
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium(0)/benzoic acid catalysis merges sequences with D2O-promoted labelling of C–H bonds†
Gianpiero
Cera
 ,
Nicola
Della Ca'
and
Giovanni
Maestri
*
,
Nicola
Della Ca'
and
Giovanni
Maestri
*
Dipartimento di Scienze Chimiche, della Vita e della Sostenibilità Ambientale, Università di Parma, Parco Area delle Scienze 17/A, 43124 Parma, Italy. E-mail: giovanni.maestri@unipr.it
First published on 25th September 2019
Abstract
The combination of a Pd(0) complex with benzoic acid in the presence of D2O enables the synthesis of valuable families of highly deuterated organics through elaborate sequential reactions. The catalytic system can convert 2-butyne fragments into the corresponding d-dienamides, which can then readily deliver labeled polycyclic quinone motifs. Propargylated tryptamines lead to formation of highly enriched tetrahydrocarbolines through the C–H activation of their unprotected indole ring. Mechanistic studies reveal the ordered series of events that regulate the outcome of these complex reactions, which include multiple, sequential and selective H/D scrambling from the cheapest and safest deuterium source.
Introduction
We report herein a catalytic method that uses unlabeled alkynes reagents (1, 3) together with deuterium oxide to directly access highly-enriched complex organics, such as d-1,3-dienamides 2 and d-tetrahydrocarbolines 4 (Scheme 1) thanks to the combination of palladium(0) and a carboxylic acid. The catalytic system triggers an ordered sequence of elementary steps that involve up to five series of isotopic scrambling/insertion and β-hydrogen elimination subsequences on their key palladium hydride intermediate.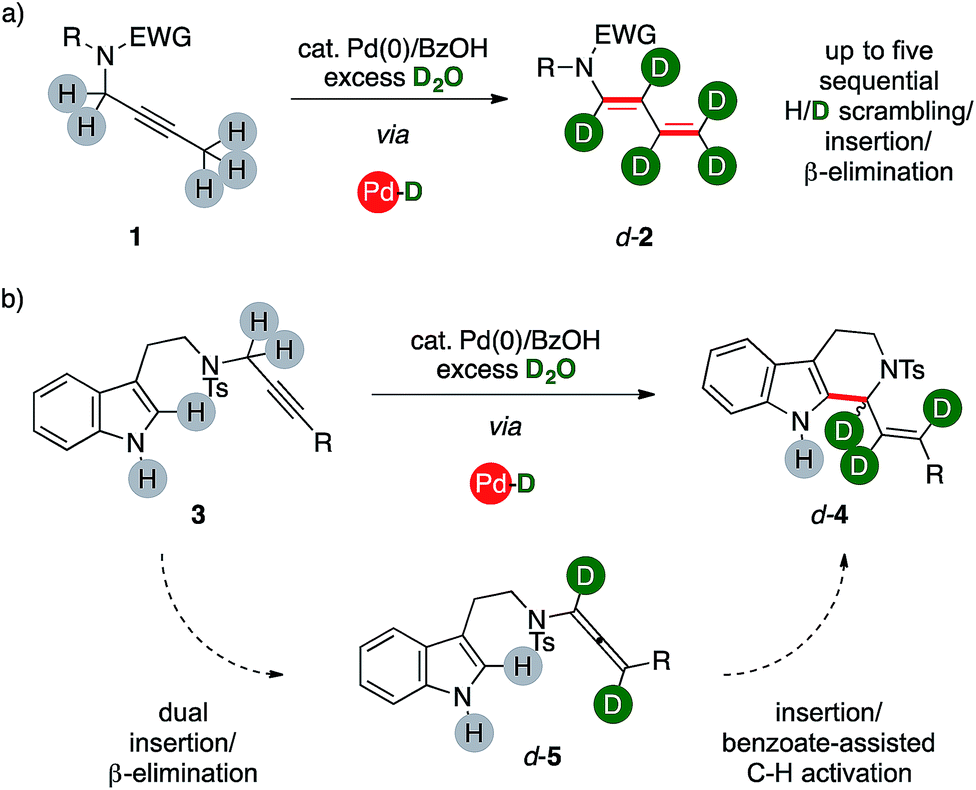 | ||
| Scheme 1 Present one-pot approach to complex, polydeuterated organics. | ||
The reaction provides a direct access to otherwise challenging polydeuterated molecular architectures, thus proving an additional synthetic tool in the growing field of studies that encompass isotopic labeling. This includes probes for reaction mechanism studies, mass-dependent analyses and, foremost, bioactivity studies.1
In this context, deuterium labelling of pharmaceuticals is surging since heavy analogues are often clinically or preclinically identical to their protio predecessor, rendering in turn the C–D bond the actual non-radioactive, bio-isosteric, -isotopic and -isoelectronic replacement for the ubiquitous C–H one.2 For these reasons, a plethora of synthetic strategies for H/D scrambling have been already developed, based either on the use of a deuterated reductant or on formal redox-neutral reaction combined with a suitable d-source.3 In this regard, transition metals-catalyzed C–H functionalizations proved to be mandatory to label non activated aromatic C(sp2)–H bonds of complex target molecules (Scheme 2).
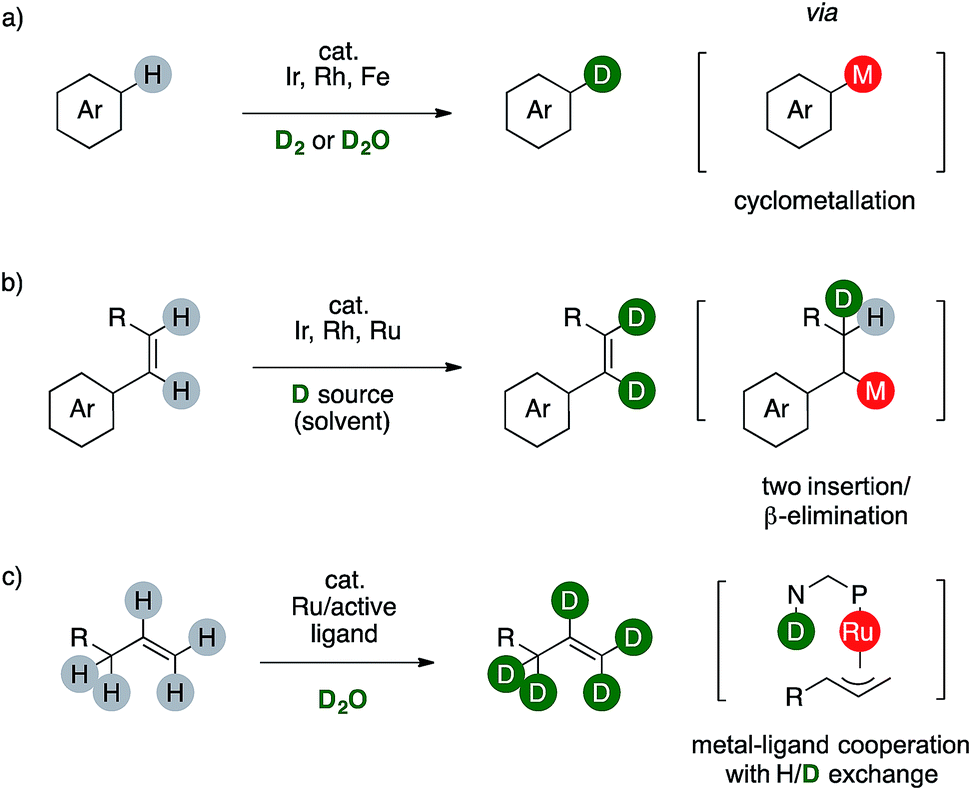 | ||
| Scheme 2 Redox neutral strategies for the deuteration of C(sp2)–H bonds. | ||
The synthesis of deuterated arenes is based on the formation of an arylmetal complex that afford a monodeuterated product through a single H/D exchange step. These strategies might employ costly catalysts or hazardous reactants such as D2 (Scheme 2a).4 The labeling of C(sp2)–H bonds of styrenes involves the formation of an M–D species, which can deliver the desired deuterated olefin via sequential insertion/β-elimination. The reaction can also occur twice using a disubstituted styrene, although the source of deuterium is usually the deuterated benzene used also as tool to observe the transformation itself. This limits in turn the synthetic viability of these elegant organometallic processes because the deuterium source is an expensive cancerogenic (Scheme 2b).5 Complementary, few examples of ruthenium catalysts are reported to promote perdeuteration of alkenes through the formation of η3-metal intermediates.6 Metal-to-ligand cooperativity exploiting the basic amino group of a bidentate N,P ligand is crucial to attain high level of deuterium incorporation on both the C(sp2) and the C(sp3) centers of an allyl fragment (Scheme 2c).7 This method affords deuterium-enriched olefins that can be useful for further transformations, eventually leading to labelled targets. Reasoning on the mechanism of these sequences prompted us to consider the possibility of a complementary redox neutral strategy by combining palladium catalysis and carboxylic acids to afford deuterium enriched products.
