasonic waves, which is fixed using nylon cable ties, covers the bottom of the nebulization cell. Water between the polyethylene membrane and the transducer transfers the ultrasonic waves. In addition, temperature-controlled heating tape (ZhengLong Electrothermal Technology Co., Ltd., China) is wound around the nebulization cell to assist in the microdroplet reaction. For comparison, all the bulk reactions were conducted at the same temperature. In the present design, no electric potential is applied but for some cases charging the microdroplets can be advantageous.
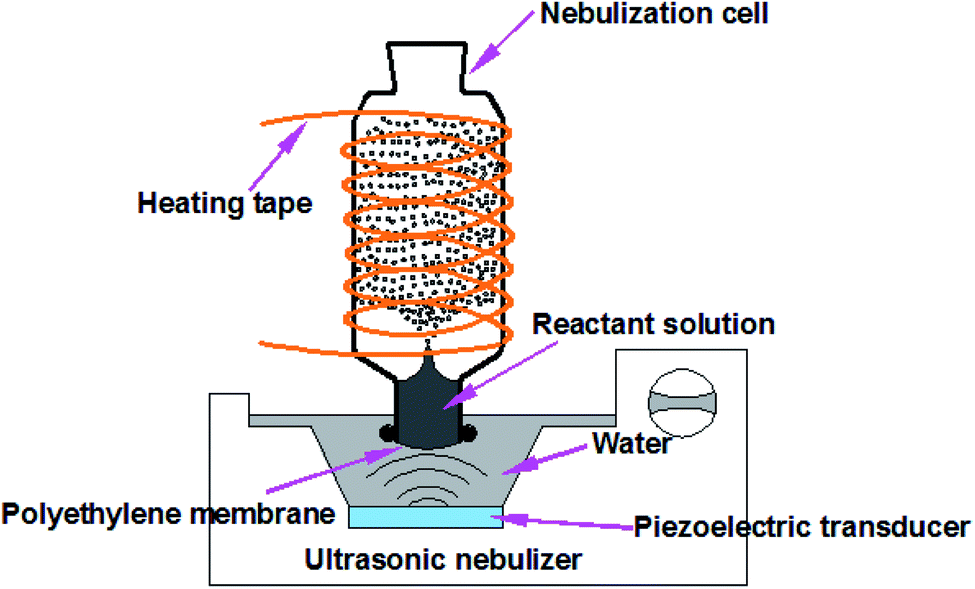 | ||
| Fig. 1 The heated ultrasonic nebulization (HUN) system used to generate continuously microdroplets in which chemical reactions are accelerated. | ||
Results and discussion
At first, the base-catalyzed Claisen–Schmidt condensation between 6-hydroxy-1-indanone 1 and benzaldehyde 2 was investigated as a model reaction (see Fig. 2). The acceleration of a similar system has been well-characterized using paper spray,23,53 ESI,3 thin film,24 and the Leidenfrost droplet reaction.22 A recent study reported that moderate heat can accelerate the solvent evaporation from microdroplets and improve the reaction rate of the ESI microdroplet reaction.54 The influence of temperature on the microdroplet reaction in the HUN system was investigated at first. As shown in Fig. S1,† temperature is quite an important factor, and the ratio of the MS product (6-hydroxy-2-benzyliden-1-indanone 3) signal at m/z 235 to the reagent (6-hydroxy-1-indanone 1) signal at m/z 147 increased markedly with elevation of the temperature from 30 °C to 50 °C. However, when increasing further the heating from 50 °C to 60 °C, no higher signal intensity of product 3 was observed and byproducts emerged. Thus, 50 °C was selected as the heating temperature for HUN microdroplet reactions, and the same temperature was used for the control experiments in the bulk phase. The effect of temperature on this reaction in the bulk phase was also investigated. As presented in Fig. S3,† the ratio of product 3 and reactant 1 increased only twofold when the temperature was increased from 30 °C to 50 °C in the bulk phase. Compared with the bulk phase, the increased temperature in HUN can reduce the size of microdroplets, which was measured using a laser diffractometer (Sympatec HELOS, Sympatec, Germany). The experimental setup and results are shown in Fig. S3.† As the temperature was increased from 30 °C to 50 °C, the average droplet size decreased from 6.9 μm to 5.4 μm, which is closer to the microdroplet size (2–3 μm) in ESI. Previous reports39,55,56 have indicated that the droplet size has a large effect on the reaction rate in microdroplets; thus we speculate that the increased reaction rate in HUN at higher temperature is caused by the smaller droplet size. The following three reactions were also conducted at 50 °C. The reaction in the nebulization cell is accelerated by the continuously atomized microdroplets from the reactant solution. The effect of reaction time on the yield of product 3 for the Claisen–Schmidt condensation reaction in HUN microdroplets was also examined (Fig. S4†). The ratio of product 