
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA transition-metal-free & diazo-free styrene cyclopropanation†
Ana G.
Herraiz
 ab and
Marcos G.
Suero
*a
ab and
Marcos G.
Suero
*a
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Av. Països Catalans 16, Tarragona, 43007, Spain. E-mail: mgsuero@iciq.es
bDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, Calle Marcel. lí Domingo, 1, Tarragona, 43007, Spain
First published on 28th August 2019
Abstract
An operationally simple and broadly applicable novel cyclopropanation of styrenes using gem-diiodomethyl carbonyl reagents has been developed. Visible-light triggered the photoinduced generation of iodomethyl carbonyl radicals, able to cyclopropanate a wide array of styrenes with excellent chemoselectivity and functional group tolerance. To highlight the utility of our photocyclopropanation, we demonstrated the late-stage functionalization of biomolecule derivatives.
Introduction
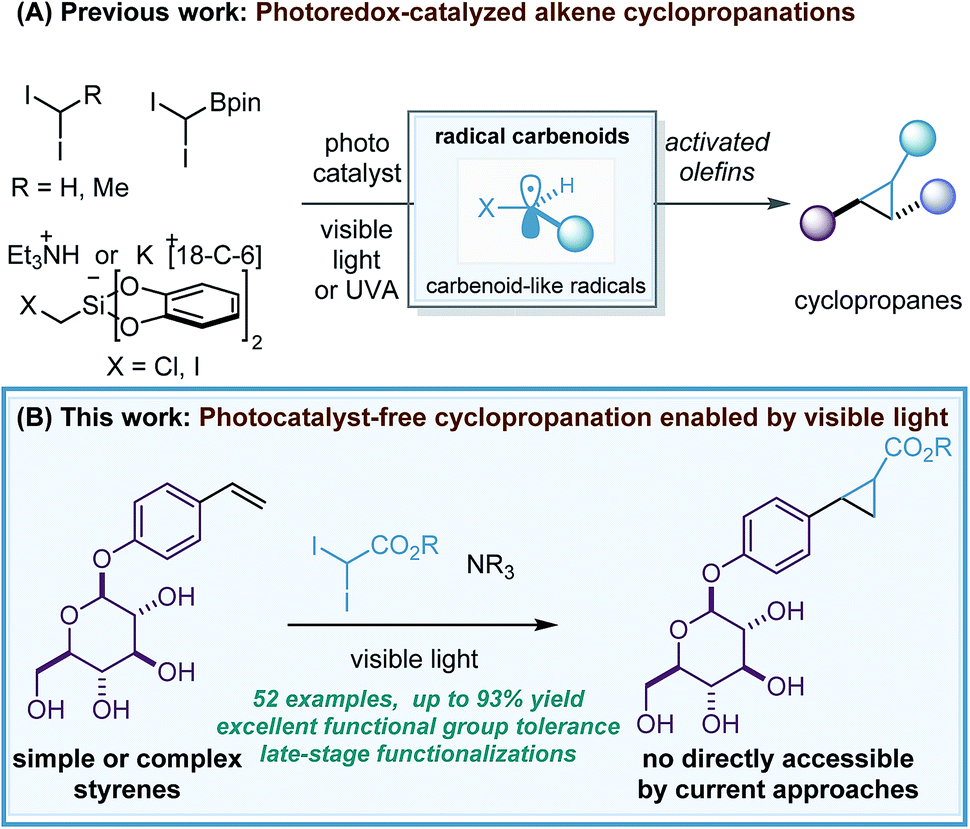
Over the last few decades, one of the main challenges in the synthesis of cyclopropane rings1 – a prevalent structural motif in pharmaceutical agents2 and natural products3 – has been to develop robust and general alkene cyclopropanations that avoid the use of explosive, highly toxic, pyrophoric or water-sensitive carbene transfer reagents (diazo reagents and organometallic carbenoids). However, although important advances have been made in this direction,4 a major limitation remaining is the broad tolerance to functional groups and consequently, the cyclopropanation of complex molecules.5In 2017, our group reported a new methodology for the stereoconvergent cyclopropanation of E/Z isomeric styrene mixtures and Michael acceptors6 by means of photoredox catalysis (Scheme 1A).7 The key to these studies was the use of the well-known [Ru(bpy)3][PF6]2 photocatalyst and photoreducible diiodomethane8 as a methylene source that enabled the generation of a carbenoid-like radical (˙)CH2I termed radical carbenoid, as a reactive cyclopropanating species. Although the method showed a broad functional group tolerance and excellent chemoselectivity, severe limitations in the styrene scope were found and an excess of diiodomethane (5 equivalents) had to be used. After this, firstly the group of Molander9 and secondly the group of Li10 reported two complementary redox-neutral cyclopropanations using photooxidizable bis(catecholato)iodomethylsilicate reagents and 4CzIPN or Ir[dF(CF3)ppy]2(dtbbpy)PF6 as photocatalysts. The Molander reaction was demonstrated in a wide variety of activated alkenes and showed an excellent functional group tolerance and chemoselectivity, however, the process was exclusively evaluated for methylene transfer via (˙)CH2I. Moreover, Charette and co-workers developed a radical borocyclopropanation enabled by flow techniques, UVA light and xanthone as photocatalyst.11 This reaction involved the generation of (˙)CH(Bpin)I and provided valuable cyclopropylboronates as mixtures of diastereoisomers.12
 | ||
| Scheme 1 New radical cyclopropanations enabled by visible-light: a valuable alternative to methods employing diazo reagents or metal-carbenoids (A and B). | ||
The novel photoredox-catalyzed radical cyclopropanations represent a clear advance in the synthesis of cyclopropane rings and a more practical approach to strategies based on diazo reagents and metal-carbenoids.13,14 However, it is clearly an underdeveloped methodology and challenges associated with alkene scope, reagent diversity or efficiency of the process (reagent equivalents) should be solved. In addition, the development of novel methodologies that avoid the use of precious Ru/Ir photocatalysts and permit late-stage functionalization of complex molecules would be highly appreciated. Here, we report the first general photocatalyst-free cyclopropanation of styrenes that use underexploited gem-diiodomethyl carbonyl compounds as cyclopropanating reagents and a simple compact fluorescence lamp (CFL) as a visible-light source (Scheme 1B). The process generates valuable cyclopropyl carboxylates that could be diversified by well-documented decarboxylative strategies.15 Its excellent functional group tolerance and chemoselectivity have enabled access to cyclopropyl carboxylates that are not possible to obtain in a direct manner by current catalytic strategies relying on metal-carbene(carbenoid) (this is because of the preference of the latter species to react with more nucleophilic functionalities than a styrene double bond).