Results and discussion
Recently, our group turned its attention on the reactivity of alkynes in the presence of palladium complexes and mild Brønsted acids.8 In this context, the combination of a mononuclear low-valent complex with a carboxylic acid is a recurrent tool for the in situ generation of palladium hydrides.9 We demonstrated the high modularity of the combination of benzoic acid with a Pd(0) precursor, which can allow a selective access to different classes of valuable unsaturated products. The catalytic system initially operates through a reversible isomerization of a propargyl fragment to the corresponding allene intermediate (5, Scheme 1). The reaction can then evolve either via base-mediated C–H activation, eventually affording tetrahydrocarbolines 4, or through a further sequential insertion/β-elimination series of steps to afford 1,3-dienes 2. Preliminary results on the mechanism of these reactions suggested the potential feasibility of incorporating isotopic labeling within a single sequence. Through a careful series of studies, we present that these palladium hydride sequences can take place in the presence of D2O, too. The system converts selectively unlabeled substrates with a propargylic arm into valuable organics with a polydeuterated fragment in one-pot. It is worth noting that the catalytic system makes use of deuterium oxide, which is the most abundant, cheapest and safest source of deuterium for chemical purposes.Optimization of the synthetic method
At the outset of our investigation, we probed our model substrate propargylamide 1a to the previously optimized catalytic conditions8a and adding a deuterated carboxylic acid (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Acid (eq.) | [D]-2a | Da, Db, Dc, Dd [%] |
| a Conditions: 1a (0.2 mmol), Pd(PPh3)4 (0.01 mmol), ligand (0.02 mmol), toluene (2.0 ml), 100 °C, yields of isolated product. b With D2O as D source (50 equiv.). c With tetrabutylammonium benzoate (0.7 mmol). d 120 °C. e Pd(PPh3)4 (0.02 mmol), PCy3 (0.04 mmol). f In 1,4-dioxane. g Without [Pd]. A = sodium 3-(diphenylphosphaneyl)benzenesulfonate. B = 2-(diphenylphosphaneyl)benzenesulfonic acid. | ||||
| 1 | PCy3 | AcOD (5) | 78 | 34, 42, 58, 50 |
| 2 | PCy3 | AcOD (10) | 72 | 32, 43, 64, 50 |
| 3 | PCy3 | AcOD (20) | 66 | 27, 68, 83, 79 |
| 4 | PCy3 | AcOD (35) | <10 | — |
| 5b | PCy3 | BzOH (0.1) | 65 | 7, 75, 91, 63 |
| 6b | A | BzOH (0.1) | <10 | — |
| 7b | B | BzOH (0.1) | — | — |
| 8b,c | PCy3 | BzOH (0.1) | 39 | 4, 65, 87, 50 |
| 9b,d | PCy3 | BzOH (0.1) | 68 | 12, 88, 89, 93 |
| 10b,d,e | PCy3 | BzOH (0.1) | 72 | 8, 75, 88, 77 |
| 11b,d,f | PCy3 | BzOH (0.1) | 53 | 6, 41, 91, 33 |
| 12b,d,g | PCy3 | BzOH (0.1) | — | — |
| 13b,d | PCy3 | — | — | — |
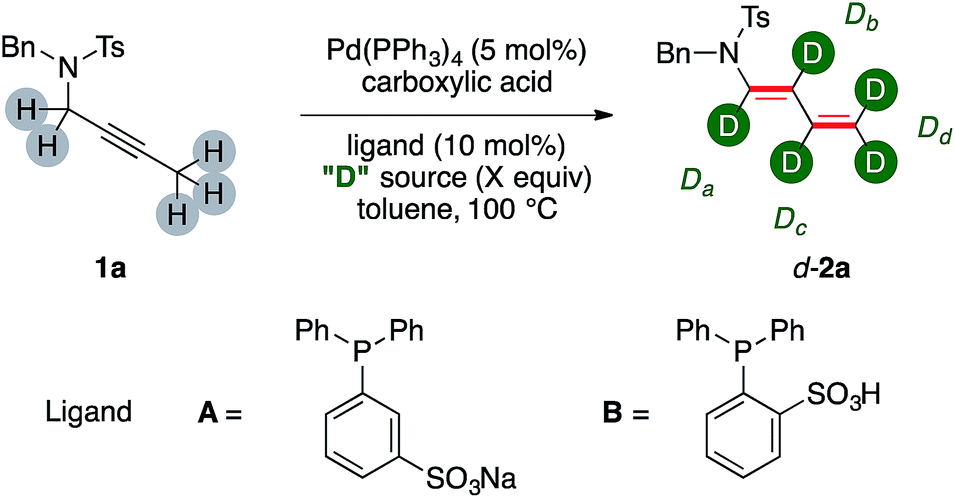
The H/D ratios in product 2a has been determined on the basis of the residual proton signals of its diene arm in the 1H NMR spectrum. Initial experiments tested 1a in the presence of increasing amounts of AcOD (Table 1, entries 1–4). The d-2a was delivered in synthetic useful yields, which however decreased fairly linearly through the series (78% and 66% for entries 1 and 3, respectively). The incorporation of deuterium moved inversely. The trend is visible comparing the labeling ratio on the carbons β-, γ- and δ- to nitrogen of d-2a from entries 1 and 3. This increases by 26%, 25% and 29%, respectively. On the contrary, labeling α- to nitrogen barely moved throughout the triad of experiments, slightly decreasing from 34% to 27%. A further increase on the molar excess of AcOD proved however detrimental for the yield of the dual isomerization (entry 4). At this point we turned our attention to the use of a smarter source of deuterium, namely D2O. We reasoned that its uncomplete miscibility with toluene might have been a tool to smooth its reactivity even using a relatively large molar excess. This, in turn, should have maybe preserved the chemical efficiency of the whole sequence, which appeared an intrinsic shortfall of the, incidentally way more expensive, AcOD labeling agent.
The use of a catalytic amount of benzoic acid lead to isolation of d-2a in good yields and deuterium incorporation (65%, entry 5). Remarkably, the distribution of deuterium among the chain was different from the outcome observed with a deuterated acid without water. In particular, the position α- to nitrogen was the least labelled one (7%), while very high values were observed for the central β- and γ- carbons (75% and 91%, respectively, for a possible rationalization, vide infra). Phosphine ligands A and B, which were already proved to be viable in phase-transfer reaction conditions,10 were not suitable for the catalytic transformations (entries 6, 7). The use of a phase-transfer reagent such as TBABz delivered d-2a with lower performance (entry 8). A higher temperature proved beneficial (68%, entry 9) and was accompanied by the highest deuterium incorporation on the terminal methylene fragment of d-2a (93%). The significant deuteration of d-2a was further confirmed by 2H NMR and MS analyses. A reaction conducted increasing the amount of catalyst to 10 mol% did not improve further the yield (72%, entry 10). Ethereal solvents such as 1,4-dioxane were suitable for the synthesis of d-2a although in diminished yields (53%, entry 11). Control experiments performed without palladium precursor or without benzoic acid did not lead to any conversion of the starting material 1a (entries 12, 13). These results highlight that both the metal and the organic acid are, together, required for the dual isomerization/multiple deuteration sequence.