3 and reactant 1 increased during the initial 120 s due to the recycling of the reactant solution and remained unchanged for longer reaction times (up to 300 s). The influence of the volume of reactant solution in the nebulization cell on the conversion efficiency was also tested. As shown in Fig. S5,† as the volume was increased from 1.2 mL to 2.4 mL, the ratio of product 3 and reactant 1 gradually decreased. It is speculated that the greater probability of recombination of microdroplets resulted in a lower reaction rate and lower conversion efficiency when higher volume of reactant solution was nebulized into larger amounts of microdroplets in the confined space of the ultrasonic nebulization cell.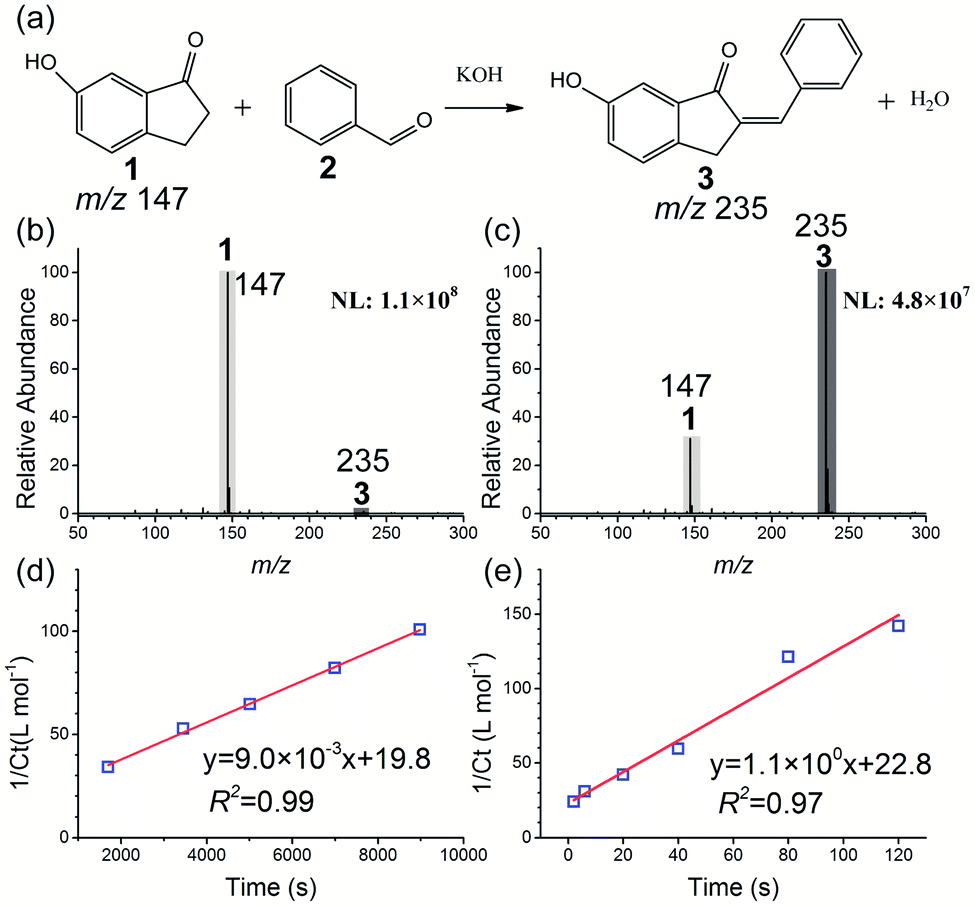 | ||
| Fig. 2 Base-catalyzed Claisen–Schmidt condensation of 6-hydroxy-1-indanone 1 with benzaldehyde 2 to form product 3: (a) reaction schematic; negative ion mode mass spectra of the condensation reaction in the (b) bulk phase and (c) HUN microdroplets after 2 min. Plots of 1/Ctvs. time for second-order reaction kinetics deriving the reaction rate constant to be (d) 9.0 × 10−3 L mol−1 s−1 in bulk and (e) 1.1 × 100 L mol−1 s−1 in HUN microdroplets. | ||
According to MS peak intensities of reactant 1 and product 3, we can calculate the remaining concentration of reactant 1 (Ct) and the yield of product 3 (Ycalculated). The reactant was found to ionize 2.2 times more efficiently than the product, resulting in a greater signal intensity for the same concentration of the material (Fig. S6†). By monitoring the reaction with time, we calculated the reaction rate constants in bulk (kbulk) and HUN microdroplets (kHUN). The ratio between kHUN and kbulk gives the acceleration factor (AF). Section 4 of the supporting information† describes the details of these calculations.
Fig. 2b and c present the mass spectra of the condensation reaction in the bulk phase and HUN microdroplets after 2 min. Compared to the 2 min yield in the bulk phase (1.6%), a much higher 2 min yield (87.6%) can be obtained in the HUN microdroplet reaction. The reaction rate constants in the bulk phase (kbulk) and HUN microdroplets (kHUN) at 50 °C were calculated by plotting the reciprocal of the remaining concentration of reactant 1 (1/Ct) against reaction time in Fig. 2d and e. The HUN microdroplet reaction showed an acceleration factor of 122 over the bulk phase reaction. An industrial nebulization plate with 10 ultrasonic nebulizers (Fig. S17†) was used for the scale-up experiment, and 37.2 mg of the product was produced within 2 minutes (ca. 19 mg min−1). Multiplexed ESI sprayer tips (25 μL min−1 per spray tip) have been used previously for the preparative synthesis of products from the Claisen–Schmidt condensation reaction in microdroplets, but the product was produced only at the rate of 0.588 mg min−1.3 The highest yield for the preparative synthesis of the product of this condensation reaction in confined volumes was ever achieved using a thin film, and only 98 mg h−1 (ca. 1.6 mg min−1) was reached.24 Thus, HUN is a promising new method for the scale-up of microdroplet chemical synthesis. Table 1 lists the results for the condensation reaction.