Results and discussion
During the development of our photoredox-catalyzed cyclopropanation, control experiments revealed that excluding the [Ru(bpy)3][PF6]2 photocatalyst in the reaction resulted in a poor yield of the corresponding cyclopropanes (15–18% yield, 18 hours).6 These interesting results encouraged us to question whether gem-diiodomethyl carbonyl reagents – activated alkyl iodides with more absorbance in the visible region − could provide an efficient photocatalyst-free cyclopropanation and deliver valuable carbonyl-substituted cyclopropane rings. Our investigations started by synthesizing 1-adamantanyl 2,2-diiodoacetate (2a) as a potential new cyclopropanating reagent (Scheme 2A). 2a is a white solid that can be prepared on a 12 gram scale from 3-(adamantan-1-yloxy)-3-oxopropanoic acid and N-iodosuccinimide (NIS). Importantly, 2a features great stability in comparison with the analogue ethyl 2,2-diiodoacetate (2b), which has to be stored in a freezer (<−20 °C) and in solution (0.5 M CH3CN). Differential scanning calorimetry (DSC) studies were performed with reagent 2a and did not show relevant exothermic decompositions, which are commonly found for diazo compounds (see ESI†).16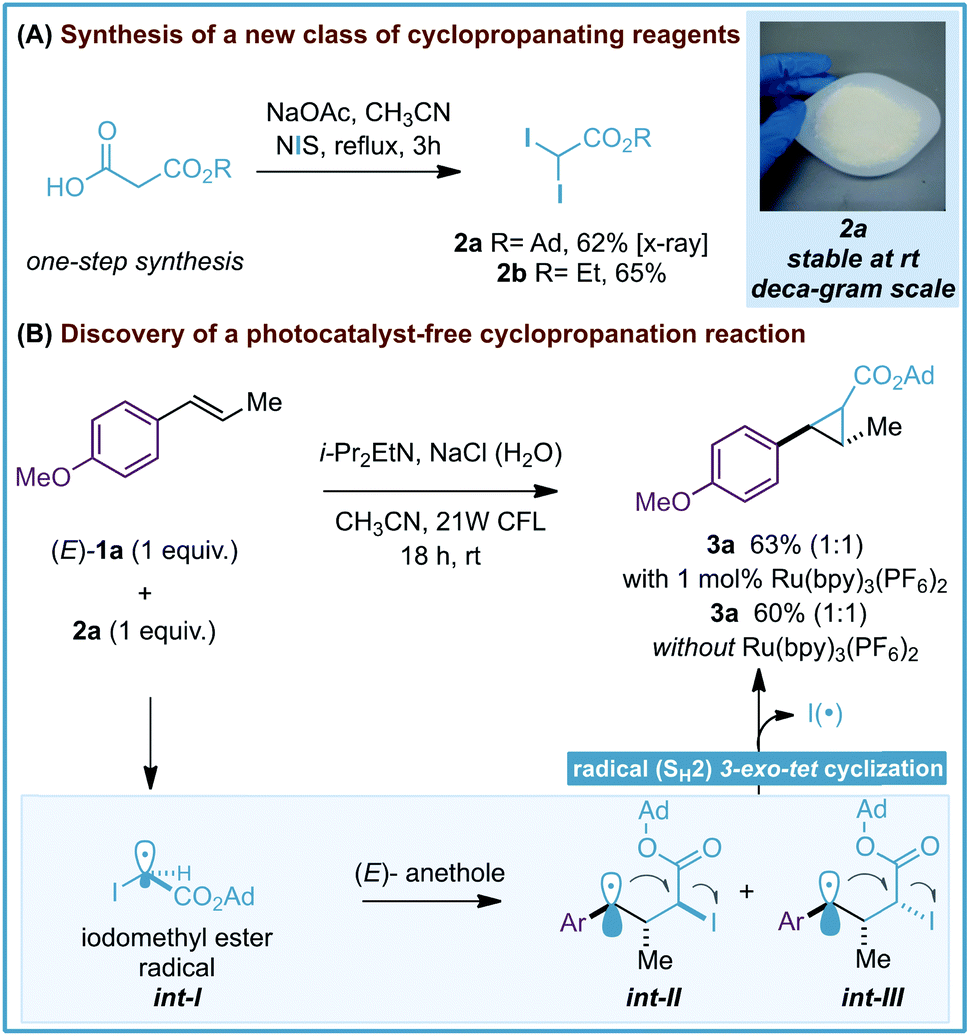 | ||
| Scheme 2 Synthesis of gem-diodomethyl carboxylate reagents 2a,b (A) and discovery of a photocatalyst-free cyclopropanation. Ad = 1-adamantyl (B). | ||
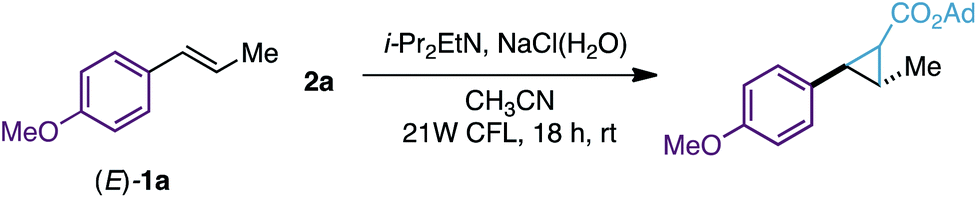
After this, we evaluated reagent 2a (1 equiv.) for the cyclopropanation of (E)-anethole (1a) with and without the [Ru(bpy)3][PF6]2 photocatalyst. We were delighted to find that both reactions led to the expected tri-substituted cyclopropane 3a in moderate yields (63–60% yield, Scheme 2B) as an equimolar isomeric mixture of diastereoisomers. To explain the formation of 3a we propose the initial generation of iodomethyl radical ester int-I as a cyclopropanating species. After this, an unbiased attack of the pyramidal sp3-hybridized radical int-I on 1a would generate benzylic radicals int-II and int-III,17 which would evolve to the corresponding cyclopropane 3a by a radical homolytic substitution (SH2) 3-exo-tet cyclization. Radical homolytic substitutions are common elementary steps in radical chemistry and in terms of frontier molecular orbitals, this cyclization involves the interaction between the high-energy singly occupied molecular orbital (SOMO) of the benzylic radical, and the lowest unoccupied molecular orbital (LUMO) of the C–I bond. A similar cyclization has been observed previously by Curran and Togo in cyclopropane synthesis using 1,3-dihaloalkanes and radical initiators/reductants.18
After these preliminary investigations, we evaluated other solvents, amines and visible-light sources and found that the best reaction conditions involve only 2 equiv. of reagent 2a, i-Pr2EtN (4 equiv.), CH3CN (1 mL), an aqueous solution of NaCl (1.25 M, 0.5 mL) and degasification of the reaction mixture prior to irradiation (91% yield, Table 1, entry 1). We were glad to find that these reaction conditions were also suitable for a 1 gram scale reaction using only 1 equiv. of 2a (75% yield). However, poorer efficiency was observed when reactions were carried out without NaCl (H2O) or H2O (17–48% yield, entries 2 and 3)19 or under air (10% yield, entry 4). No conversion to 3a was observed without i-Pr2EtN or the CFL (entries 5 and 6).
|
|
||
|---|---|---|
| Entry | Deviation of the reaction conditions | Yieldb3a, % |
| a Reaction conditions: 1a (0.10 mmol), 2a (0.10 mmol), i-PrEt2N (0.20 mmol), CH3CN (1 mL), NaCl (1.25 M in H2O, 0.5 mL). Reactions were degassed prior to irradiation. b 1H-NMR yields calculated using 1,2-dimethoxyethane as the internal standard. c Yield of the isolated product adding additional 2a (0.10 mmol, 1 equiv.) and i-PrEt2N (0.20 mmol, 2 equiv.) after 4 hours. d Yield of the isolated product using 1 gram of (E)-1a and 1 equiv. of 2a. See the ESI for the evaluation of other solvents, amines, or visible-light sources. | ||
| 1 | None | 91c(75)d |
| 2 | Without NaCl (H2O) | 17 |
| 3 | Without NaCl | 48 |
| 4 | Under air | 10 |
| 5 | Without i-Pr2EtN | 0 |
| 6 | In the dark | 0 |
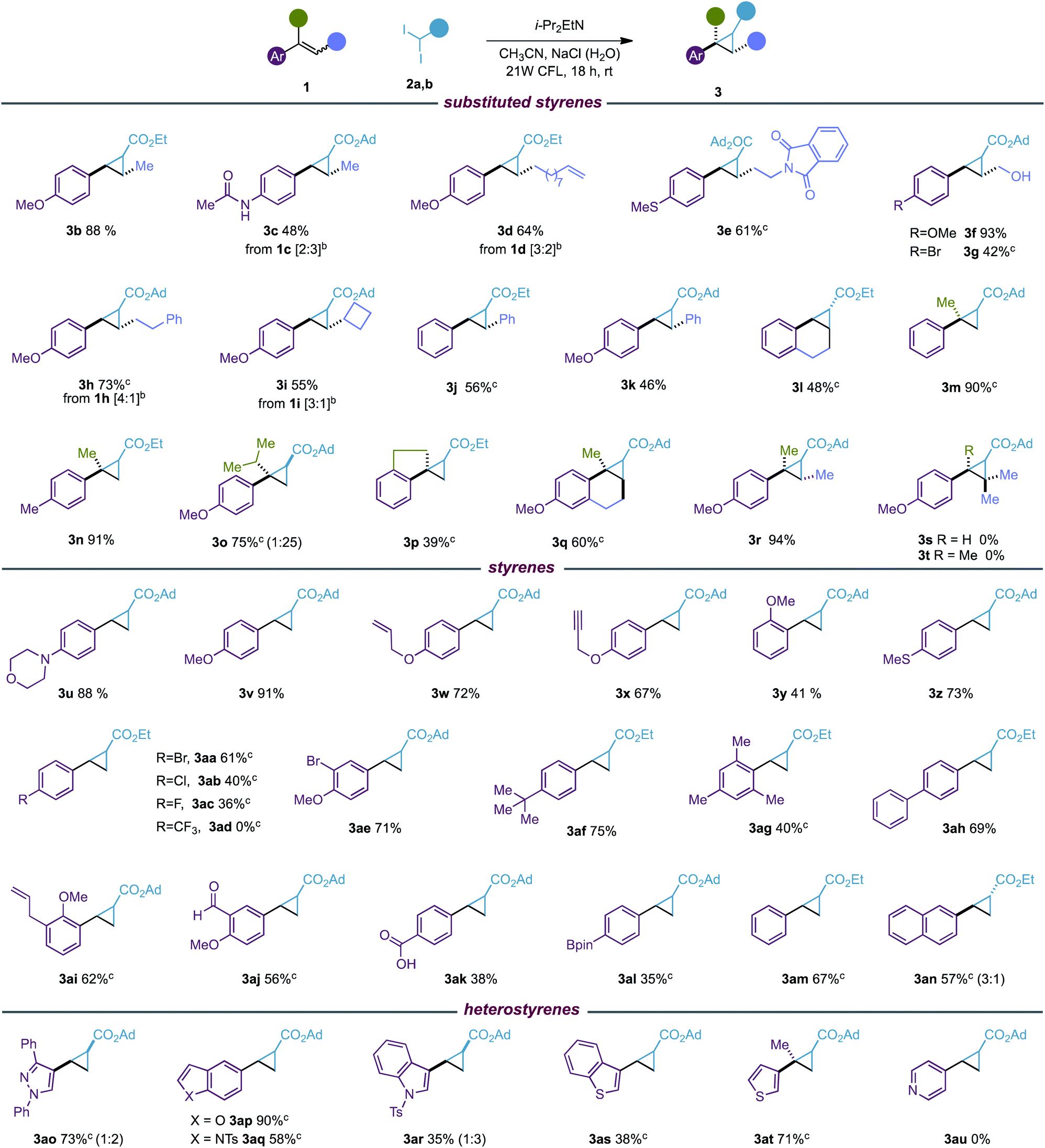
With the optimized reaction conditions in hand, we evaluated the scope of the new photocyclopropanation using 46 styrene derivatives and reagents 2a, b.20 As shown in Table 2, our protocol was effective for a variety of diversely substituted styrenes (3b–r), substituted at the β position with methyl (3b, c) or alkyl groups functionalized with an olefin (3d), phthalimide (3e), alcohol (3f–g) and phenyl (3h) or cyclobutyl rings (3i). We were glad to find that our photocyclopropanation worked well for stilbenes (3j, k), cyclic (3l, q) and α-substituted styrenes (3m–q), which failed in our previous photoredox cyclopropanation.6a In addition, whereas the more substituted acyclic and cyclic trisubstituted styrenes were well tolerated (3q, r), our process did not work for styrenes with a sterically demanding β carbon center (3s, t). It is worth highlighting that although the reaction generates mixtures of diastereoisomers at the α-carbonyl center, which are easily separable by column chromatography, the reaction proceeded with absolute stereoconvergence for E/Z isomeric mixtures of di and tri-substituted styrenes (1c, d, h, i, r).
a Reaction conditions: 1 (0.20 mmol), 2 (0.40 mmol), i-PrEt2N (0.80 mmol), CH3CN (2 mL), NaCl (1.25 M in H2O, 1 mL); yields of the isolated product. Diastereomeric ratios were between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1.5:1 and determined by 1H NMR analysis.
b
E/Z-alkene mixtures 1 are shown in brackets.
c Reaction conditions 1 (0.40 mmol), 2 (0.20 mmol), i-PrEt2N (0.40 mmol), CH3CN (2 mL), NaCl (1.25 M in H2O, 1 mL). :1 and 1.5:1 and determined by 1H NMR analysis.
b
E/Z-alkene mixtures 1 are shown in brackets.
c Reaction conditions 1 (0.40 mmol), 2 (0.20 mmol), i-PrEt2N (0.40 mmol), CH3CN (2 mL), NaCl (1.25 M in H2O, 1 mL).
|
|---|
|
We then decided to investigate the scope of terminal (hetero)styrenes (3u–as). These substrates provided poor conversion to the cyclopropane in our methylenation reaction,6a and instead open-chain dimeric products were found from the homocoupling of benzylic radical intermediates. We were delighted to find that the new photocatalyst-free cyclopropanation was able to cyclopropanate a broad variety of styrenes functionalized with amine (3u), alkoxide (3v–x), sulfide (3z), halogen (3aa–3ac, 3ae), alkyl (3af, ag), phenyl (3ah), allyl (3ai), aldehyde (3aj), carboxylic acid (3ak), pinacol boronic ester (3al), unsubstituted (3am) or naphthalene (3an) groups. Unfortunately, styrenes substituted with strong electron-withdrawing groups such as CF3 did not yield the desired cyclopropane 3ad. Furthermore, we observed that while our protocol was able to functionalize heterostyrenes containing imidazole (3ao), benzofurane (3ap), indole (3aq, 3ar), benzothiophene (3as) and thiophene (3at), no cyclopropanation reaction was found for pyridine derivatives (3au).
The excellent functional group tolerance and chemoselectivity observed towards the styrene double bond are in sharp contrast to current catalytic methods that cyclopropanate styrenes involving electrophilic metal-carbenes from diazoacetates. These methods may suffer from chemo- and site-selectivity in challenging substrates bearing additional alkenes (3d, w, ai),21 or functionalities able to intercept the corresponding metal-carbene intermediate such as alcohol (3f, g),22 aldehyde (3aj),23 alkyne (3x),24 carboxylic acid (3ak)25 or sulfide (3e, z) groups.26
On the other hand, although our process was successfully extended to other gem-diiodomethyl esters (3au, av) or ketone reagents (4a–c, Scheme 3A), the corresponding analogues substituted with amides (4d) or other electron-withdrawing groups did not provide the expected cyclopropane ring (5a–c). The synthetic potential of our photocyclopropanation was further illustrated with the late-stage functionalization of biomolecule derivatives (Scheme 3B). Our process was able to functionalize styrenes derived from estrone (6a), L-valine (6b) and β-D-glucose pentaacetate (6c).27 In addition, we were delighted to find that the process could also work with the unprotected glucose derivative (6d).28 This final example clearly highlights the potential of our methodology to be applicable in alternative complex scenarios.29
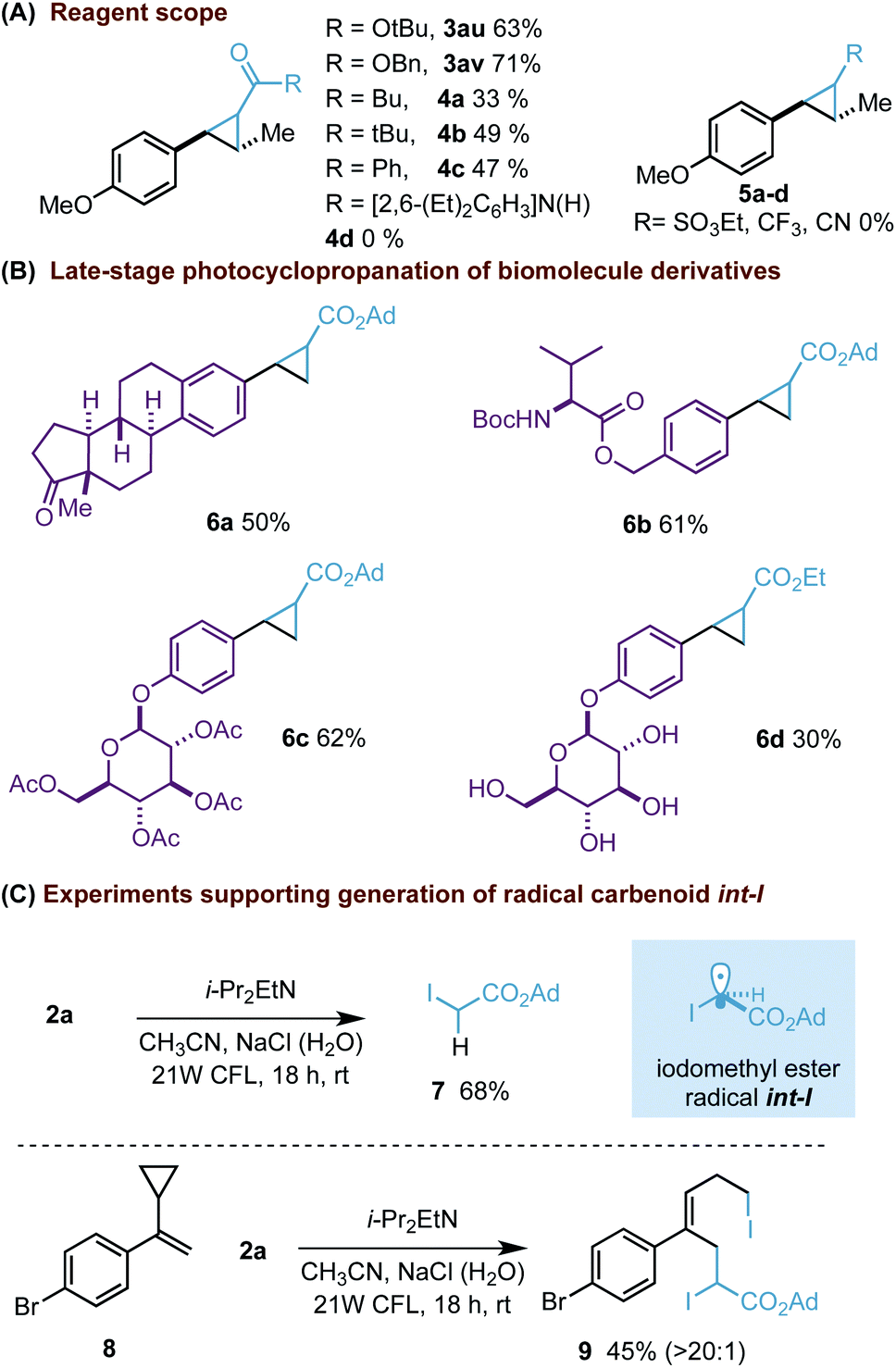 | ||
| Scheme 3 Reagent scope (A), late-stage functionalization (B) and radical trapping experiments (C). Diastereomeric ratios of cyclopropanes were between 1:1 and 1.5:1 and determined by 1H NMR analysis. | ||
Moreover, we wanted to prove the formation of radical int-I.30 Firstly, an experiment carried out without styrene resulted in the isolation of iodoacetate 7 (Scheme 3C). Its formation can be explained involving abstraction of a hydrogen atom with int-I. An additional experiment carried out with the classic cyclopropyl radical probe 8 also supports the formation of int-I as an intermediate by forming an atom-transfer radical addition (ATRA) product 9.
Finally, our current hypotheses for the photogeneration of radical carbenoid int-I are depicted in Scheme 4. Considering that our reagent 2a absorbs in the visible region (tail of absorption: 460 nm at 0.1 M, see ESI†), we believe that one of the possible scenarios could involve the photogeneration of the excited state [2a]* and its evolution to int-I and I(˙) by homolysis (Scheme 4A).31 In this particular case, i-Pr2EtN might be acting as a quencher of the iodine species, I(˙) or I2, formed during the reaction.32 Recently, the group of Aggarwal hypothesized a homolytic cleavage of the C–I bond in α-iodo ketones by visible light as a suitable pathway for the generation of electrophilic alkyl radicals, analogous to int-I.33
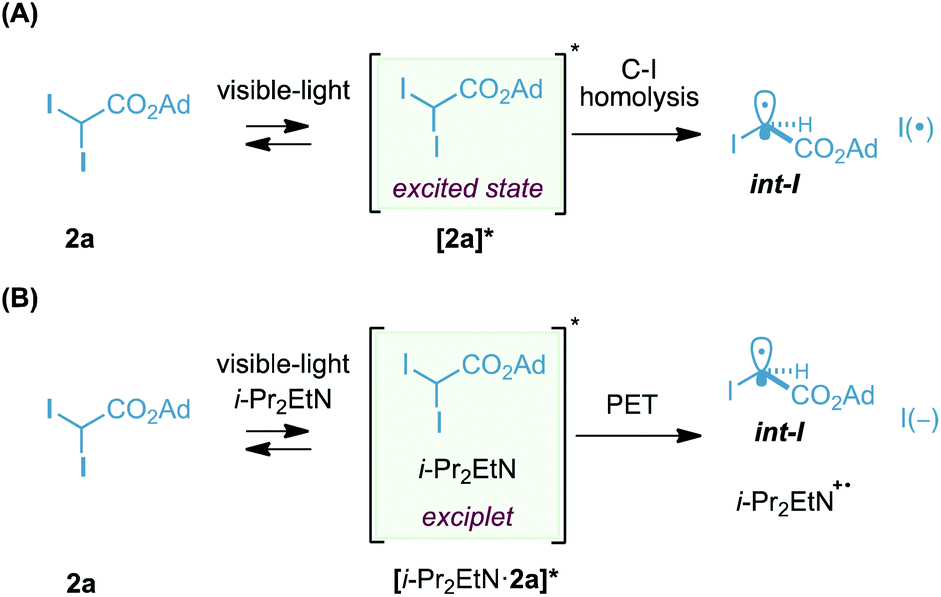 | ||
| Scheme 4 Mechanistic hypotheses for the photogeneration of int-I (A and B). | ||
On the other hand, another possible scenario involves the formation of a short-lived exciplet [i-Pr2EtN·2a]* that undergoes photoinduced electron transfer and generates int-I.34 Exciplet analogues have been proposed recently in the generation of radicals from inactivated alkyl iodides.35
The quantum yield measured for the model photocyclopropanation of (E)-1a with 2a was found to be 1.5 (λ = 410 nm in CH3CN/H2O, using potassium ferrioxalate as the actinometer), suggesting that if a radical-chain is operating, it is very inefficient. At this point, this result does not allow us to discriminate between one of the two mechanistic hypotheses depicted in Scheme 4.
Conclusions
In summary, we have developed a robust, scalable, and safe photochemical cyclopropanation reaction with gem-diiodomethyl carbonyl reagents. Our new cyclopropanation features an excellent functional group tolerance that permitted the functionalization of a broad range of functionalized styrenes and biomolecules.Conflicts of interest
A patent application describing the results presented in this manuscript has been filled.Acknowledgements
The ICIQ Starting Career Programme, Agencia Estatal de Investigación (AEI) of the Ministerio de Ciencia, Innovación y Universidades (CTQ2016-75311-P, AEI/FEDER-EU), the CELLEX Foundation through the CELLEX-ICIQ high-throughput experimentation platform and the CERCA Programme (Generalitat de Catalunya) are gratefully acknowledged for financial support. We thank the CELLEX Foundation for the pre-doctoral fellowship (to A. G. H.) and the ICIQ Research support area. We also thank Y. YuChao for preliminary experiments, P. Sarró for proofreading and Profs. R. Martin and P. Melchiorre for insightful discussions.Notes and references
- H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed.
- T. T. Talele, J. Med. Chem., 2016, 59, 8712 CrossRef CAS PubMed.
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651 CrossRef CAS PubMed.
- For a review: (a) W. Wu, Z. Lin and H. Jiang, Org. Biomol. Chem., 2018, 16, 7315 RSC. For selected examples, see: (b) C. D. Papageorgiou, M. A. Cubillo De Dios, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 4641 CrossRef CAS PubMed; (c) B. Moreau and A. B. Charette, J. Am. Chem. Soc., 2005, 127, 18014 CrossRef CAS PubMed; (d) R. K. Kunz and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3240 CrossRef CAS PubMed; (e) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034 CrossRef CAS PubMed; (f) C. R. Solorio-Alvarado, Y. Wang and A. M. Echavarren, J. Am. Chem. Soc., 2011, 133, 11952 CrossRef CAS PubMed; (g) B. Morandi and E. M. Carreira, Science, 2012, 335, 1471 CrossRef CAS; (h) J. Barluenga, N. Quiñones, M. Tomás-Gamasa and M. P. Cabal, Eur. J. Org. Chem., 2012, 2312 CrossRef CAS; (i) P. Cotugno, A. Monopoli, F. Ciminale, A. Milella and A. Nacci, Angew. Chem., Int. Ed., 2014, 53, 13563 CrossRef CAS PubMed; (j) T. Piou and T. Rovis, J. Am. Chem. Soc., 2014, 136, 11292 CrossRef CAS PubMed; (k) T. Den Hartog, J. M. S. Toro and P. Chen, Org. Lett., 2014, 16, 1100 CrossRef CAS PubMed; (l) S. A. Künzi, J. M. Sarria Toro, T. den Hartog and P. Chen, Angew. Chem., Int. Ed., 2015, 54, 10670 ( Angew. Chem. , 2015 , 127 , 10817 ) CrossRef PubMed; (m) M. J. González, J. González, L. A. López and R. Vicente, Angew. Chem., Int. Ed., 2015, 54, 12139 ( Angew. Chem. , 2015 , 127 , 12307 ) CrossRef PubMed; (n) S. Manna and A. P. Antonchick, Angew. Chem., Int. Ed., 2015, 54, 14845 ( Angew. Chem. , 2015 , 127 , 15058 ) CrossRef CAS PubMed; (o) Y. Y. Zhou and C. Uyeda, Angew. Chem., Int. Ed., 2016, 55, 3171 ( Angew. Chem. , 2016 , 128 , 3223 ) CrossRef CAS PubMed; (p) J. Xu, N. B. Samsuri and H. A. Duong, Chem. Commun., 2016, 52, 3372 RSC; (q) B. Herle, P. M. Holstein and A. M. Echavarren, ACS Catal., 2017, 7, 3668 CrossRef CAS; (r) J. Werth and C. Uyeda, Chem. Sci., 2018, 9, 1604 RSC; (s) J. Werth and C. Uyeda, Angew. Chem., Int. Ed., 2018, 57, 13902 ( Angew. Chem. , 2018 , 130 , 14098 ) CrossRef CAS; (t) M. Mato, B. Herlé and A. M. Echavarren, Org. Lett., 2018, 20, 4341 CrossRef CAS PubMed; (u) M. Mato and A. M. Echavarren, Angew. Chem., Int. Ed., 2019, 58, 2088 ( Angew. Chem. , 2019 , 131 , 2110 ) CrossRef CAS.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC.
- (a) A. M. del Hoyo, A. G. Herraiz and M. G. Suero, Angew. Chem., Int. Ed., 2017, 56, 1610 ( Angew. Chem. , 2017 , 129 , 1632 ) CrossRef CAS PubMed; (b) A. M. del Hoyo and M. G. Suero, Eur. J. Org. Chem., 2017, 2017, 2122 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed.
- For pioneering examples in photoredox carbon–halogen bond cleavage: (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (b) D. A. Nagib, M. E. Scott and D. W. C. Macmillan, J. Am. Chem. Soc., 2009, 2, 10875 CrossRef PubMed; (c) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756 CrossRef CAS PubMed; (d) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed; (e) C. J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef CAS PubMed.
- J. P. Phelan, S. B. Lang, J. S. Compton, C. B. Kelly, R. Dykstra, O. Gutierrez and G. A. Molander, J. Am. Chem. Soc., 2018, 140, 8037 CrossRef CAS.
- T. Guo, L. Zhang, X. Liu, Y. Fang, X. Jin, Y. Yang, Y. Li, B. Chen and M. Ouyang, Adv. Synth. Catal., 2018, 360, 4459 CrossRef CAS.
- M. Sayes, G. Benoit and A. B. Charette, Angew. Chem., Int. Ed., 2018, 57, 13514 CrossRef CAS PubMed.
- T. Ohtani, Y. Tsuchiya, D. Uraguchi and T. Ooi, Org. Chem. Front., 2019, 6, 1734 RSC.
- A. G. Herraiz and M. G. Suero, Synthesis, 2019, 51, 2821 CrossRef CAS.
- For photoredox cyclopropanations involving diazo compounds: (a) F. J. Sarabia and E. M. Ferreira, Org. Lett., 2017, 19, 2865 CrossRef CAS PubMed; (b) P. Li, J. Zhao, L. Shi, J. Wang, X. Shi and F. Li, Nat. Commun., 2018, 9, 1972 CrossRef PubMed. For alternative cyclopropane synthesis by means of visible-light photoredox catalysis: (c) Y. Zhang, R. Qian, X.-L. Zheng, Y. Zeng, J. Sun, Y. Chen, A. Ding and H. Guo, Chem. Commun., 2015, 51, 54 RSC; (d) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430 ( Angew. Chem. , 2018 , 130 , 15656 ) CrossRef CAS PubMed.
- For selected recent examples: (a) S. Ventre, F. R. Petronijevic and D. W. C. Macmillan, J. Am. Chem. Soc., 2015, 137, 5654 CrossRef CAS PubMed; (b) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (c) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322 CrossRef CAS PubMed; (d) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213 CrossRef CAS PubMed; (e) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson and A. K. Chatterjee, et al. , Science, 2017, 356, 1045 CAS; (f) J. A. Kautzky, T. Wang, R. W. Evans and D. W. C. Macmillan, J. Am. Chem. Soc., 2018, 140, 6522 CrossRef CAS; (g) Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83 CrossRef CAS PubMed; (h) S. Ni, N. M. Padial, C. Kingston, J. C. Vantourout, D. C. Schmitt, J. T. Edwards, M. M. Kruszyk, R. R. Merchant, P. K. Mykhailiuk and B. B. Sanchez, et al. , J. Am. Chem. Soc., 2019, 141, 6726 CrossRef CAS PubMed; (i) M. Montesinos-Magraner, M. Costantini, R. Ramírez-Contreras, M. E. Muratore, M. J. Johansson and A. Mendoza, Angew. Chem., Int. Ed., 2019, 58, 5930 CrossRef CAS PubMed. For a review: (j) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735 CrossRef CAS.
- J. D. Clark, A. S. Shah, J. C. Peterson, L. Patelis, R. J. A. Kersten, A. H. Heemskerk, M. Grogan and S. Camden, Thermochim. Acta, 2002, 386, 65 CrossRef CAS.
- This hypothesis is in line with the rationalization of Macmillan to explain the lack of diastereocontrol of a radical attack on a pi-system: (a) J. L. Jeffrey, F. R. Petronijević and D. W. C. Macmillan, J. Am. Chem. Soc., 2015, 137, 8404 CrossRef CAS PubMed; see also (b) D. Mazzarella, G. E. M. Crisenza and P. Melchiorre, J. Am. Chem. Soc., 2018, 140, 8439 CrossRef CAS.
- (a) D. P. Curran and A. E. Gabarda, Tetrahedron, 1999, 55, 3327 CrossRef CAS; (b) T. Ohkita, Y. Tsuchiya and H. Togo, Tetrahedron, 2008, 64, 7247 CrossRef CAS.
- We believe that the role of aqueous NaCl is to quench possible water-soluble sub-products formed during the reaction, whose absorption reduces the number of available photons and the progress of the photocyclopropanation. The sub-products could come from the decomposition of Hünig's base. A similar observation has been recently found by Bach et al.: A. Böhm and T. Bach, Chem.–Eur. J., 2016, 22, 15921 CrossRef PubMed.
- During the evaluation of the scope, we observed that a reverse stoichiometry provided better yields for some styrenes. See footnote c in Table 2 for experimental details.
- H. M. L. Davies and S. A. Panaro, Tetrahedron, 2000, 56, 4871 CrossRef CAS.
- C. Chen, S. F. Zhu, B. Liu, L. X. Wang and Q. L. Zhou, J. Am. Chem. Soc., 2007, 129, 12616 CrossRef CAS PubMed.
- M. P. Doyle, D. C. Forbes, M. N. Protopopova, S. A. Stanley, M. M. Vasbinder and K. R. Xavier, J. Org. Chem., 1997, 62, 7210 CrossRef CAS PubMed.
- (a) Y. Lou, M. Horikawa, R. A. Kloster, N. A. Hawryluk and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 8916 CrossRef CAS PubMed; (b) A. Suárez and G. C. Fu, Angew. Chem., Int. Ed., 2004, 43, 3580 CrossRef.
- M. C. M. Van Oers, L. K. E. A. Abdelmohsen, F. P. J. T. Rutjes and J. C. M. Van Hest, Chem. Commun., 2014, 50, 4040 RSC.
- A. Padwa and S. F. Hornbuckle, Chem. Rev., 1991, 91, 263 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- Two experiments carried out with ethyldiazoacetate (2 equiv.), Rh2(OAc)4 (1 mol%) and CH2Cl2 or CH3CN as solvents did not provide the corresponding cyclopropane analogue to 6d.
- For examples of biocompatible alkene cyclopropanation reactions with diazo compounds, see: (a) P. S. Coelho, E. M. Brustard, A. Kannan and F. H. Arnold, Science, 2013, 339, 307 CrossRef CAS; (b) M. Bordeaux, V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2015, 54, 1744 ( Angew. Chem. , 2015, , 127 , 1764 ) CrossRef CAS; (c) S. Wallace and E. P. Balskus, Angew. Chem., Int. Ed., 2015, 54, 7106 ( Angew. Chem. , 2015 , 127 , 7212 ) CrossRef CAS; (d) A. Tinoco, V. Steck, V. Tyagi and R. Fasan, J. Am. Chem. Soc., 2017, 139, 5293 CrossRef CAS PubMed; (e) A. M. Knight, S. B. J. Kan, R. D. Lewis, O. F. Brandenberg, K. Chen and F. H. Arnold, ACS Cent. Sci., 2018, 4, 372 CrossRef CAS PubMed.
- A radical intermediate analogous to int-I has been recently proposed and detected in a cyclopropanation reaction using ethyl diazoacetate and I2/Ru(bpy)3Cl2 catalysts under visible-light irradiation. See ref. 14b.
- W. G. McGimpsey and J. C. Scaiano, Can. J. Chem., 1988, 66, 1474 CrossRef CAS.
- (a) Diiodine is a well-known radical inhibitor of radical chain reactions: D. P. Curran and C.-T. Chang, Tetrahedron Lett., 1990, 31, 933 CrossRef CAS; (b) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS.
- M. Silvi, C. Sandford and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 5736 CrossRef CAS.
- The possibility of formation of a photoactive ground-state charge-transfer complex between i-Pr2EtN and 2a may be excluded: UV/vis absorption analysis of an equimolar mixture of i-Pr2EtN and 2a did not show the characteristic bathochromic shift expected for a charge-transfer complex (see ESI†).
- (a) Y. Shen, J. Cornella, F. Juliá-Hernández and R. Martin, ACS Catal., 2017, 7, 409 CrossRef CAS. For related photochemical generation of radical species with activated alkyl halides via charge-transfer or electron donor–acceptor complexes see: (b) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750 CrossRef CAS; (c) X. Sun, W. Wang, Y. Li, J. Ma and S. Yu, Org. Lett., 2016, 18, 4638 CrossRef CAS; (d) Y. Wang, J. Wang, G. X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442 CrossRef CAS ; and ref. 19.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1915890. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02749a |
| This journal is © The Royal Society of Chemistry 2019 |