Synthesis of polydeuterated 1,3-dienes
We next probed the generality of the methodology submitting to the optimized reaction conditions a family of propargylic amides (Fig. 1).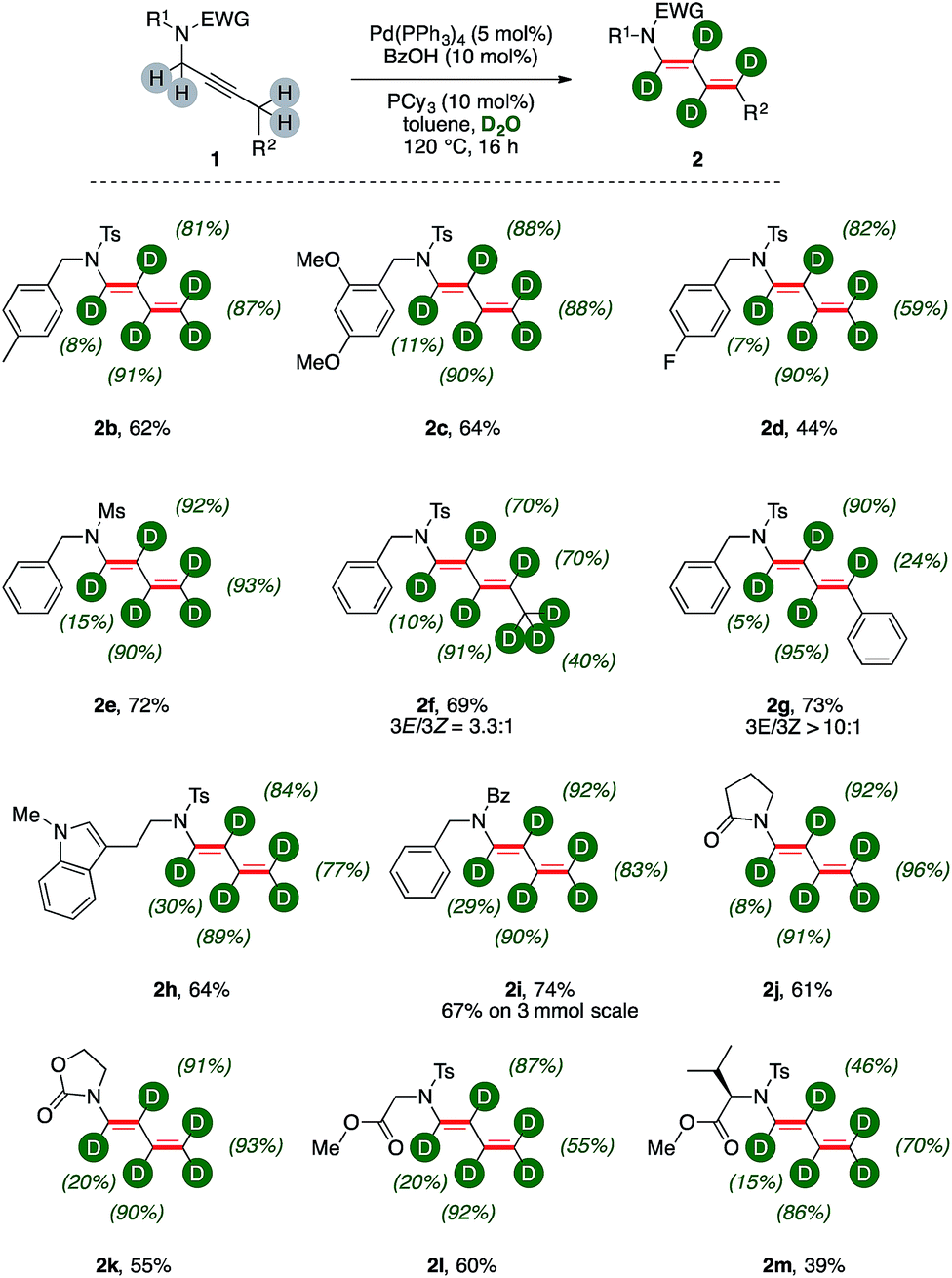 | ||
| Fig. 1 Catalytic synthesis of d-dienes 2. | ||
Tandem isomerization/deuteration was applicable to different N-protected sulphonamides in synthetically useful yields and high levels of deuterium incorporation (Fig. 1, 2b–2e). Pentyne fragment 1f delivered the corresponding deuterated 1,3-diene 2f with complete chemoselectivity and moderate regioselectivity (3E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3Z = 3:1) observing partial deuteration at the terminal methyl unit (40%). The substitution with a phenyl ring on the butyne moiety led to an enhanced regiocontrol (3E:3Z > 10:1), affording conjugated diene 2g in good yields. Aromatic C–H bonds of the substituent are left untouched in the disubstituted diene 2g, and the benzylic carbon has little deuterium content (24%). This is curiously in contrast to 2f, in which the diene has an aliphatic substituent extensively labeled and significant d-content on the carbon δ- to nitrogen (70%). Hence, tryptamine derivative 2h was synthesized (64%) highlighting the tolerance of the methodology with respect to electron-rich heterocycles. Propargylic amides and cyclic carbamates 1i–1k could be transformed to the corresponding products with comparable levels of deuteration and good yields (55–74%). Particularly, the reaction showed high robustness with deuterated dienamide 2i synthesized with good yields (67%) on a 3 mmol scale reaction. Finally, N,O-protected amino acids derivatives 1l–1m were converted to the corresponding products with high chemoselectivity (60% and 39%, respectively). Indeed, their acidic C–H bonds were found untouched while the isomerization of the propargylic unit produced conjugated 1,3 dienes 2l–2m with good deuterium incorporation. Among all these products, deuterium incorporation on the carbon α- to nitrogen is the lowest of the chain, ranging from 7% to 30%, while the labelling efficiency on β-, γ-, and δ- carbons is generally above 80%.
:3Z = 3:1) observing partial deuteration at the terminal methyl unit (40%). The substitution with a phenyl ring on the butyne moiety led to an enhanced regiocontrol (3E:3Z > 10:1), affording conjugated diene 2g in good yields. Aromatic C–H bonds of the substituent are left untouched in the disubstituted diene 2g, and the benzylic carbon has little deuterium content (24%). This is curiously in contrast to 2f, in which the diene has an aliphatic substituent extensively labeled and significant d-content on the carbon δ- to nitrogen (70%). Hence, tryptamine derivative 2h was synthesized (64%) highlighting the tolerance of the methodology with respect to electron-rich heterocycles. Propargylic amides and cyclic carbamates 1i–1k could be transformed to the corresponding products with comparable levels of deuteration and good yields (55–74%). Particularly, the reaction showed high robustness with deuterated dienamide 2i synthesized with good yields (67%) on a 3 mmol scale reaction. Finally, N,O-protected amino acids derivatives 1l–1m were converted to the corresponding products with high chemoselectivity (60% and 39%, respectively). Indeed, their acidic C–H bonds were found untouched while the isomerization of the propargylic unit produced conjugated 1,3 dienes 2l–2m with good deuterium incorporation. Among all these products, deuterium incorporation on the carbon α- to nitrogen is the lowest of the chain, ranging from 7% to 30%, while the labelling efficiency on β-, γ-, and δ- carbons is generally above 80%.
Substrate 1n, which bears a tertiary carbon α- to nitrogen, lead selectively to deuterated diene d-2n in good yields (76%, Scheme 3). In this case, the whole ethyl fragment of the substrate undergoes extensive deuteration (76% and 46%, respectively), the alkyne carbons are the most abundantly labeled sites (89% and 91%, respectively), and no H/D exchange took place on the hindered position alpha to nitrogen. Furthermore, dienamide isomer 2n′ was not observed at all. This shows that the directionality of the isomerization was likely influenced by steric demands.