| Reaction type | k bulk | k HUN | AF | Y calculated | Isolated amounta | Y isolated |
|---|---|---|---|---|---|---|
| a The isolated amount is obtained by using 10 reactors based on an industrial nebulization plate. All the reactions were conducted at 50 °C. The reaction times for the Claisen–Schmidt condensation, the oximation reaction, the two-phase reaction and the Eschenmoser coupling reaction are 2 min, 2 min, 3 min and 3.5 min, respectively. The reactant solution volumes for the Claisen–Schmidt condensation, the oximation reaction, the two-phase reaction and the Eschenmoser coupling reaction are 1.2 mL, 1 mL, 1.1 mL, and 1 mL, respectively. | ||||||
| Claisen–Schmidt condensation | 9.0 × 10−3 L mol−1 s−1 | 1.1 × 100 L mol−1 s−1 | 122 | 87.6% | 37.2 mg | 78.8% |
| Oximation reaction | 9.1 × 10−4 s−1 | 2.1 × 10−2 s−1 | 23 | 92.4% | 41.5 mg | 84.3% |
| Two-phase reaction | 1.1 × 10−6 s−1 | 7.3 × 10−3 s−1 | 6536 | 75.4% | 93 mg | 68.4% |
| Eschenmoser coupling reaction | 1.4 × 10−3 L mol−1 s−1 | 8.7 × 10−2 L mol−1 s−1 | 62 | 78% | 140 mg | 67% |
The oximation reaction of benzophenone 4 with hydroxylamine 5 (Fig. 3a) was also investigated to yield the corresponding benzophenone oxime 6, which is an important reaction material for amide synthesis by the Beckman rearrangement reaction.57 This reaction has previously been investigated using an ESI sprayer (10 μL min−1) and showed significant rate acceleration.58 However, the rate of product production obtained by the microdroplet reaction was too small (less than 50 μg min−1).58Fig. 3b and c show, respectively, the HPLC chromatograms of the oximation reaction in the bulk phase and HUN microdroplets after 2 min. The concentration of hydroxylamine 5 in this experiment was 20 times higher than that of benzophenone 4; thus, the oximation reaction followed pseudo first-order kinetics. Fig. 3d and e present plots of ln(Ct/C0) vs. time (Ct and C0 represent the remaining and initial concentrations of reactant 4) of this reaction in the bulk phase and HUN microdroplets, respectively. As listed in Table 1, the reaction rate constant in HUN microdroplets (kHUN) is 23-fold higher than that in bulk (kbulk). For preparative synthesis, 41.5 mg of benzophenone oxime was synthesised from benzophenone within 2 minutes (ca. 21 mg min−1) using the setup shown in Fig. S17.†
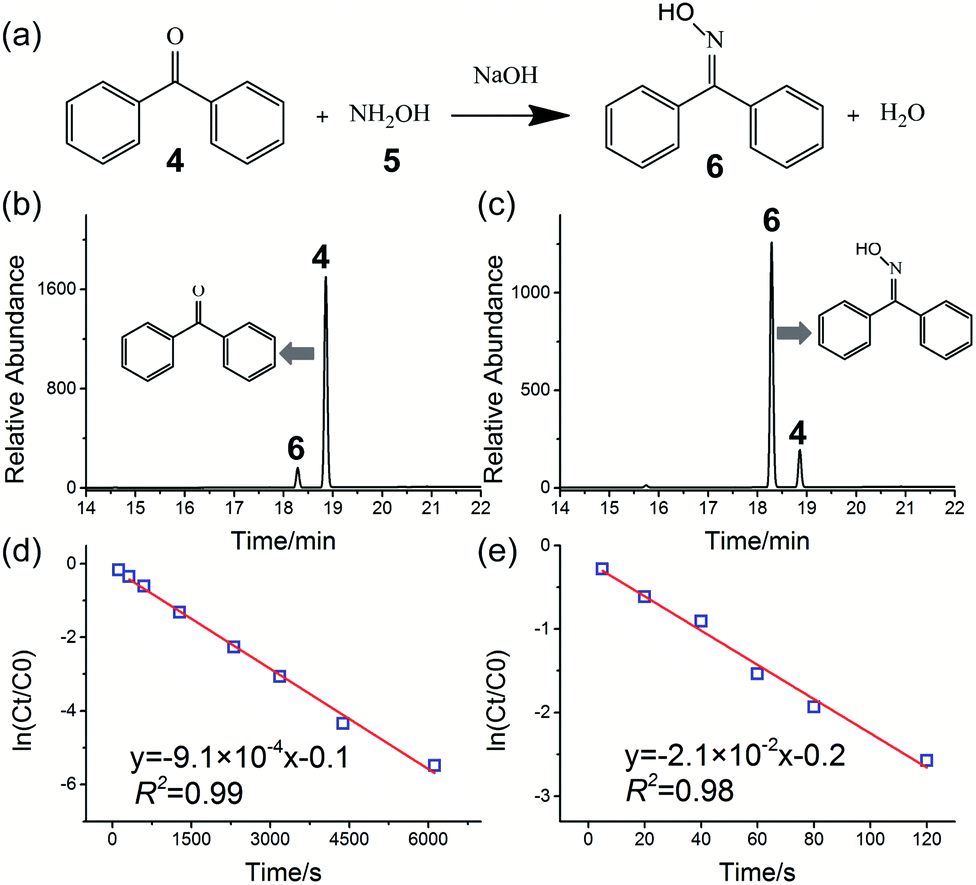 | ||
| Fig. 3 Oximation reaction of benzophenone 4 with hydroxylamine 5 to form benzophenone oxime 6: (a) reaction schematic; HPLC chromatograms of oximation reaction in the bulk phase (b) and HUN microdroplets (c) after 2 min. A higher yield of product 6 was obtained in HUN microdroplets. Plots of ln(Ct/C0) vs. time deriving the pseudo first-order reaction rate constant to be (d) 9.1 × 10−4 s−1 in bulk and (e) 2.1 × 10−2 s−1 in HUN microdroplets. | ||
When a bulk liquid–liquid two-phase system is nebulized as small microdroplets, the interfacial area of the two-phases can be increased by many orders of magnitude. Thus, the two-phase reaction was readily facilitated without the use of a phase-transfer-catalyst (PTC) in ESI microdroplets,16,54 which meets the requirement of green chemistry and sustainable synthesis. In this study, the two-phase reaction of 4-methoxybenzyl alcohol 7 in ethyl acetate (EtOAc) with aqueous NaClO (Fig. 4a) was examined without using PTC.
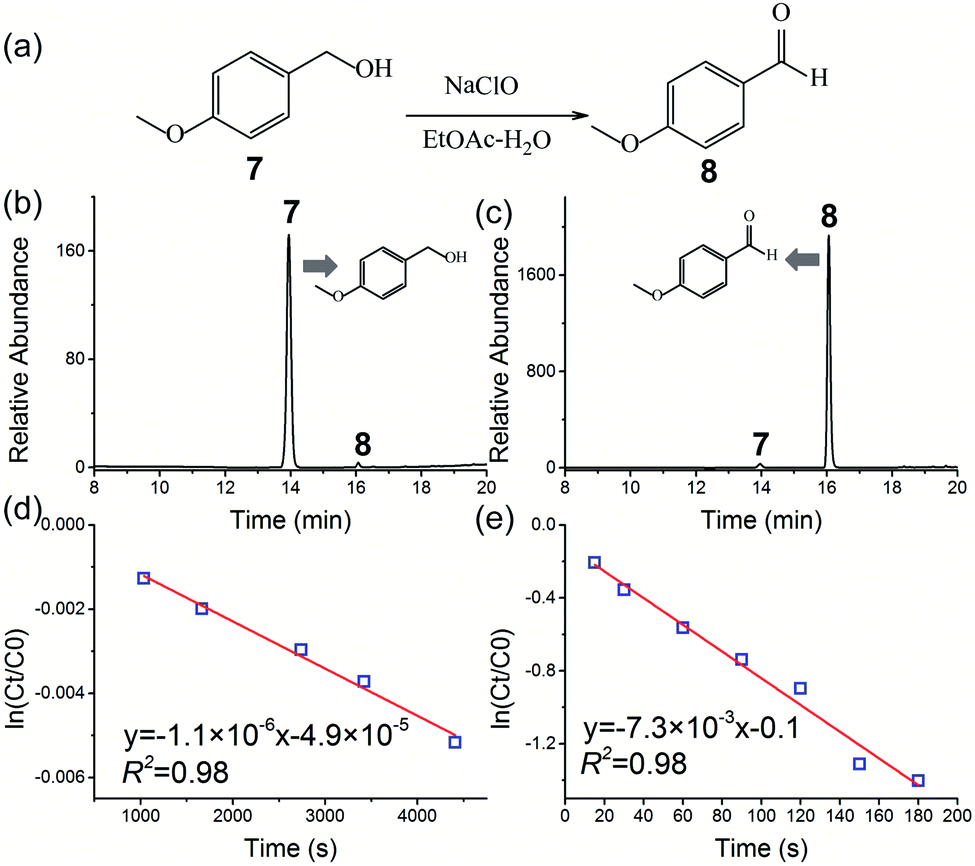 | ||
| Fig. 4 Synthesis of 4-methoxybenzaldehyde 8 by a two-phase oxidation reaction between 4-methoxybenzyl alcohol 7 in ethyl acetate (EtOAc) and aqueous NaClO without the use of PTC: (a) reaction schematic; HPLC chromatograms of the two-phase oxidation reaction in the (b) bulk phase and (c) HUN microdroplets after 3 min. Plots of ln(Ct/C0) vs. time deriving the pseudo first-order reaction rate constant to be (d) 1.1 × 10−6 s−1 in the “initial-rate region” of the bulk phase and (e) 7.3 × 10−3 s−1 in HUN microdroplets. | ||
Fig. 4b and c show, respectively, the HPLC chromatograms of the two-phase reaction in the bulk phase and HUN microdroplets after 3 min. In sharp contrast with that (0.1%) in the bulk phase, a strikingly higher yield (75.4%) of the product (4-methoxybenzaldehyde 8) can be obtained from the HUN microdroplet reaction. The bulk reaction was also investigated for a longer reaction time, and we found that there were abundant byproducts (such as 4-chloroanisole and 3-chloro-4-methoxybenzaldehyde) in addition to the oxidation reaction product 8 in the two-phase reaction without the use of PTC in the bulk phase (Fig. S11†). Gas chromatography mass spectrometry and nuclear magnetic resonance (NMR) enabled the identification of these byproducts (Fig. S12 and S13†). The bulk reaction was monitored by HPLC for 7 h, and Fig. S14† presents the temporal peak area profiles of reactants and products. According to previous studies,59,60 the possible reaction pathway for the two-phase reaction in the bulk phase is proposed to be as shown in Scheme S1.† In the initial stage, reaction of 4-methoxybenzyl alcohol with NaClO resulted in 4-chloroanisole through ipso substitution in addition to the oxidation product (4-methoxybenzaldehyde), which is consistent with a previous report.60 Then the chlorination reaction of the newly formed 4-methoxybenzaldehyde with NaClO generated 3-chloro-4-methoxybenzaldehyde.59 After 7 h of bulk reaction, the yield of 4-methoxybenzaldehyde 8 was determined to be only 11.3% (owing to the formation of byproducts) by using a standard calibration curve of 4-methoxybenzaldehyde. These results also indicate that in addition to rate acceleration, highly selective synthesis of 4-methoxybenz-aldehyde 8 was facilitated by HUN microdroplets. As noted in previous reports,5,20 the acceleration of reaction rate in microdroplets can be largely attributed to the accumulation of reactants on the surface of microdroplets. For this reaction, the concentrated NaClO on HUN microdroplets can also change the pH on the microdroplet surface. According to a previous report,60 the byproduct, 4-chloroanisole, formed by ipso substitution of 4-methoxybenzyl alcohol with hypochlorite (ClO−) is strongly affected by pH. Thus, we speculate that the special pH microenvironment in HUN microdroplets inhibited the ipso substitution and at the same time, the production of another product, 3-chloro-4-methoxybenzaldehyde, was avoided by the short reaction time in HUN microdroplets.