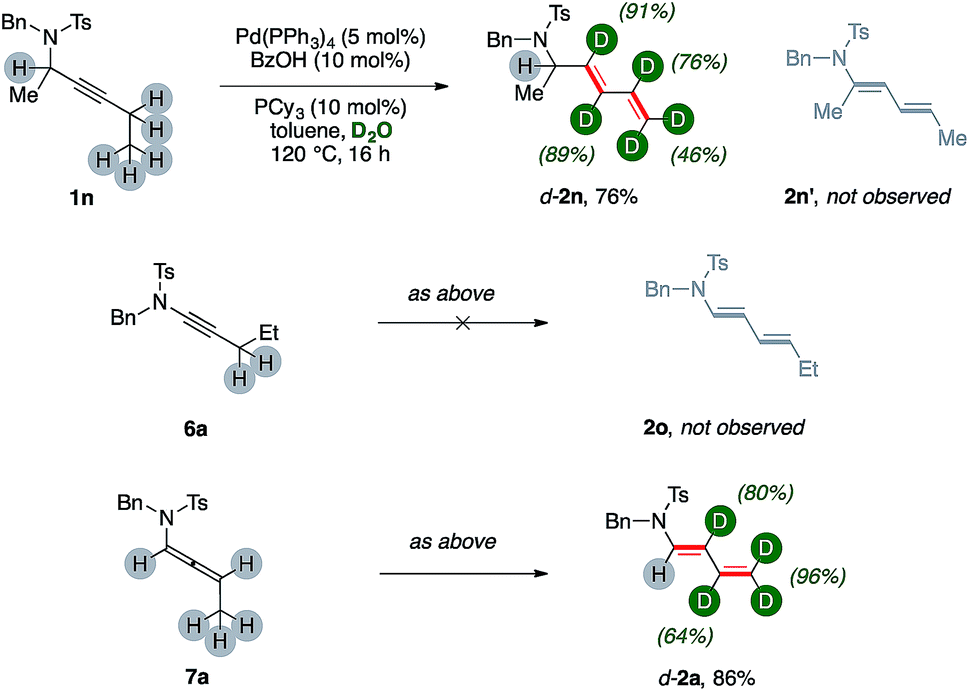 | ||
| Scheme 3 Directionality of the dual isomerization to d-2. | ||
The ynamide derivative 6a was not prone to any transformation, even in the absence of D2O. This result shows that a propargylamide fragment is crucial for the synthesis of (deuterated) 1,3-dienes from carbon–carbon triple bonds using present catalytic system. However, allenamide 7a can deliver the desired product d-2a, too, under optimized conditions (86%), showing extensive deuteration in its terminal arm (up to 96%) coupled with a negligible one on its position α- to nitrogen. This result strongly suggests that allenamides are likely intermediates in present sequences.
Access to polydeuterated polycycles
The synthetic utility of d-1,3-amido dienes was further reflected by the expedient access to highly decorated and deuterium-enriched polycyclic molecules (Scheme 4).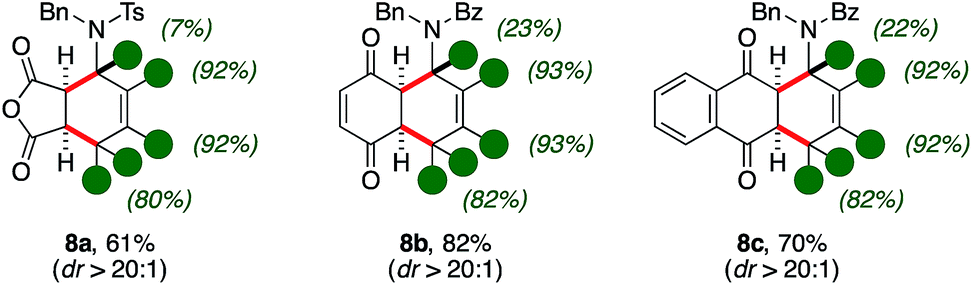 | ||
| Scheme 4 Access to polycycles d-8via Diels–Alder cycloaddition from d-2. | ||
Indeed, thermal Diels–Alder reactions were found to be accessible in the presence of activated dienophiles such as maleic anhydride.11 In this case, bicycle 8a was isolated with moderate yields, high diastereocontrol (61%, dr > 20:1) and up to excellent deuterium content on its aminocyclohexene ring. Importantly, quinones were found suitable patterns for the [4 + 2] cycloaddition as well. The reaction with benzoquinone and naphtoquinone led to the formation of d-tetrahydronaphtoquinone and d-tetrahydroantraquinone 8b–c in good yields and diastereoselectivity (82% and 70%, respectively). It is worth-noting that the scaffold of aminoanthraquinone derivatives is recurrent in a variety of bioactive natural products and its assembly often carry intrinsic synthetic challenges.12 Furthermore, similar protio analogues of d-8 were already probed to have bioactive properties with respect to different human tumor cell lines.13
Synthesis of polydeuterated carbolines
We then wondered whether it could have been possible to extend this multiple isomerization/deuteration method to C–C forming sequences, too (Fig. 2).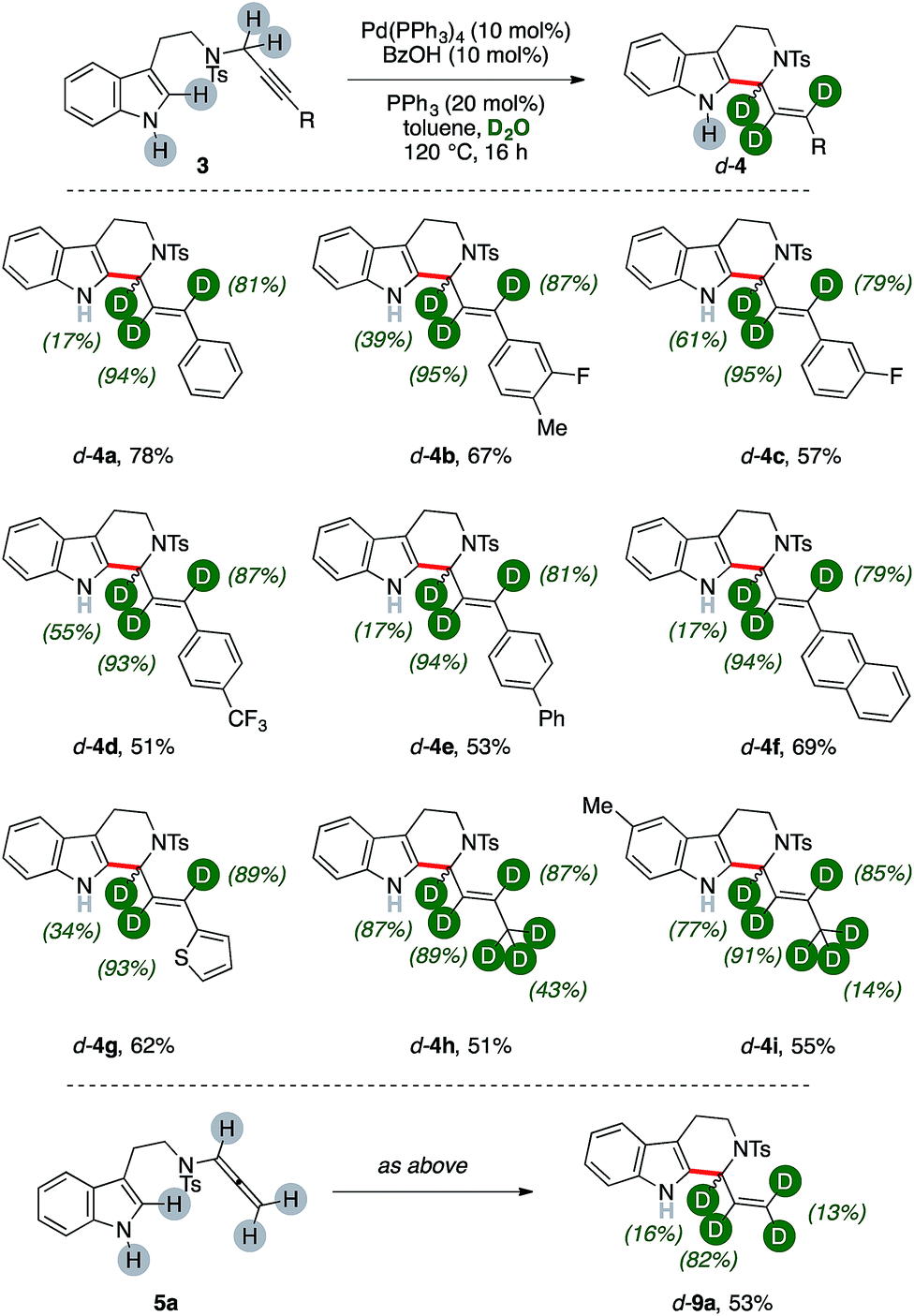 | ||
| Fig. 2 Catalytic synthesis of d-carbolines 4. | ||
The propargylic fragment of NH-free tryptamine propargylic derivative 3 could deliver 1-alkenyltetrahydrocarboline (THCs) 4 under Pd/benzoic acid catalysis.8b Intrigued by the potential of the combined isomerization/deuteration process, we submitted substrates to the optimized catalytic system in the presence of PPh3 as the ligand, which was already proved to be beneficial in the process.