In this two-phase reaction, the concentration of sodium hypochlorite (NaClO) was much higher than that of 4-methoxybenzyl alcohol 7. Consequently, this reaction is presumed to proceed as a pseudo first-order reaction, which was confirmed by the kinetic analysis with reactant 7 at different initial concentrations (Fig. S9†). The reaction rates in bulk and HUN microdroplets were derived by plotting ln(Ct/C0) of reactant 7vs. time, as shown in Fig. 4d and e. The acceleration factor for this reaction in HUN microdroplets was determined to be 6536. In the HUN system, 93 mg of the product (4-methoxybenzaldehyde 8) of the two-phase reaction with 68.4% yield was produced within 3 min (31 mg min−1), which is a great advance for the scale-up of microdroplet reactions compared with that (1.2 mg min−1)16 obtained by a previous multiplexed ESI sprayer method (15 μL min−1 per spray tip).
Encouraged by these results, we further examined the applicability of this new method for a new reaction, i.e., the preparative synthesis of enaminone by the Eschenmoser coupling reaction, which has not been examined previously using the microdroplet reaction. Enaminones are important building blocks in organic synthesis and the Eschenmoser coupling reaction has been extensively employed in the construction of enaminones.61,62 However, the coupling reaction often requires a long reaction time.63–65
Fig. 5a shows the reaction schematic of the Eschenmoser coupling reaction between 1-methylpyrrolidine-2-thione 9 and diethyl bromomalonate 10 to form the enaminone product 11. The HPLC chromatograms of the Eschenmoser coupling reaction in the bulk phase and HUN microdroplets after 3.5 min are shown in Fig. 5b and c, respectively. In comparison with the 3.5 min yield in the bulk phase (6.2%), the 3.5 min yield (78%) obtained from the HUN microdroplet reaction is much higher. The initial concentration of diethyl bromomalonate 10 (C100) in this experiment was 1.5 times higher than that of 1-methylpyrrolidine-2-thione 9 (C90) and thus this reaction proceeds as a second order reaction with reactants at different initial concentrations. The reaction rate constants in the bulk phase (kbulk) and HUN microdroplets (kHUN) were calculated by plotting 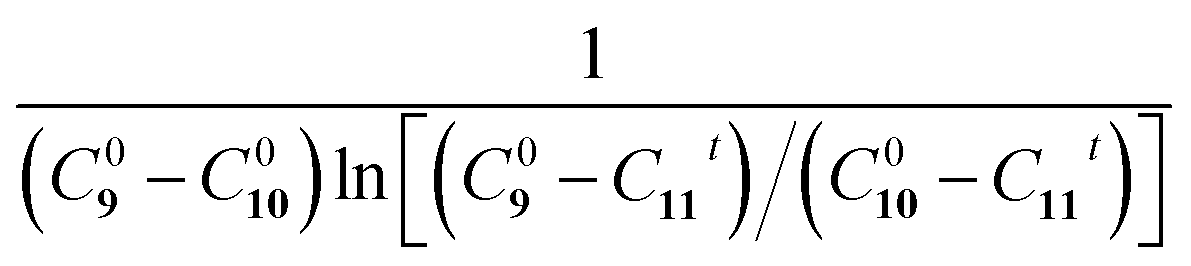 vs. reaction time (C11t represents the concentrations of product 11) in Fig. 5d and e. The acceleration factor for this reaction in HUN microdroplets was calculated to be 62. By using the HUN scale-up setup, 140 mg of the product was produced within 3.5 min with 67% yield (40 mg min−1). Its 1H NMR spectrum is provided after purification (Fig. S16†).
vs. reaction time (C11t represents the concentrations of product 11) in Fig. 5d and e. The acceleration factor for this reaction in HUN microdroplets was calculated to be 62. By using the HUN scale-up setup, 140 mg of the product was produced within 3.5 min with 67% yield (40 mg min−1). Its 1H NMR spectrum is provided after purification (Fig. S16†).
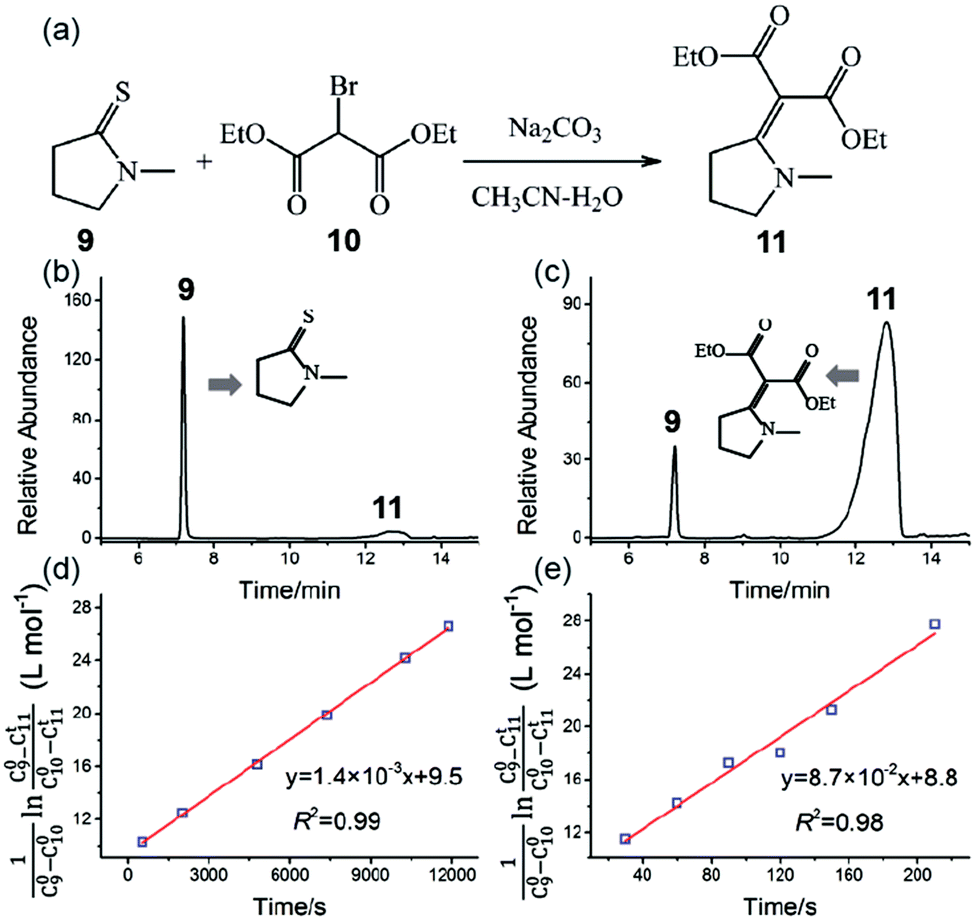 | ||
Fig. 5 The Eschenmoser coupling reaction of 1-methylpyrrolidine-2-thione 9 with diethyl bromomalonate 10 to form the enaminone product 11: (a) reaction schematic; HPLC chromatograms of the Eschenmoser coupling reaction in the (b) bulk phase and (c) HUN microdroplets after 3.5 min. Plots of 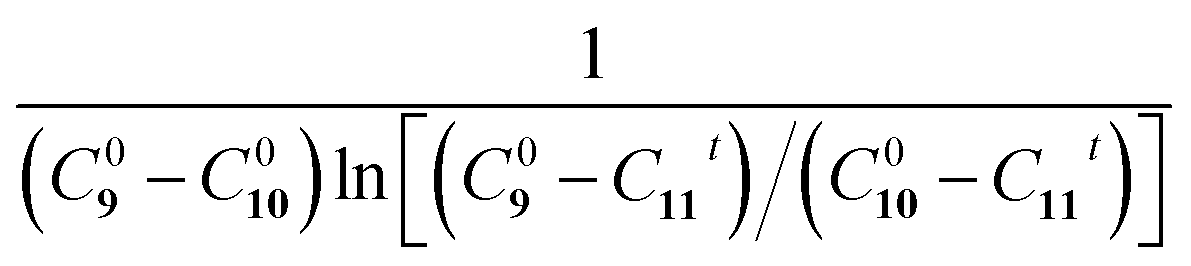 vs. time deriving the second-order reaction rate constant to be (d) 1.4 × 10−3 L mol−1 s−1 in the bulk phase and (e) 8.7 × 10−2 L mol−1 s−1 in HUN microdroplets. vs. time deriving the second-order reaction rate constant to be (d) 1.4 × 10−3 L mol−1 s−1 in the bulk phase and (e) 8.7 × 10−2 L mol−1 s−1 in HUN microdroplets. | ||
Conclusions
In summary, we have developed a new method for the scale-up of microdroplet reactions based on the HUN system. Four reactions including a base-catalyzed Claisen–Schmidt condensation, an oximation reaction, a two-phase reaction without the use of a phase transfer catalyst, and an Eschenmoser coupling reaction were examined. The rates of these reactions can be markedly accelerated in HUN microdroplets by one to three orders of magnitude higher than those of the corresponding bulk reactions. In addition we demonstrated that these reactions could be scaled up using an industrial nebulization plate with multiple nebulizers. By this means, products at the rate of 19–40 mg min−1 were synthesized in HUN microdroplets. The above results indicate that microdroplet chemistry shows promise for the semi-preparative synthesis of many different compounds.Experimental
For the Claisen–Schmidt condensation reaction, 50 mM 6-hydroxy-1-indanone, 50 mM benzaldehyde and 1.8 M KOH were mixed in equal volumes and methanol was the solvent. For the oximation reaction, 50 mM benzophenone and 1 M hydroxylamine dissolved in 1 M NaOH were mixed in equal volumes and methanol was used as the solvent. For the two-phase oxidation reaction, 200 mM 4-methoxybenzyl alcohol in EtOAc and aqueous NaClO (11–14%) were mixed at a volume ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6. For the Eschenmoser coupling reaction, 87 mM 1-methylpyrrolidine-2-thione, 130 mM diethyl bromomalonate and 87 mM Na2CO3 were mixed and CH3CN/H2O (v/v = 9:1) was used as the solvent.
:6. For the Eschenmoser coupling reaction, 87 mM 1-methylpyrrolidine-2-thione, 130 mM diethyl bromomalonate and 87 mM Na2CO3 were mixed and CH3CN/H2O (v/v = 9:1) was used as the solvent.