To our delight, deuterated THC d-4a was isolated in good yields (78%, Fig. 2) and with high deuterium incorporation on its allylic arm (17%, 94% and 81% labelling ratios, respectively). It is noteworthy that the process is highly selective for the deuteration of the former propargylic fragment while aromatic C–H bonds and the unprotected N–H one remains virtually untouched during the sequence. We next probed the generality of the process with propargylic tryptamine substrates bearing different substitution patterns on the aryl ring. The reaction was well tolerated in the presence of fluoro- and trifluoromethyl moieties with THCs d-4b–d isolated in good yields (67%, 57% and 51%, respectively) and deuterations (39–95%). Extended aromatic systems, namely biphenyl and naphthalenes, were smoothly converted (53% and 69% for d-4e and d-4f, respectively) as well as heterocycles such as thiophene (d-4g, 62%). The protocol was found selective in the presence of extended aliphatic chains, such as a butyne fragment. The synthesis of THCs d-4h–j was accomplished in synthetically useful yields (51–55%). Furthermore, these examples showed the highest labeling on the carbon atom alpha to nitrogen (87% and 77%, respectively) and, in analogy with the pentyne fragment of diene d-2f (Fig. 1), partial deuteration was observed at their terminal methyl group (43% and 14%, respectively), too. The replacement of the propargylic arm of 3 with the corresponding allenamide (5a) did not hamper the reaction, leading to the recovery of the deuterated carboline d-9a in 53% yield but with a lower isotopic labeling, in particular for its terminal methylene (13%).
Mechanistic studies
In order to study if the high levels of deuteration observed could have been ascribed to an ordered series of chemical events, we performed the model experiment running five parallel reactions. Each of them has been warmed for the indicated period, rapidly cooled down to room temperature, diluted with dichloromethane, passed through a short pad of silica and eventually analysed by NMR with an internal standard (Fig. 3, details in ESI†).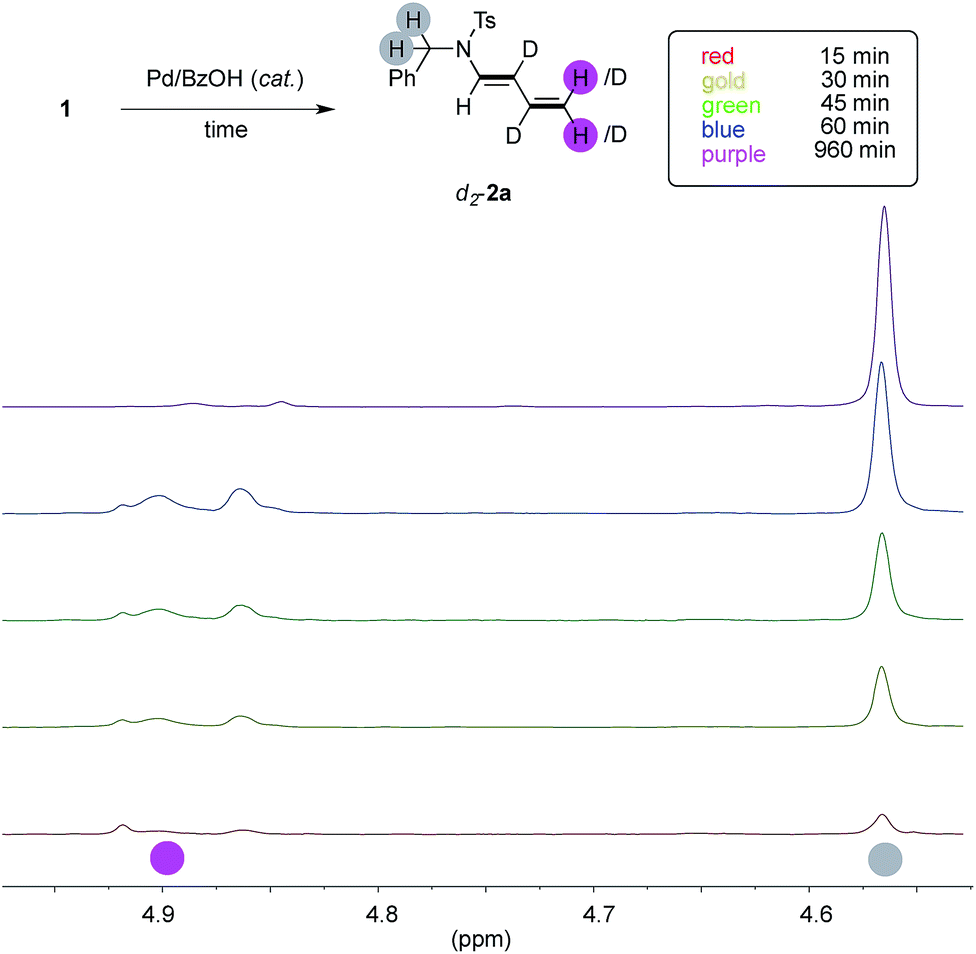 | ||
| Fig. 3 Key 1H-NMR resonances through time. | ||
We looked at experiments taken from fifteen minutes up to one hour (red, gold, green and blue spectra, respectively), and we observed that the conversion of 1a through time (up to 32%, blue spectrum) has a linear slope during this period and it has not yet reached its plateau. The 1H NMR signal of the benzylic protons of d-2a increases linearly, too, throughout this interval (highlighted in gray). However, the trend of deuteration showed that the β- and γ- carbons only were labeled in the initial stage of the reaction (see ESI, Table S1 and Fig. S1–S3† for details). On the contrary, the α- and the δ- ones remained almost untouched in this timeframe, as witnessed by the relative intensity of the C(sp2)–H signals of d-2a (highlighted in pink). This surprised us because extensive deuteration of the terminal methylene group of products 2 was usually observed (55–96%, Fig. 1). Furthermore, the proton resonances due to the unlabeled terminal methylene of d-2a are no longer observed when experiments are performed overnight (purple line).
These results let us think that unlabeled products, too, should have been able to undergo at least partial deuteration. We thus submitted various unlabeled products 2 and 4 to the optimized reaction conditions (Scheme 5).
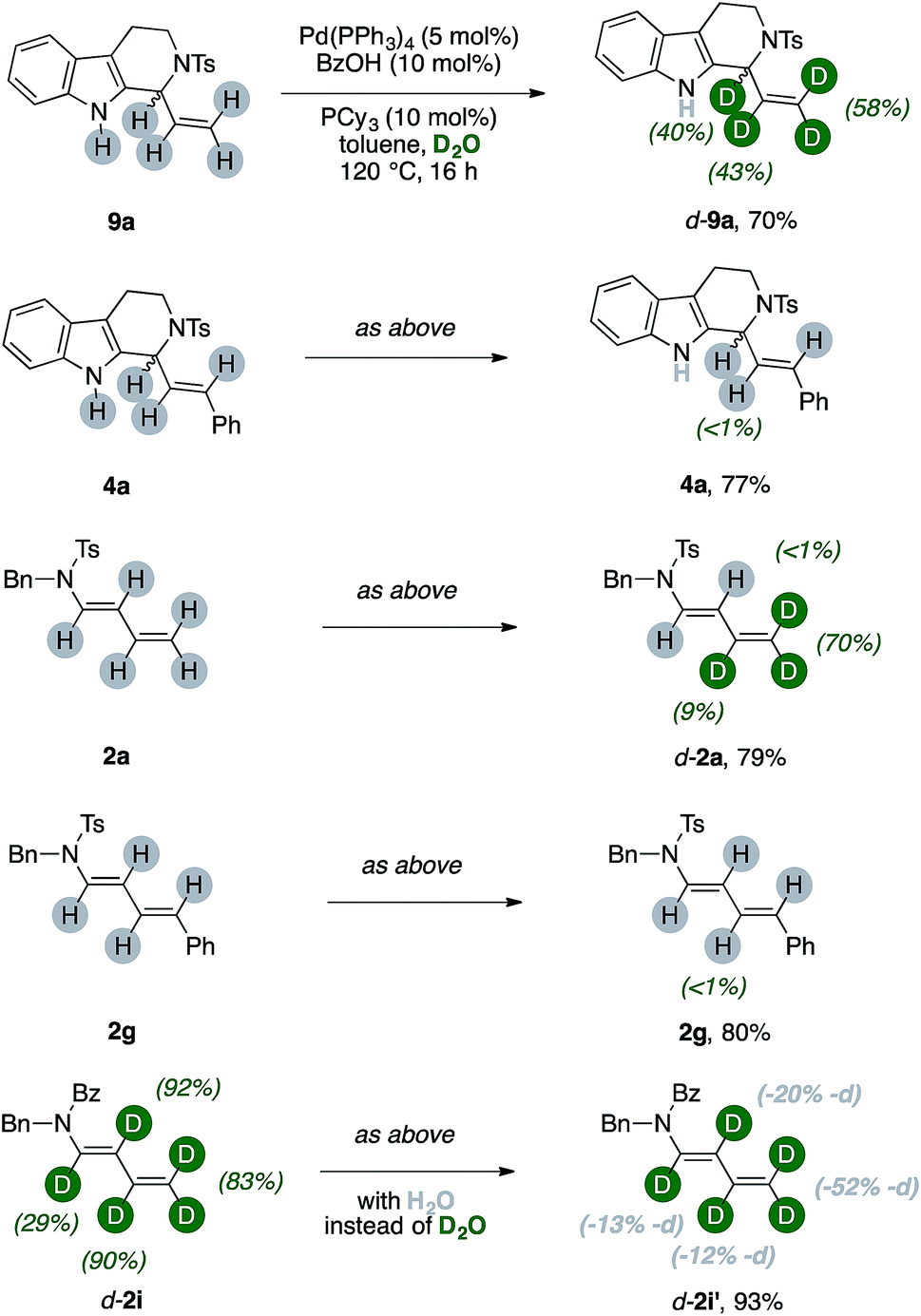 | ||
| Scheme 5 Reactivity of unlabeled products under reaction conditions. | ||
The protio-9a, which has a terminal vinyl group, can be selectively labeled upon submission to the optimized reaction conditions. We observed partial incorporation on its entire allylic fragment (40%, 43% and 58%, respectively). On the contrary, the THC 4a, which has a disubstituted alkene motif, did not undergo any H/D scrambling.14 We then tested diene [H]-2a. Interestingly, deuteration of the substrate occurred and this reaction was selective for the terminal methylene unit of the conjugated system (up to 70%, Scheme 5). Replacement of the terminal vinyl group of 2a with a styryl arm, such as in 2g, shut down completely any H/D scrambling under otherwise identical conditions. The d-2i can undergo an erosion of its deuterium content in the presence of the catalytic system and 50 equiv. of H2O. This effect is pronounced on its terminal methylene and becomes more nuanced along the diene chain (−13, −20, −12 and −52% deuterium labeling for the carbons α-, β-, γ- and δ- to nitrogen, respectively).15
The results of Scheme 5 suggested us that a terminal vinyl group, too, could have been activated and successfully undergo H/D scrambling up to meaningful levels with present catalytic system. We therefore tested this hypothesis on a simple alkene, such as 4-methylstyrene 10a (Scheme 6a). Indeed, extensive deuteration of the terminal methylene unit was observed, thus probing the generality of palladium(0)/carboxylic acid catalysis as valuable labeling method for generic terminal olefins. Preliminary results on styrenes showed that extensive trideuteration of their vinyl arms can be readily achieved by performing the reaction in toluene/D2O mixture and replacing BzOH with (+)mandelic acid (30 mol%). This enabled also simpler purification of these broadly used, volatile alkenes, and can for instance lead to the isolation of d-10a in good yields, high labelling content and purity (Scheme 6b). The method can be applied to 4-chlorostyrene (d-10b, 65%), delivering excellent deuteration of its terminal methylene while the α- position underwent H/D exchange to a lower extent (26%). Higher isolated yields were retrieved with less volatile styrenes (d-10c–d, 80% and 82%, respectively). These products showed extensive labelling of their vinyl C–H bonds (23–97%) and revealed that the method tolerates various heterocycles such as carbamates and triazoles.16
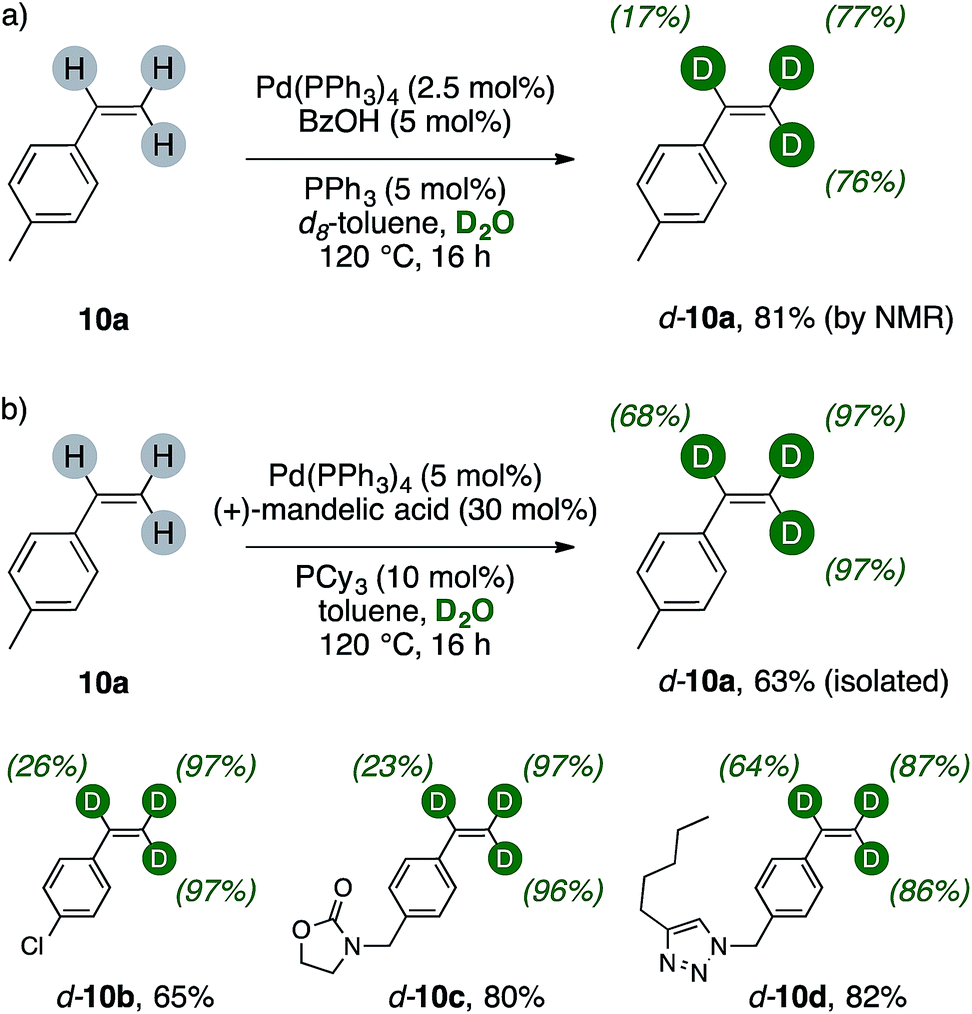 | ||
| Scheme 6 Deuteration of substituted styrenes. | ||
Taken together, these findings coupled with extensive DFT studies8 strongly suggest that products form through an ordered sequence of events, such as those presented in Scheme 7.
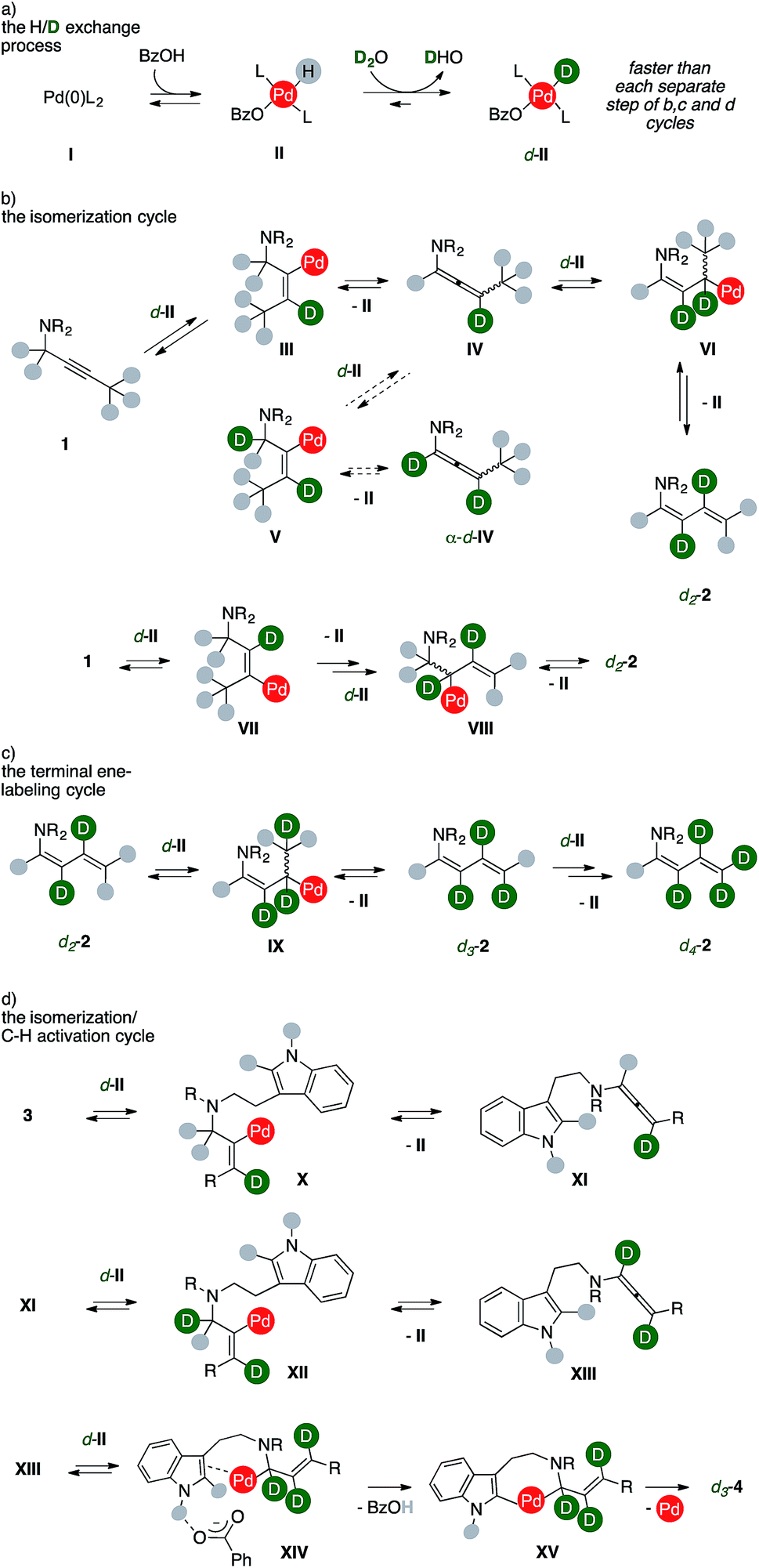 | ||
| Scheme 7 Proposed mechanistic rationale. | ||
A Pd(0) species, such as I, can reversibly react with benzoic acid to deliver the corresponding Pd(II)-hydride complex II.9 While water is not sufficiently acidic to directly react with I (Table 1, entry 13), isotopic scrambling with D2O can take place on the metal hydrides complex II, giving d-II (Scheme 7, cycle a). This process has to remain relatively faster than any other intermediate steps of the sequences in order to ensure a high deuterium incorporation. Upon substrate coordination, alkyne insertion into the Pd–D bond afford the vinylpalladium complex III (Scheme 7, cycle b). Despite a limited rotational flexibility, the intermediate can undergo β-hydrogen elimination to regenerate the Pd-hydride II, which has to reenter the H/D exchange process (cycle a), together with the formation of an allenamide (IV). This intermediate could evolve through two different routes depending on the regioselectivity of the allene insertion into the Pd–D bond of another d-II species.
According to the usually lower deuteration of carbons alpha to nitrogen in products, the least favorable pathway for the insertion of d-II into IV leads to complex V, which then eliminates II liberating allenamide α-d-IV. The alternative insertion delivers allylic complex VI, which could liberate the d2-2 upon β-elimination from the terminal methyl group. This correlates with the trend of deuterium incorporation observed through time (Fig. 3). The dideuterated diene d2-2 can alternatively result from an equivalent isomerization process. The vinyl complex VII can be formed from the first alkyne insertion into a Pd–D bond. This manifold can then proceed analogously to the previous one, and the product can be formed via β-hydrogen elimination from complex VIII. The d2-2 accumulates as long as the concentration of alkyne 1 remains high. Once it fades, the diene can more easily compete with phosphines for metal coordination on the key intermediate d-II, thus re-entering the scene (Scheme 7, cycle c). A further sequential insertion/β-hydrogen elimination via complex IX regenerates then II from d-II together with a molecule of d3-2. Steered by mass effects, one more series of isotopic scrambling/insertion/β-hydrogen elimination can efficiently take place to deliver a fully deuterated terminal methylene group, such as in d4-2.
Analogously, the sequence delivering labeled carbolines d-4 presents at least three productive series of H/D exchange (Scheme 7, cycle d). Insertion of substrate 3 into the Pd–D bond of d-II can yield complex X, which can undergo β-hydrogen elimination to provide II and allenamide XI. A second insertion of the latter on d-II could then provide vinylpalladium complex XII. Sequential β-hydrogen elimination leads to a dideuterated allene (XIII). The lack of deuteration observed using 4a (Scheme 5) shows that these two sub-cycles took place prior to the eventual annulation. Upon a third H/D exchange process, this intermediate can insert into the Pd–D bond of d-II and then undergo the crucial C–H activation step, which is assisted by the benzoate anion in XIV. This step occurs likely through an outer-sphere CMD mechanism that is favored by the hydrogen bonding tethering the carboxylate anion and the unprotected indole N–H group.8b A C–C forming reductive elimination from metallacycle XV eventually liberates the trideuterated carboline d3-4 together with palladium(0). Remarkably, the extensive deuteration observed in the terminal methyl group of d-4h and d-4j shows that this ordered series of events can be extended to the entire chain that decorates the tryptamine reagent.
Conclusions
We reported that the synthesis of valuable organics can be coupled with extensive and chemoselective deuteration using benign D2O. These reactions use readily available reagents, offering a convenient tool to access families of labelled compounds thanks to the combination of a Pd(0) precursor with benzoic acid. An ordered series of up to sixteen productive elementary steps is responsible for the formation of the d-dienes in the present dual isomerization/polydeuteration sequences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors dedicate this manuscript to Prof. Marta Catellani on the occasion of her 75th anniversary. We warmly thank support from UniPr (Grant RADI-CAT) and MIUR (Grant AROMA-TriP and Departments of Excellence framework through the COMP-Hub Lab). Use of modeling facilities was provided by ICSN-CNRS (UPR2301, Gif s/Yvette, France).Notes and references
- E. M. Isin, C. S. Elmore, G. N. Nilsson, R. A. Thompson and L. Weidolf, Chem. Res. Toxicol., 2012, 25, 532–542 Search PubMed.
- T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS PubMed.
- Selected recent examples of catalytic deuteration reactions: (a) T. R. Puleo, A. J. Strong and J. S. Bandar, J. Am. Chem. Soc., 2018, 141, 1467–1472 CrossRef PubMed; (b) V. Soulard, G. Villa, D. P. Vollmar and P. Renaud, J. Am. Chem. Soc., 2018, 140, 155–158 CrossRef CAS PubMed; (c) D. M. Kaphan, R. C. Klet, F. A. Perras, M. Pruski, C. Yang, A. J. Kropf and M. Delferro, ACS Catal., 2018, 8, 5363–5373 CrossRef CAS; (d) M. Han, Y. Ding, H. Li, S. Luo, A. Adijiang, Y. Ling and J. An, Org. Lett., 2018, 20, 3010–3013 CrossRef CAS PubMed; (e) Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. R. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182–1187 CrossRef CAS; for reductivedeuteration of alkynes, see: (f) M. Han, Y. Ding, Y. Yan, H. Li, S. Luo, A. Adijiang, Y. Ling and J. An, Org. Lett., 2018, 20, 3010–3013 CrossRef CAS PubMed; (g) K. T. Neumann, S. Klimczyk, M. N. Burhardt, B. Bang-Andersen, T. Skrydstrup and A. T. Lindhardt, ACS Catal., 2016, 6, 4710–4714 CrossRef CAS; (h) Y. Yabe, Y. Sawama, Y. Monguchi and H. Sajiki, Chem.–Eur. J., 2013, 19, 484–488 CrossRef CAS; (i) E. Shirakawa, H. Otsukaa and T. Hayashi, Chem. Commun., 2005, 41, 5885–5886 RSC.
- A review, see: (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed. Selected recent examples: (b) W. J. Kerr, D. M. Lindsay, P. K. Owens, M. Reid, T. Tuttle and S. Campos, ACS Catal., 2017, 7, 7182–7186 CrossRef CAS; (c) R. P. Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195–199 CrossRef; (d) S. Ma, G. Villa, P. S. Thuy-Boun, A. Homs and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 734–737 CrossRef CAS; (e) M. H. Emmert, J. B. Gary, J. M. Villalobos and M. S. Sanford, Angew. Chem., Int. Ed., 2010, 49, 5884–5886 CrossRef CAS PubMed.
- (a) A. Bechtoldt and L. Ackermann, ChemCatChem, 2019, 11, 435–438 CrossRef CAS; (b) M. Hatano, T. Nishimura and H. Yorimitsu, Org. Lett., 2016, 18, 3674–3677 CrossRef CAS PubMed; (c) A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente, F. J. Lahoz and L. A. Oro, Chem.–Eur. J., 2014, 20, 8391–8403 CrossRef CAS PubMed; (d) A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente, F. J. Lahoz, V. Polo and L. A. Oro, Angew. Chem., Int. Ed., 2011, 50, 3938–3942 CrossRef CAS PubMed; (e) J. Zhou and J. F. Hartwig, Angew. Chem., Int. Ed., 2008, 47, 5783–5787 CrossRef CAS PubMed; (f) C. M. Yung, M. B. Skaddan and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 13033–13043 CrossRef CAS PubMed; (g) S. R. Klei, J. T. Golden, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 2092–2093 CrossRef CAS.
- G. Erdogan and D. B. Grotjahn, J. Am. Chem. Soc., 2009, 131, 10354–10355 CrossRef CAS PubMed.
- A. V. Smarun, M. Petković, M. S. Shchepinov and D. Vidović, J. Org. Chem., 2017, 82, 13115–13120 CrossRef CAS PubMed.
- (a) G. Cera, M. Lanzi, F. Bigi, R. Maggi, M. Malacria and G. Maestri, Chem. Commun., 2018, 54, 14021–14024 RSC; (b) G. Cera, M. Lanzi, D. Balestri, N. Della Cà, R. Maggi, F. Bigi, M. Malacria and G. Maestri, Org. Lett., 2018, 20, 3220–3224 CrossRef CAS PubMed; for the combination of carboxylic acids with multinuclear palladium complexes, see: (c) M. Lanzi, T. Caneque, L. Marchiò, R. Maggi, F. Bigi, M. Malacria and G. Maestri, ACS Catal., 2018, 8, 144–147 CrossRef CAS; (d) C. Cecchini, M. Lanzi, G. Cera, M. Malacria and G. Maestri, Synthesis, 2019, 51, 1216–1224 CrossRef CAS.
- (a) P. Zheng, C. Wang, Y.-C. Chen and G. Dong, ACS Catal., 2019, 9, 5515–5521 CrossRef CAS; (b) A. M. Haydl, B. Breit, T. Lang and M. J. Krische, Angew. Chem., Int. Ed., 2017, 56, 11312–11325 CrossRef CAS PubMed; (c) D. A. Petrone, I. Franzoni, J. Ye, J. F. Rodriguez, A. Poblador-Bahamonde and M. Lautens, J. Am. Chem. Soc., 2017, 139, 3546–3557 CrossRef CAS PubMed; (d) M. D. Peacock, C. B. Roos and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 647–652 CrossRef; (e) D. J. Vyas, E. Larionov, C. Besnard, L. Guenee and C. Mazet, J. Am. Chem. Soc., 2013, 135, 6177–6183 CrossRef CAS PubMed.
- (a) A. Monfredini, V. Santacroce, L. Marchiò, R. Maggi, F. Bigi, G. Maestri and M. Malacria, ACS Sustainable Chem. Eng., 2017, 5, 8205–8212 CrossRef CAS; (b) A. Monfredini, V. Santacroce, P.-A. Deyris, R. Maggi, F. Bigi, G. Maestri and M. Malacria, Dalton Trans., 2016, 45, 15786–15790 RSC; (c) A. Nakamura, T. Kageyama, H. Goto, B. P. Carrow, S. Ito and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 12366–12369 CrossRef CAS PubMed; (d) T. M. J. Anselment, C. A. Anderson, B. Rieger, M. B. Boeddinghaus and T. F. Fässler, Dalton Trans., 2011, 40, 8304–8313 RSC; (e) A review, see: K. H. Shaughnessy, Chem. Rev., 2009, 109, 643–710 CrossRef CAS PubMed.
- In a typical experiment, a toluene solution of the desired dienophile (0.3 mmol, 1 M) and the deuterated diene (1.1 equiv.) was heated for 24 hours, eventually affording target deuterated polycycles upon chromatography; see the ESI† for Experimental details.
- (a) T. Caneque, F. Gomes, T. T. Trang, G. Maestri, M. Malacria and R. Rodriguez, Nat. Chem., 2015, 7, 744–751 CrossRef CAS PubMed; (b) N. Maugel and B. B. Snider, Org. Lett., 2009, 11, 4926–4929 CrossRef CAS.
- M. Chigr, H. Fillion, A. Rougny, M. Berlion, J. Riondel and H. Beriel, Chem. Pharm. Bull., 1990, 38, 688–691 CrossRef CAS.
- As suggested by a referee, the lack of reactivity showed by 4a and 4g could stem from the beta-styryl unit that is much less susceptible to migratory insertion into a Pd–H or Pd–D bond.
- We warmly thank a reviewer for this worthy suggestion.
- Extension of this reactivity to terminal olefins is currently underway.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization of products, copies of NMR spectra. See DOI: 10.1039/c9sc03682b |
| This journal is © The Royal Society of Chemistry 2019 |