Microdroplet reactions were conducted in a nebulization cell with the temperature of the heating tape set at 50 °C. For comparison, bulk reactions were conducted in a 50 °C water bath. Upon completion of the reaction, the products were collected and diluted for subsequent analysis by using a LTQ XL Orbitrap hybrid mass spectrometer (Thermo Fisher Scientific, Germany) or an Agilent 1260 high-performance liquid chromatography (HPLC) (Agilent, USA) system. The size of microdroplets in the nebulization cell was measured using a laser diffractometer (Sympatec HELOS, Sympatec, Germany). Additional experimental details on each reaction system can be found in the supporting information (Sections S1 and S2†). Preparative synthesis of the microdroplet reactions was facilitated by using an industrial nebulization plate with 10 ultrasonic nebulizers (A10D48-001, Dongguan RunYang Electronic Co., Ltd., China).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Yan Xia for useful discussions and comments. This work was supported by the Scientific Research Startup Foundation (Grant IDH1615113) of Fudan University. C. Y. Liu is thankful for the funding from the China Postdoctoral Science Foundation (No. 2018M640329).Notes and references
- X. Yan, R. M. Bain and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 12960–12972 CrossRef CAS.
- M. Girod, E. Moyano, D. I. Campbell and R. G. Cooks, Chem. Sci., 2011, 2, 501–510 RSC.
- T. Muller, A. Badu-Tawiah and R. G. Cooks, Angew. Chem., Int. Ed., 2012, 51, 11832–11835 CrossRef.
- R. M. Bain, C. J. Pulliam and R. G. Cooks, Chem. Sci., 2015, 6, 397–401 RSC.
- Z. Zhou, X. Yan, Y.-H. Lai and R. N. Zare, J. Phys. Chem. Lett., 2018, 9, 2928–2932 CrossRef CAS PubMed.
- N. Sahota, D. I. AbuSalim, M. L. Wang, C. J. Brown, Z. Zhang, T. J. El-Baba, S. Cook and D. E. Clemmer, Chem. Sci., 2019, 10, 4822–4827 RSC.
- T. Kenderdine, Z. Xia, E. R. Williams and D. Fabris, Anal. Chem., 2018, 90, 13541–13548 CrossRef CAS PubMed.
- D. N. Mortensen and E. R. Williams, Anal. Chem., 2014, 87, 1281–1287 CrossRef PubMed.
- D. N. Mortensen and E. R. Williams, J. Am. Chem. Soc., 2016, 138, 3453–3460 CrossRef CAS PubMed.
- G. I. Ioannou, T. Montagnon, D. Kalaitzakis, S. A. Pergantis and G. Vassilikogiannakis, Org. Biomol. Chem., 2017, 15, 10151–10155 RSC.
- G. I. Ioannou, T. Montagnon, D. Kalaitzakis, S. A. Pergantis and G. Vassilikogiannakis, ChemPhotoChem, 2018, 2, 860–864 CrossRef CAS PubMed.
- S. Banerjee and R. N. Zare, Angew. Chem., Int. Ed., 2015, 54, 14795–14799 CrossRef CAS PubMed.
- J. K. Lee, S. Kim, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2015, 201503689 Search PubMed.
- J. K. Lee, S. Banerjee, H. G. Nam and R. N. Zare, Q. Rev. Biophys., 2015, 48, 437–444 CrossRef CAS.
- E. T. Jansson, Y. H. Lai, J. G. Santiago and R. N. Zare, J. Am. Chem. Soc., 2017, 139, 6851–6854 CrossRef CAS PubMed.
- X. Yan, H. Cheng and R. N. Zare, Angew. Chem., Int. Ed., 2017, 129, 3616–3619 CrossRef.
- B. Sharma, Y. Takamura, T. Shimoda and M. Biyani, Sci. Rep., 2016, 6, 26257 CrossRef CAS PubMed.
- A. Fallah-Araghi, K. Meguellati, J. C. Baret, A. El Harrak, T. Mangeat, M. Karplus, S. Ladame, C. M. Marques and A. D. Griffiths, Phys. Rev. Lett., 2014, 112, 028301 CrossRef.
- M. Guardingo, F. Busqué and D. Ruiz-Molina, Chem. Commun., 2016, 52, 11617–11626 RSC.
- S. Mondal, S. Acharya, R. Biswas, B. Bagchi and R. N. Zare, J. Chem. Phys., 2018, 148, 244704 CrossRef PubMed.
- H. Wei, E. P. Vejerano, W. Leng, Q. Huang, M. R. Willner, L. C. Marr and P. J. Vikesland, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7272–7277 CrossRef CAS PubMed.
- R. M. Bain, C. J. Pulliam, F. Thery and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 10478–10482 CrossRef CAS PubMed.
- Y. Li, X. Yan and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 3433–3437 CrossRef CAS PubMed.
- Z. Wei, M. Wleklinski, C. Ferreira and R. G. Cooks, Angew. Chem., Int. Ed., 2017, 129, 9514–9518 CrossRef.
- K. Holmberg, Curr. Opin. Colloid Interface Sci., 2003, 8, 187–196 CrossRef CAS.
- K. Holmberg, Adv. Colloid Interface Sci., 1994, 51, 137–174 CrossRef CAS.
- L. García-Río, J. R. Leis, J. C. Mejuto and M. Pérez-Lorenzo, Pure Appl. Chem., 2007, 79, 1111–1123 Search PubMed.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 7336–7356 CrossRef CAS.
- J. Wang, G. Sui, V. P. Mocharla, R. J. Lin, M. E. Phelps, H. C. Kolb and H. R. Tseng, Angew. Chem., Int. Ed., 2006, 45, 5276–5281 CrossRef CAS.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS PubMed.
- K. Churski, P. Korczyk and P. Garstecki, Lab Chip, 2010, 10, 816–818 RSC.
- A. J. Demello, Nature, 2006, 442, 394 CrossRef CAS PubMed.
- K. Geyer, T. Gustafsson and P. H. Seeberger, Synlett, 2009, 2009, 2382–2391 CrossRef.
- S. Taghavi-Moghadam, A. Kleemann and G. Golbig, Org. Process Res. Dev., 2001, 5, 652–658 CrossRef CAS.
- D. R. Acke and C. V. Stevens, Org. Process Res. Dev., 2006, 10, 417–422 CrossRef CAS.
- X. Zhang, S. Stefanick and F. J. Villani, Org. Process Res. Dev., 2004, 8, 455–460 CrossRef CAS.
- I. Nam, J. K. Lee, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12396–12400 CrossRef CAS.
- D. Gao, F. Jin, X. Yan and R. N. Zare, Chem.–Eur. J., 2019, 25, 1466–1471 CrossRef CAS.
- Y. H. Lai, S. Sathyamoorthi, R. M. Bain and R. N. Zare, J. Am. Soc. Mass Spectrom., 2018, 29, 1036–1043 CrossRef CAS PubMed.
- I. Nam, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 36–40 CrossRef CAS PubMed.
- R. W. Wood and A. L. Loomis, Philos. Mag., 1927, 4, 417–436 CAS.
- M. A. Tarr, G. Zhu and R. F. Browner, Anal. Chem., 1993, 65, 1689–1695 CrossRef CAS.
- M. A. M. da Silva, V. L. A. Frescura and A. J. Curtius, Spectrochim. Acta, Part B, 2000, 55, 803–813 CrossRef.
- J. Shiea, D. Y. Chang, C. H. Lin and S. J. Jiang, Anal. Chem., 2001, 73, 4983–4987 CrossRef CAS PubMed.
- D.-Y. Chang, C.-C. Lee and J. Shiea, Anal. Chem., 2002, 74, 2465–2469 CrossRef CAS PubMed.
- L. Zhu, G. Gamez, H. Chen, K. Chingin and R. Zenobi, Chem. Commun., 2009, 559–561, 10.1039/b818541g.
- C. Liu, Y. Zhu, Z. Zhou, J. Yang, F. Qi and Y. Pan, Anal. Chim. Acta, 2015, 891, 203–210 CrossRef CAS PubMed.
- C. Liu, J. Yang, J. Wang, Y. Hu, W. Zhao, Z. Zhou, F. Qi and Y. Pan, J. Am. Soc. Mass Spectrom., 2016, 27, 1597–1605 CrossRef CAS PubMed.
- Y. T. Didenko and K. S. Suslick, J. Am. Chem. Soc., 2005, 127, 12196–12197 CrossRef CAS PubMed.
- S. E. Skrabalak and K. S. Suslick, J. Am. Chem. Soc., 2006, 128, 12642–12643 CrossRef CAS PubMed.
- J. H. Bang and K. S. Suslick, Adv. Mater., 2010, 22, 1039–1059 CrossRef CAS PubMed.
- E. A. Crawford, C. Esen and D. A. Volmer, Anal. Chem., 2016, 88, 8396–8403 CrossRef CAS PubMed.
- R. M. Bain, C. J. Pulliam, X. Yan, K. F. Moore, T. Müller and R. G. Cooks, J. Chem. Educ., 2014, 91, 1985–1989 CrossRef CAS.
- W. Zhang, H. Cheng and J. Liu, ACS Sustainable Chem. Eng., 2018, 6, 8125–8129 CrossRef CAS.
- X. Yan, Y.-H. Lai and R. N. Zare, Chem. Sci., 2018, 9, 5207–5211 RSC.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed.
- L. Guy Donaruma and W. Z. Heldt, Org. React., 2004, 11, 1–156 Search PubMed.
- W. Zhang, S. Yang, Q. Lin, H. Cheng and J. Liu, J. Org. Chem., 2018, 84, 851–859 CrossRef PubMed.
- A. T. Lebedev, G. M. Shaydullina, N. A. Sinikova and N. V. Harchevnikova, Water Res., 2004, 38, 3713–3718 CrossRef CAS PubMed.
- H. V. Carrillo, A. Y. Rodriguez, R. G. Landolt and W. H. Hendrickson, Synlett, 2011, 22, 2069–2071 Search PubMed.
- K. Shiosaki, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Flemming, Pergamon Press, Oxford, U.K., 1991, vol. 2, ch. 3.7, p. 865 Search PubMed.
- N. D. Koduri, H. Scott, B. Hileman, J. D. Cox, M. Coffin, L. Glicksberg and S. R. Hussaini, Org. Lett., 2011, 14, 440–443 CrossRef PubMed.
- S. R. Hussaini and M. G. Moloney, Org. Biomol. Chem., 2006, 4, 2600–2615 RSC.
- J. A. Campbell and H. Rapoport, J. Org. Chem., 1996, 61, 6313–6325 CrossRef CAS PubMed.
- N. D. Koduri, B. Hileman, J. D. Cox, H. Scott, P. Hoang, A. Robbins, K. Bowers, L. Tsebaot, K. Miao and M. Castaneda, RSC Adv., 2013, 3, 181–188 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03701b |
| ‡ C. Y. Liu and J. Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |