
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a DUB-selective fluorogenic substrate†
Lorina
Gjonaj
a,
Aysegul
Sapmaz
a,
Dennis
Flierman‡
a,
George M. C.
Janssen
b,
Peter A.
van Veelen
b and
Huib
Ovaa
 *a
*a
aOncode Institute, Department of Cell and Chemical Biology, Leiden University Medical Center, Einthovenweg 20, 2333 ZC, Leiden, The Netherlands. E-mail: H.Ovaa@lumc.nl
bCentre for Proteomics and Metabolomics, Leiden University Medical Center, Albinusdreef 2, 2333 ZA, Leiden, The Netherlands
First published on 30th September 2019
Abstract
Ubiquitination is a post-translational modification that is involved in a plethora of cellular processes. Target proteins can be specifically modified with a single ubiquitin (Ub) molecule or with complex chains. In recent years, research has focused on deubiquitinating enzymes (DUBs) as potential therapeutic candidates in various diseases. USP16 is an emerging target due to its involvement in mitosis and stem cell self-renewal. Generally, activity-based probes (ABPs) used to study DUBs are based on the ubiquitin scaffold, thus lacking target selectivity. To overcome this issue, we designed a Ub-based activity probe bearing specific mutations to achieve selectivity for USP16, by combining structural modelling and analysis and mutational calculation predictions. We develop a fluorogenic substrate, the first of its kind, that is processed exclusively by USP16, which allows us to monitor USP16 activity in complex samples.
Introduction
Ubiquitin (Ub) is a small protein that plays an essential role in cellular processes and is highly conserved in eukaryotic cells.1 This protein is covalently attached via an isopeptide bond to an amino group (lysine residue or at the N-terminal site) of a target protein through an enzymatic cascade involving three enzymes. Different combinations of these enzymes can result in various mono- or polymeric ubiquitin adducts.2 Deubiquitinases (DUBs) catalyse the release of ubiquitin from the substrate and degradation of polyubiquitin chains.3 This class of enzymes regulates many biological processes that are under Ub control.4 DUBs are considered important drug targets due to their involvement in various diseases.5 Therefore, more insight is needed into their biological function. Different probes are currently available to study DUB-activity, but they are generally based on the ubiquitin wild-type sequence, making it difficult to achieve selectivity. The most common tools used by the scientific community are activity-based probes (ABP) and fluorogenic substrates.6–8 An ABP reacts in a covalent fashion with DUBs and is generally used to detect or isolate active enzymes. Fluorogenic substrates for DUBs are usually ubiquitin-based reagents, modified with reporter groups at the C-terminus of ubiquitin. One of the most commonly used substrates is Ub-AMC (7-amido-4-methylcoumarin). Once the substrate is bound to a DUB, the amide bond between the last glycine residue of Ub and the fluorogenic leaving group will be cleaved. The fluorescence signal obtained upon hydrolysis of the substrate is used to monitor enzyme activity.Engineered Ub variants (UbV) have been shown to strongly bind and target various members of the DUB and E3 ligase families.9 USP2, USP8, USP21, USP7, USP10 and recently USP15 are a few of the DUBs targeted using ubiquitin mutants.10–13 We have previously established a platform for the design of active-site directed probes with high affinity for the target USP7.14 We envisioned that our work could be applied to other DUB targets of interest as well as expanding it to designing fluorogenic substrates, even though the mode of action is different compared to APB reagents. Ubiquitin has a robust and flexible structure. For this reason, it can still maintain its function upon introduction of various mutations. Therefore, Ub mutants can be used to address the above-mentioned selectivity issue.
Ubiquitin-specific proteases (USPs) are the largest class within the DUB family, with 56 proteins out of approximately 100 known human DUBs. USP16, a member of the USP family, could be considered a multifaceted therapeutic target and the elucidation of the full extent of its biological role is therefore of great importance. This DUB deubiquitinates histone H2A and is essential in cell cycle progression.15 In addition, USP16 was shown to be a key regulator of hematopoietic stem cell differentiation.16 It has previously been reported that downregulation of USP16 has contributed to hepatocellular carcinoma pathogenesis.17 Furthermore, during DNA damage, there is an increase of ubiquitinated DNA-damage foci, that is negatively regulated by USP16.18 USP16 is one of the genes located on chromosome 21, of which partial or full trisomy results in Down syndrome (DS). USP16 is known to decrease the self-renewal ability of somatic stem cells that contributes to DS pathogenesis. It has been shown that by negatively regulating this enzyme it is possible to rescue proliferation anomalies in human DS fibroblasts and alleviate some of the pathologies associated with Down's syndrome.19 For these reasons USP16 is a target of interest for the development of inhibitors and as such it is important to have good tools to better understand its function. In order to shed light on the activity of USP16 and elucidate possible upregulation/downregulation mechanisms essential for its function, we aimed to generate a USP16-selective activity-based probe (ABP) and a matching selective fluorogenic substrate.
We have previously reported an ABP highly selective for USP7, modified at the N-terminus with a fluorescent tag, rhodamine (Rh) and at the C-terminus with a reactive alkyne functionality (PA).14 Mass spectrometry experiments showed that this probe reacts to a minor extent with two other USPs, USP15 and USP16. We hypothesized that this covalent reagent can be fine-tuned to only react with USP16 by modulating the primary structure of ubiquitin. In order to disturb specific interactions between the Ub structure and USP7 and USP15, we incorporated mutations in the sequence of this mutant, (M6).
To discover the mutations that are necessary to obtain the desired selectivity, we took advantage of data from structural analysis of these DUBs in complex with ubiquitin and FoldX-based mutational predictions20 (Scheme 1). This algorithm can calculate the theoretical relative binding energies of a DUB with ubiquitin, in which each position is in silico mutated towards the 20 genetically encoded amino acids. Various point mutation combinations were applied during the design and screening process to find selective ABPs for USP16. From almost 40 new synthetic Ub mutants that we synthesized, the best mutant proved to be mutant M20 (Rh-M20-PA), showing high reactivity and selectivity for the target USP16. In addition, we synthesized an AMC fluorogenic substrate based on the sequence of this ubiquitin mutant. Here, we report a USP16-selective ABP and a matching fluorogenic substrate which is selectively recognized and processed only by USP16.
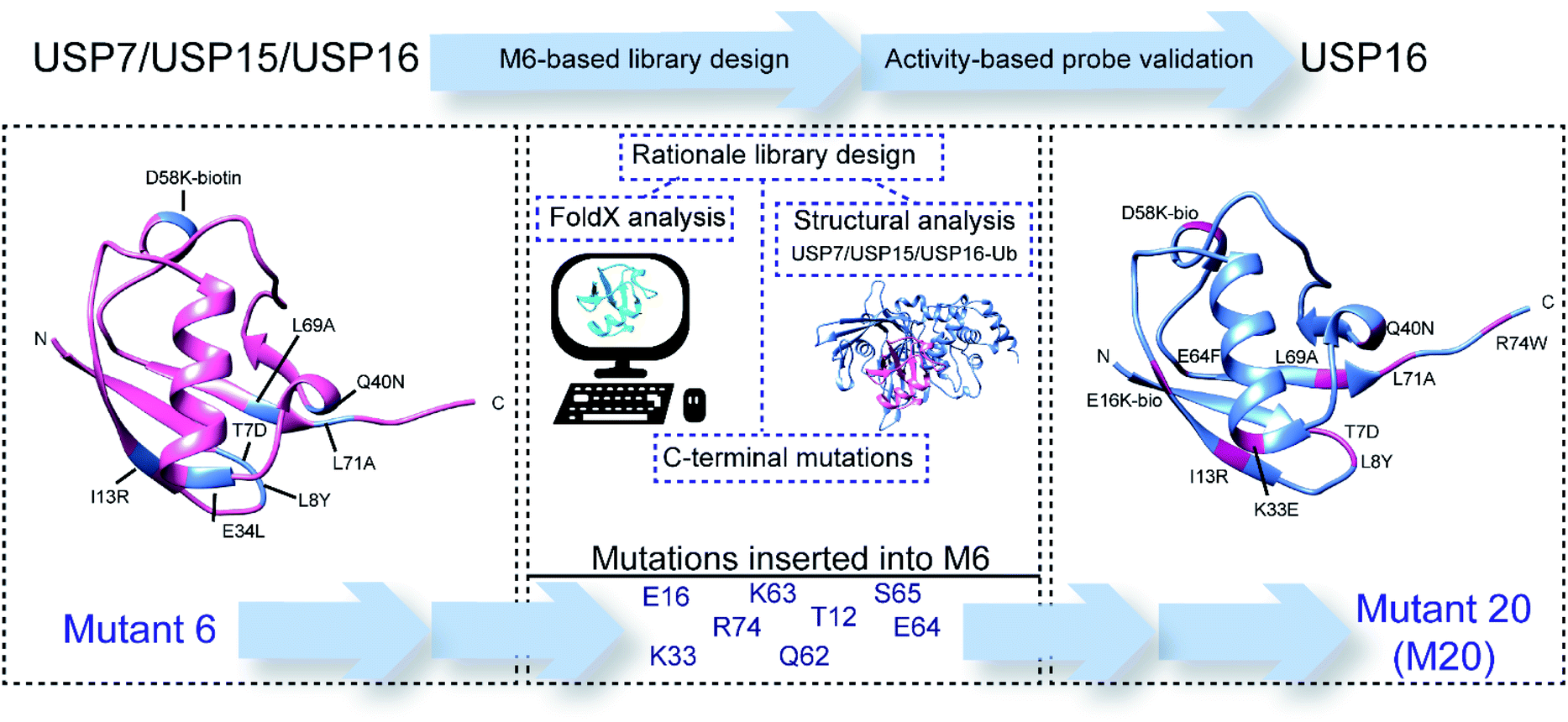 | ||
| Scheme 1 Schematic representation of the rationale and design process towards USP16-selective ubiquitin mutant 20 (M20). | ||
Results and discussion
Rationale behind library design
To study and evaluate the interactions between USP16/USP7/USP15 and ubiquitin, crystal structures of these DUBs are desirable. The structures of USP16 and USP15, however, were not available at the time. We therefore used in silico modelling to generate models of these USPs. Swiss-Model is one of several tools used to build three-dimensional protein structures based on structural homology to closely related proteins.21 After structurally evaluating the models obtained with Swiss-Model, we selected the model generated using the template of USP4 (PDB ID: 2Y6E) as a structural model for USP15. USP4 is the closest related member from the USP family to USP15 (57% sequence identity). For USP16 we chose the model based on the USP2–Ub complex (PDB ID: 2HD5), which has a sequence identity of 34%. Subsequently, we aligned the structures of USP7, USP15, and USP16 using UCSF Chimera (Fig. 1).22 We generated a model of USP15 (Fig. S1†) and USP16 (Fig. S2†) in complex with ubiquitin, based on the position of ubiquitin from a known USP7–Ub complex (PDB ID: 1NBF). Recently, the crystal structure of the catalytic domain of USP15 has been reported,23 and it superimposes very well with the model generated by us with Swiss-Model (Fig. S3†), validating our efforts in modelling and our assumptions made based on these structures.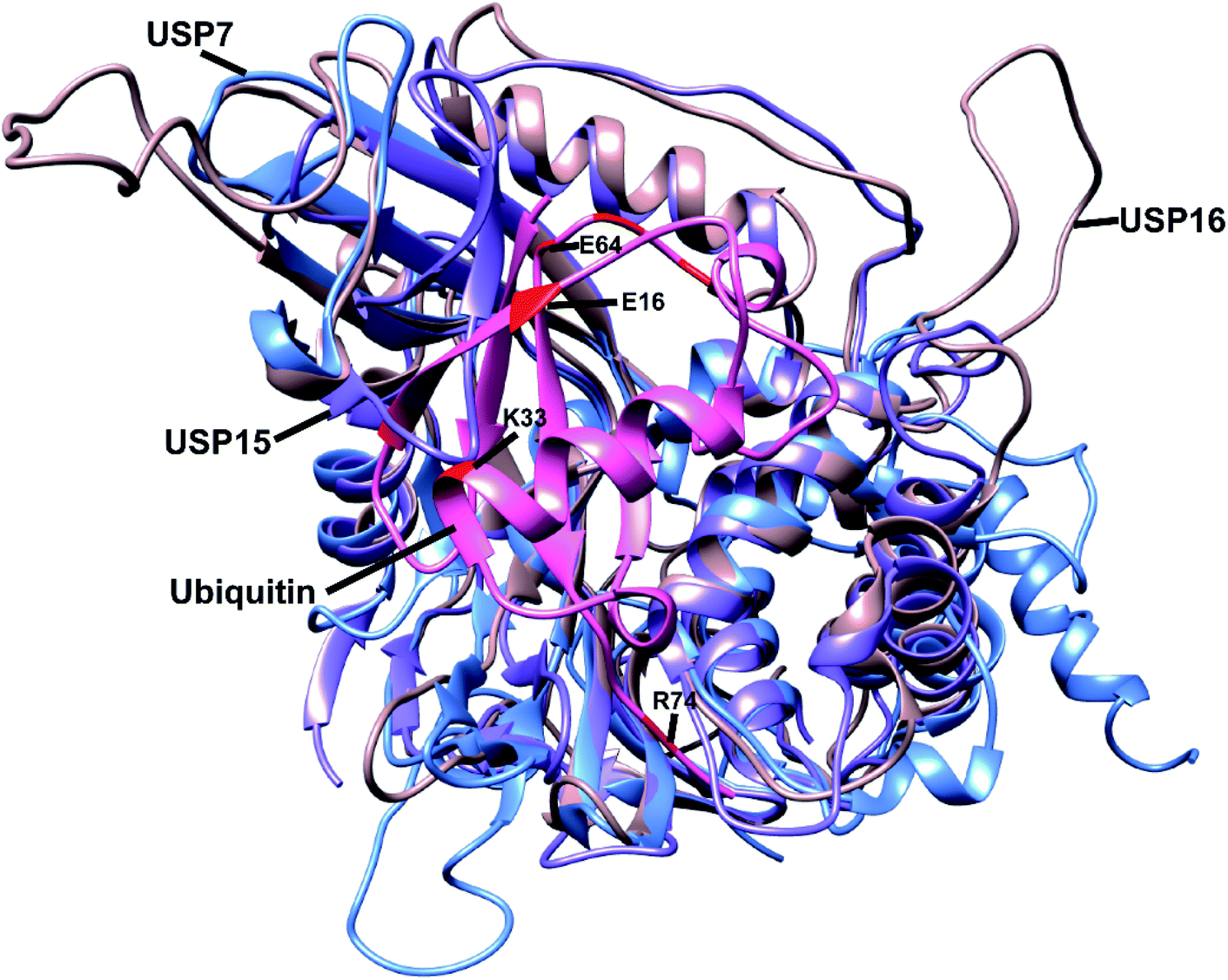 | ||
| Fig. 1 Alignment of the structure of USP7 (blue), USP15 (purple) and USP16 (light brown). Ubiquitin (pink) position is based on the USP7–Ub complex (PDB ID: 1NBF). Some of the selected residues considered in the library design are shown in red. The most important residues are labelled. | ||
The modelled USP-Ub structures were then used to identify the residues that are involved in the interaction between these three DUBs and Ub (Fig. S4†). In the case of USP15, we could deduce that upon binding to Ub, structural changes were necessary to accommodate ubiquitin close to the catalytic site (Fig. S4b†). To our knowledge, so far USP7 and USP15 are the only DUBs with a misaligned catalytic triad, in a non-active conformation. We hypothesized that we could insert mutations in specific regions of the Ub structure that would either lead to steric hindrance, resulting in disturbance of the interactions between Ub and these two DUBs, or prevent the necessary conformational changes within the USP15 structure. Structural analysis led to the identification of possible favourable positions (e.g. glutamate 16 and lysine 33) in the ubiquitin sequence that might increase the affinity of the reagents towards USP16 (Fig. 1).
We recently successfully used the FoldX software package to perform structure-based computational predictions to identify possible mutations, which would alter the binding affinity between Ub and USP7.14 FoldX-based calculations were performed also on the structures modelled here, whereby we selected possible mutations which would enhance the affinity for USP16 and negatively affect the affinity for either USP7 or USP15 or both (ESI data 1†).
By combining the information acquired from the approaches described above, we selected residues of Ub in positions 12, 15, 16, 33 and 62–65 (Fig. 1). Our rationale for selecting these residues and their mutations is as follows: E16 in the Ub structure does not interact with any amino acids present in the structure of USP16 and by mutating it into a bulky group (e.g. lysine–biotin) we might be able to introduce steric hindrance or interrupt possible conformational changes, which are crucial for USP15 (Fig. 1 and S4†). K33 is part of the interaction site between ubiquitin and both USP15 and USP7. The mutation K33E, as shown in Fig. S5a,† would lead to the loss of the hydrogen bonds formed between this lysine and aspartate (D376) in case of USP7 and possibly an electrostatic repulsion. A similar effect can be hypothesized for USP15, once conformational changes take place upon ubiquitin binding (E804 in USP15, Fig. S5b†). The mutation of E64 for a phenylalanine or histidine residue was based on FoldX calculations. And finally, we also considered randomly mutating the C-terminus of ubiquitin which is generally known to be an important part in the recognition and activity of DUBs. All these data together were applied in the library design.
Developing a USP16-selective ABP reagent
Our main objective was to generate a selective fluorogenic substrate for a single DUB. As a proof of principle, we first synthesized an ABP mutant library and tested this library using a previously developed method.14 Such an ABP reagent, having a rhodamine moiety attached at the N-terminus, enables a quick assay read-out, while the propargyl moiety reacts covalently and traps the targeted DUBs.24 With this convenient screening process, we could determine the preference in labelling specific DUBs for each synthesized mutant probe. This information can subsequently be used in the selection step and applied in case new mutants are needed to be designed with increased affinity for the desired target. Moreover, using this type of probe we could capture the targeted DUBs and identify them in a straightforward manner, using mass spectrometry.Mainly based on the sequence of the best mutant from our previous study, mutations were incorporated in multiple sites of the ubiquitin sequence and we synthesized 40 new synthetic N-terminally rhodamine tagged ABPs. The synthetic ubiquitin mutants were obtained via Fmoc-based solid-phase peptide synthesis as previously described.24
To assess the reactivity of each mutant probe, HAP1 cell lysate was labelled with the different mutants and analysed using an in-gel fluorescence-based assay (Fig. S6†). Our main interest was to investigate the difference in the labelling of USP16 obtained with the library. However, due to the low endogenous levels of USP16 and USP15, difficulties in distinguishing USP16 from USP5, which is very abundant and with a similar molecular weight to USP16, and the lack of a specific antibody against USP16 (commercial antibodies used by us proved not to be specific), different types of experiments were necessary. We decided to overexpress Flag-tagged USP16 in HeLa cells followed by western blotting as a read-out of the reactivity of the most interesting mutants (Fig. S7†). From 40 ubiquitin reagents, we identified mutant M20 which seemed to label USP16 in a selective manner (Fig. S7†).
Next, experiments were performed only with this mutant M20 (Rh-M20-PA) and wild-type (WT) Ub probe (Rh-Ub-PA) as the control, in order to assess the selectivity of this reagent. Labelling experiments were performed with HeLa cells lysate overexpressing Flag-USP5, Flag-USP7, Flag-USP15 or Flag-USP16 and their catalytically inactive versions (Fig. 2). In Fig. 2, it is shown that only Flag-USP16 reacts with M20, while all the other DUBs are labelled only by the wild-type probe. As expected, both reagents showed no labelling with HeLa lysate overexpressing the catalytic inactive version of the above-mentioned DUBs. We observed that only one endogenous DUB (approx. 100 kDa) was labelled in the HeLa lysates. As anticipated, by varying the sequence of M6, the reagent showed a decreased affinity for USP7 and USP15, while still maintaining its reactivity towards USP16.
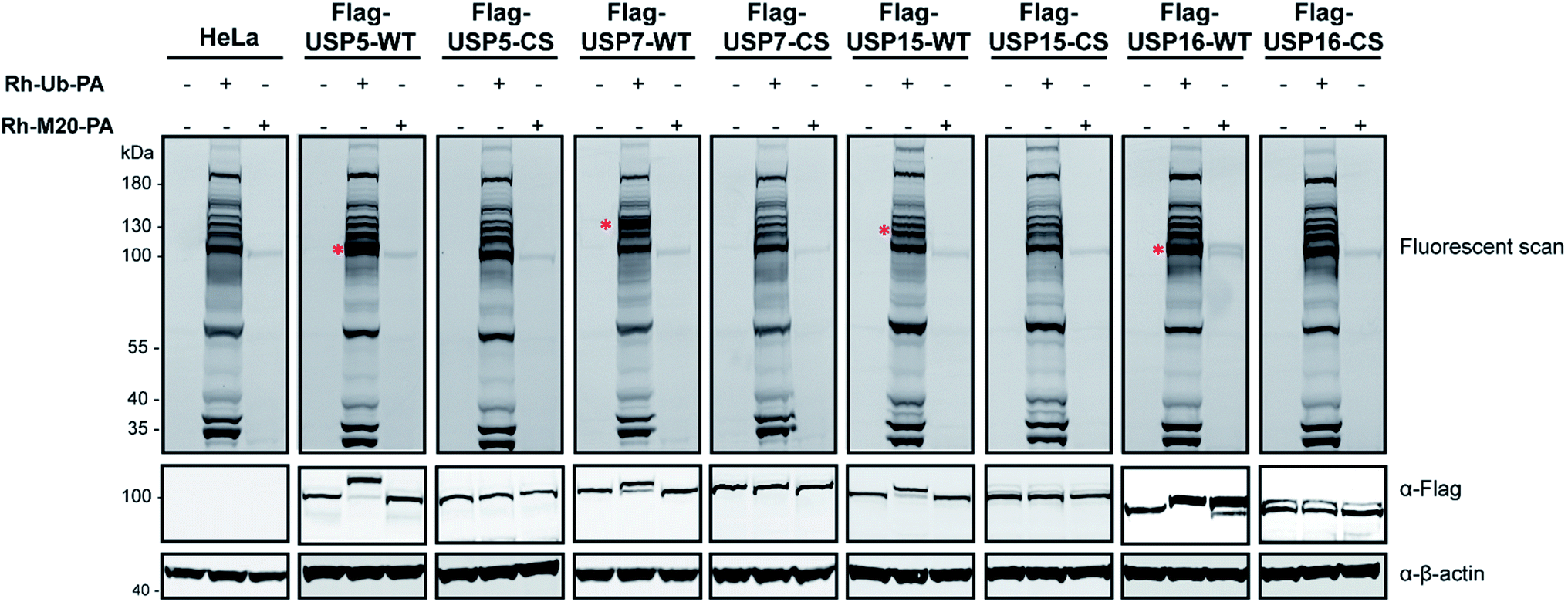 | ||
| Fig. 2 Labelling experiment with the Rh-Ub-PA and Rh-M20-PA. Wild-type and catalytically inactive versions of Flag-tagged USP5, USP7, USP15 and USP16 were overexpressed in HeLa cells. Flag antibody is used to follow the levels of labelling. β-actin is used as a loading control. | ||
To address whether the band we observed in the labelling assay was indeed USP16 (HAP1 and HeLa lysate with M20, respectively Fig. S6† and 2), and to further confirm the selectivity and reactivity of the mutant probe M20, we carried out labelling experiments with HAP1 WT and HAP1 USP16KO lysate (knockout USP16 from HAP1 cells). Due to the lack of a functional commercial antibody for USP16, we initially performed a pull-down experiment using Biotin-Ub-PA, in order to verify the levels of USP16 in both HAP1 cell lines. This reagent covalently reacted with the pool of DUBs present in the lysate obtained from the two different cell lines. N-terminal biotin-PEG was used as a handle for pull-down experiments, followed by mass spectrometry analysis (Fig. S8, ESI data 2†). As expected, no USP16 was identified in the USP16KO cells.
The total levels of the DUBs labelled with the wild-type ubiquitin reagent and mutant M20 were compared for the above mentioned HAP1 lysates (Fig. 3). In the case of the labelling experiment with USP16KO lysate, no DUBs were reactive with the M20 probe. On the other hand, only one DUB was labelled, once this probe was added to the HAP1 WT lysate. Taken together, these results indicate the high selectivity of the mutant M20 for USP16, the only DUB which is absent in the USP16KO cell line (Fig. 3). To investigate the selective reactivity of mutant M20 with USP16, we carried out another set of pull-down experiments taking advantage of the biotin handles (in positions 16 and 58) (Fig. 4, ESI data 3†). USP16 is the only DUB that we isolated from a pool of proteins present in the HAP1 WT lysate.
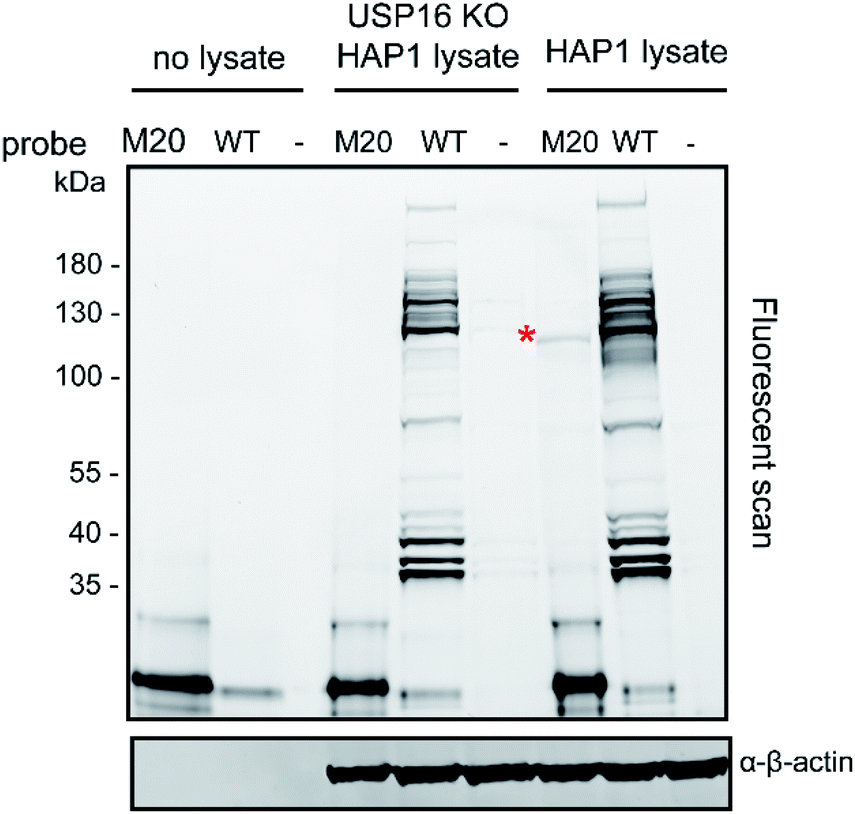 | ||
| Fig. 3 Labelling experiment of Rh-Ub-PA (WT) or Rh-M20-PA (M20) with HAP1 WT and USP16KO HAP1 lysate. β-actin is used as a loading control. | ||
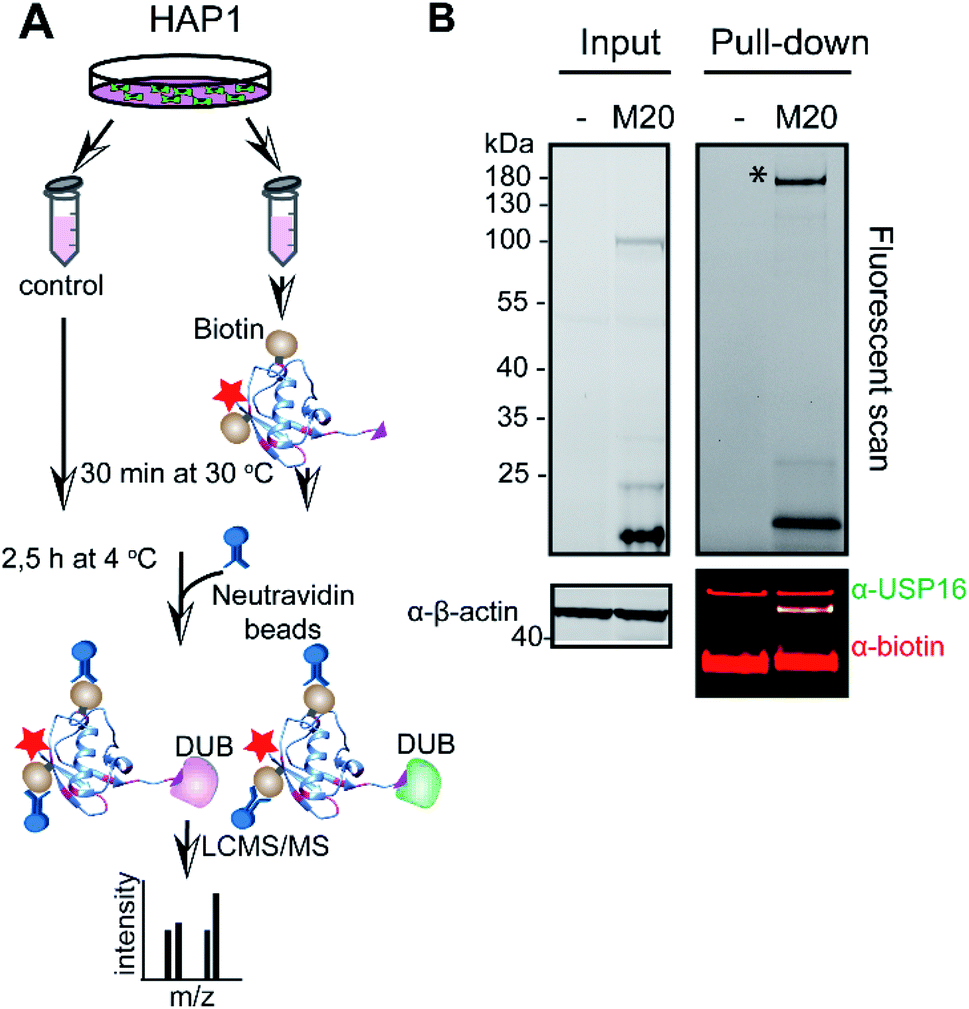 | ||
| Fig. 4 Pull-down experiment using M20 mutant (biotin handle in position 16 and 58). (A) Schematic representation of the pull-down experiment. HAP1 cell lysate was incubated either with DMSO or Rh-M20-PA probe (M20) for 30 min at 30 °C. DUBs that bound to the probe were precipitated by neutravidin beads and analysed using LC-MS/MS. (B). Fluorescent scan (top panel) and western blot analysis (bottom panel) for a representative pull-down experiment. Cell lysate incubated with either DMSO (−) or the selective probe was used for western blot analysis and immunostained with anti-USP16 antibody and biotin. β-Actin is used as a loading control. See also ESI data 3.† * shows the gel selection analysed using LC-MS/MS. | ||
Developing a USP16-selective fluorogenic substrate
For a better comprehension of cellular processes depending on the activity of USP16, it would be optimal to develop an AMC fluorogenic substrate which is processed only by USP16. We need to underline that the mode of action of these two types of reagents (ABP and fluorogenic substrate) towards DUBs is different. ABPs react covalently with the catalytic cysteine whilst fluorogenic reagents act as a substrate and are processed upon binding to the catalytic site. It would be very interesting to see if we can maintain the selectivity by designing a synthetic AMC fluorogenic substrate based on the sequence of mutant M20. These reagents were produced following a previously published procedure.25 Ubiquitin WT or the M20 mutant (residues 1–75) were synthesized using Fmoc-based solid phase peptide synthesis, followed by cleavage from the trityl resin using a mixture of 20% (v/v) hexafluoroisopropanol in methylene chloride. GlyAMC was coupled at the C-terminus using a standard peptide coupling procedure. Finally, M20-AMC and Ub-AMC were obtained after global deprotection followed by HPLC purification. We tested these reagents using fluorescent activity assays, first with purified recombinant DUBs, followed by HAP1 lysate experiments. Following the assay we can monitor in time the catalytic activity of the DUBs. The fluorescent signal is due to the release of free AMC upon hydrolysis of the substrate. If the substrate is not being processed by the enzymes, no increase in the fluorescent signal will be measured.Both reagents were tested towards USP5, USP7, USP15, and USP16 (Fig. 5). All these enzymes were highly reactive towards Ub-AMC, shown by the fluorescence increment. On the other hand, USP16 was the only USP to process M20-AMC, proving that the selectivity could be preserved between different types of reagents. Furthermore, similar experiments were performed with HAP1WT and HAP1 USP16KO lysate (Fig. 6). Ub-AMC was processed at the same rate in both cases, while M20-AMC showed an increase in fluorescent signal, due to the release of the AMC functional group, only for the HAP1 WT lysate. No fluorescent signal could be determined for the USP16KO lysate, related to the absence of USP16 in the lysate. M20-AMC can be used to follow the activity of USP16 in lysate, a useful tool to study USP16 biology.
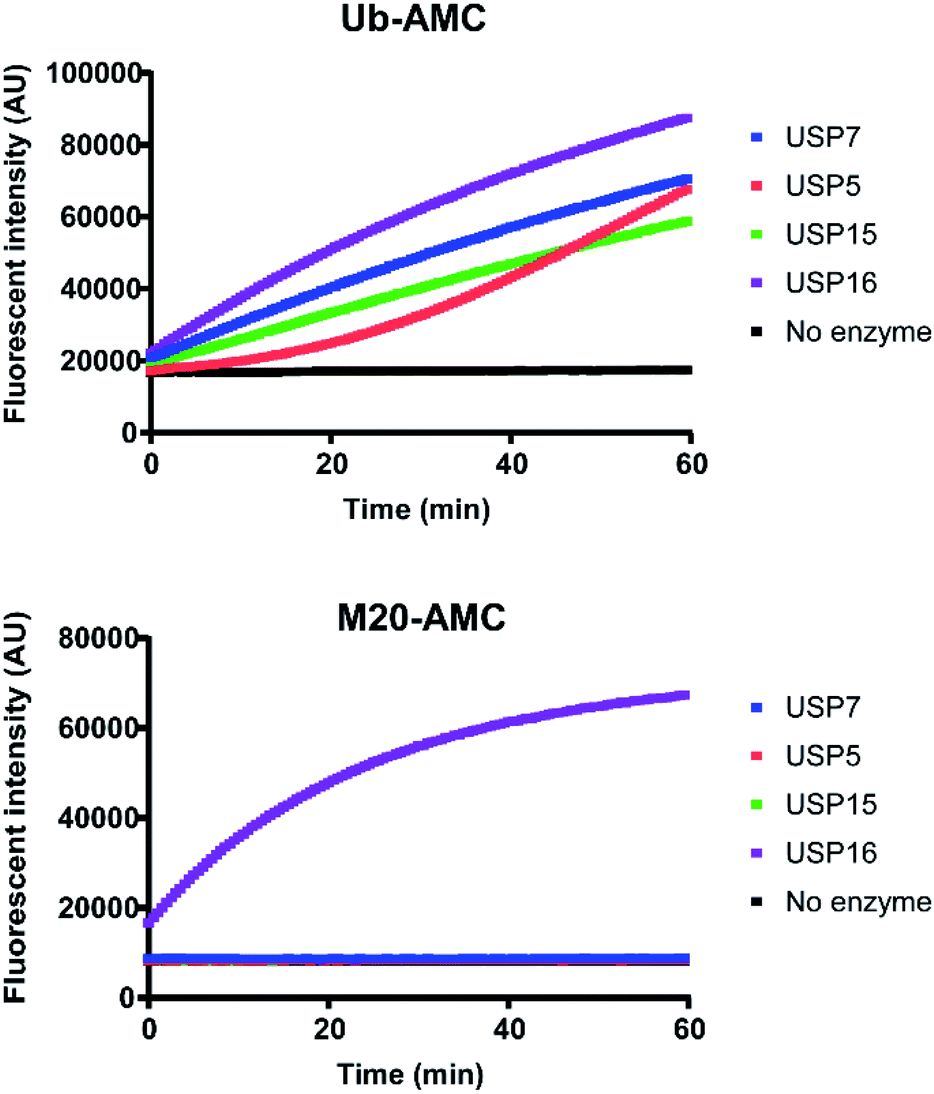 | ||
| Fig. 5 Fluorescent activity assay with purified recombinant DUBs incubated with either Ub-AMC or M20-AMC reagent. Fluorescent signal obtained from Ub-AMC (top graph) or M20-AMC (bottom graph) for each measurement was detected every 30 s. The assay was carried out in duplicate. | ||
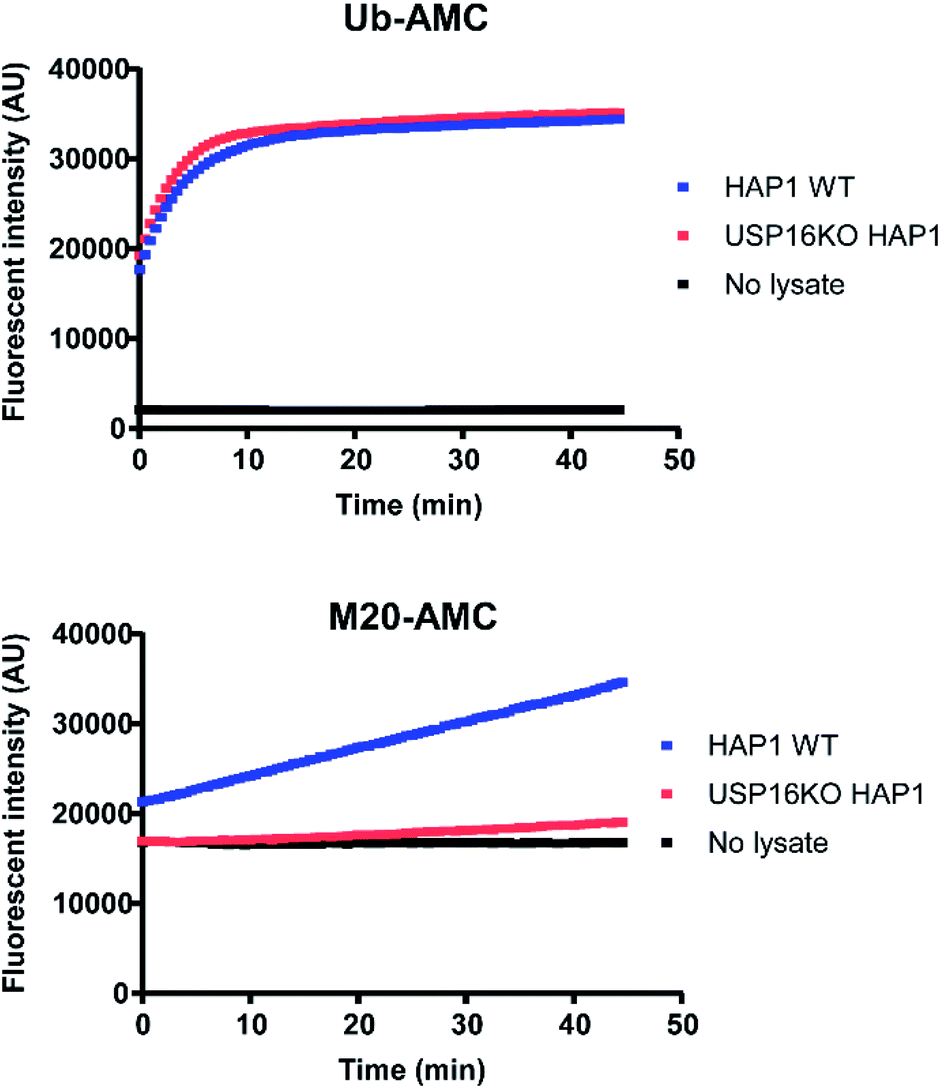 | ||
| Fig. 6 Fluorescent activity assay in the presence of HAP1 WT and USP16KO HAP1 lysate incubated with either Ub-AMC or M20-AMC substrate. Fluorescent signal obtained from Ub-AMC (top graph) or M20-AMC (bottom graph) for each measurement was detected every 30 s. The assay was carried out in duplicate. | ||
For these two reagents, we also determined affinity (KM) and catalytic turnover (kcat) using the Michaelis–Menten equation (Fig. 7 and S9†). DUBs were incubated with different substrate concentrations. The slope (initial rates) for each of the concentrations was plotted and fitted using the Michaelis–Menten equation. USP16 demonstrated a higher affinity (two-fold) for the wild-type substrate compared to M20. USP7 and USP15 did not process M20-AMC even at high substrate concentration, no KM could be determined. We hypothesize that the selectivity of the reagent for USP16 can possibly be explained by affinity differences towards other DUBs. In contrast, we observed a for USP16 four-fold increase in catalytic turnover for M20 compared to the wild-type AMC reagent. The difference in kcat might be due to a slightly altered active site placement of the C-terminal Ub tail making the hydrolysis mechanistically more favourable.
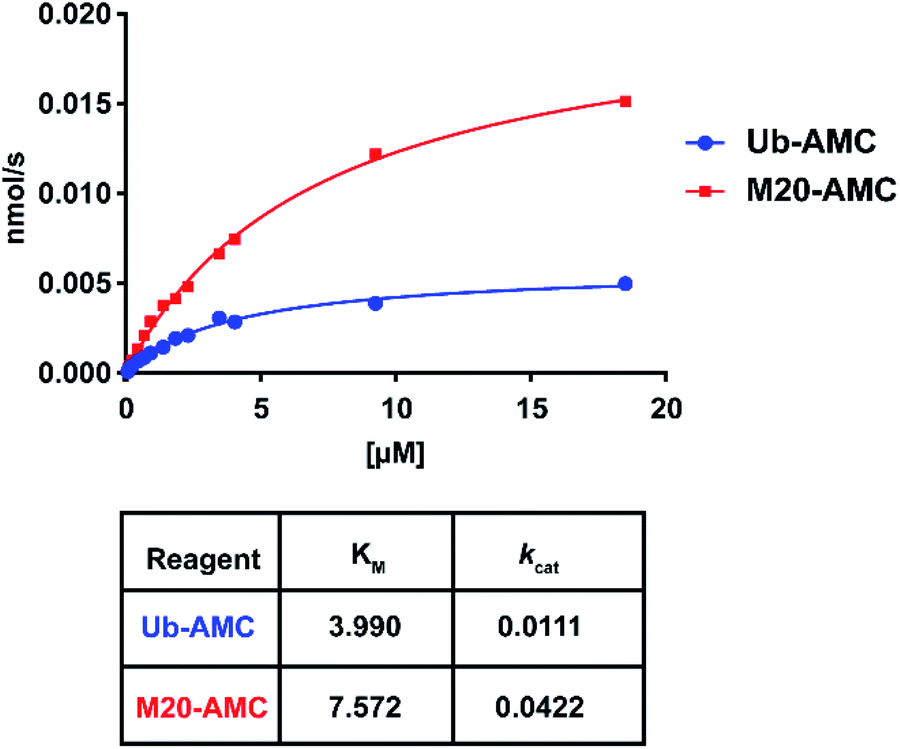 | ||
| Fig. 7 Kinetic measurements of Ub-AMC and M20-AMC in presence of USP16 using Michaelis–Menten fit achieved by plotting the initial rates (V0) at different AMC reagent concentrations and a final concentration of USP16 of 0.53 nM. The measurements were performed in four independent replicates. | ||
Conclusions
In summary, through an elaborate design and screening process, we developed an ABP and a fluorogenic substrate highly selective for the DUB USP16. This study illustrates the possibility of designing reagents for a target protein with an unknown structure by combining modelling, structural analysis, and computational prediction. Here, we reported the first example of a DUB-selective fluorogenic substrate. The activity-based probe facilitates the detection of active levels of USP16. Rh-M20-PA is a double-functionalized reagent which enables for a quick assay read-out using the fluorescent tag. Secondly, the biotin handle can be used for pull-down experiments (co-immunoprecipitation) followed by mass spectrometry analysis. The USP16-selective ubiquitin-based probe and fluorogenic substrate can be valuable tools for the development of small molecules for therapeutic purposes. This work contributes to offering the necessary tools to study USP16 and related biology.Conflicts of interest
H. O. is shareholder of the company UbiQ.Acknowledgements
We would like to thank Robbert Q. Kim for discussions and protein production. We would like to thank Dris El Atmioui for peptide synthesis and Patrick Celie for protein production. We would also like to thank Jimmy J. Akkermans for USP7 constructs. This work was financially supported by a VICI grant from the Netherlands Organization for Scientific Research N.W.O. (724.013.002).Notes and references
- K. N. Swatek and D. Komander, Cell Res., 2016, 26, 399–422 CrossRef CAS PubMed.
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS PubMed.
- M. J. Clague, S. Urbe and D. Komander, Nat. Rev. Mol. Cell Biol., 2019, 20(6), 338–352 CrossRef CAS PubMed.
- T. E. T. Mevissen and D. Komander, Annu. Rev. Biochem., 2017, 86, 159–192 CrossRef CAS PubMed.
- J. A. Harrigan, X. Jacq, N. M. Martin and S. P. Jackson, Nat. Rev. Drug Discovery, 2018, 17, 57–78 CrossRef CAS PubMed.
- P. Gopinath, S. Ohayon, M. Nawatha and A. Brik, Chem. Soc. Rev., 2016, 45, 4171–4198 RSC.
- D. S. Hewings, J. A. Flygare, M. Bogyo and I. E. Wertz, FEBS J., 2017, 284, 1555–1576 CrossRef CAS PubMed.
- D. S. Hameed, A. Sapmaz and H. Ovaa, Bioconjugate Chem., 2017, 28, 805–815 CrossRef CAS PubMed.
- M. Gorelik and S. S. Sidhu, Bioeng. Transl. Med., 2017, 2, 31–42 CrossRef CAS PubMed.
- A. Ernst, G. Avvakumov, J. Tong, Y. Fan, Y. Zhao, P. Alberts, A. Persaud, J. R. Walker, A. M. Neculai, D. Neculai, A. Vorobyov, P. Garg, L. Beatty, P. K. Chan, Y. C. Juang, M. C. Landry, C. Yeh, E. Zeqiraj, K. Karamboulas, A. Allali-Hassani, M. Vedadi, M. Tyers, J. Moffat, F. Sicheri, L. Pelletier, D. Durocher, B. Raught, D. Rotin, J. Yang, M. F. Moran, S. Dhe-Paganon and S. S. Sidhu, Science, 2013, 339, 590–595 CrossRef CAS PubMed.
- Y. Zhang, L. Zhou, L. Rouge, A. H. Phillips, C. Lam, P. Liu, W. Sandoval, E. Helgason, J. M. Murray, I. E. Wertz and J. E. Corn, Nat. Chem. Biol., 2013, 9, 51–58 CrossRef CAS PubMed.
- W. Zhang, M. A. Sartori, T. Makhnevych, K. E. Federowicz, X. Dong, L. Liu, S. Nim, A. Dong, J. Yang, Y. Li, D. Haddad, A. Ernst, D. Heerding, Y. Tong, J. Moffat and S. S. Sidhu, J. Mol. Biol., 2017, 429, 3546–3560 CrossRef CAS PubMed.
- J. Teyra, A. U. Singer, F. W. Schmitges, P. Jaynes, S. Kit Leng Lui, M. J. Polyak, N. Fodil, J. R. Krieger, J. Tong, C. Schwerdtfeger, B. B. Brasher, D. F. J. Ceccarelli, J. Moffat, F. Sicheri, M. F. Moran, P. Gros, P. J. A. Eichhorn, M. Lenter, G. Boehmelt and S. S. Sidhu, Structure, 2019, 27(4), 590–605 CrossRef CAS PubMed.
- L. Gjonaj, A. Sapmaz, R. Gonzalez-Prieto, A. C. O. Vertegaal, D. Flierman and H. Ovaa, Chem. Commun., 2019, 55, 5075–5078 RSC.
- H. Y. Joo, L. Zhai, C. Yang, S. Nie, H. Erdjument-Bromage, P. Tempst, C. Chang and H. Wang, Nature, 2007, 449, 1068–1072 CrossRef CAS PubMed.
- Y. Gu, A. E. Jones, W. Yang, S. Liu, Q. Dai, Y. Liu, C. S. Swindle, D. Zhou, Z. Zhang, T. M. Ryan, T. M. Townes, C. A. Klug, D. Chen and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E51–E60 CrossRef CAS PubMed.
- Y. Qian, B. Wang, A. Ma, L. Zhang, G. Xu, Q. Ding, T. Jing, L. Wu, Y. Liu, Z. Yang and Y. Liu, Sci. Rep., 2016, 6, 33039 CrossRef CAS PubMed.
- Z. Zhang, H. Yang and H. Wang, J. Biol. Chem., 2014, 289, 32883–32894 CrossRef CAS PubMed.
- M. Adorno, S. Sikandar, S. S. Mitra, A. Kuo, B. Nicolis Di Robilant, V. Haro-Acosta, Y. Ouadah, M. Quarta, J. Rodriguez, D. Qian, V. M. Reddy, S. Cheshier, C. C. Garner and M. F. Clarke, Nature, 2013, 501, 380–384 CrossRef CAS PubMed.
- J. Schymkowitz, J. Borg, F. Stricher, R. Nys, F. Rousseau and L. Serrano, Nucleic Acids Res., 2005, 33, W382–W388 CrossRef CAS PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. Gallo Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252–W258 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- S. J. Ward, H. E. Gratton, P. Indrayudha, C. Michavila, R. Mukhopadhyay, S. K. Maurer, S. G. Caulton, J. Emsley and I. Dreveny, J. Biol. Chem., 2018, 293, 17362–17374 CrossRef CAS PubMed.
- R. Ekkebus, S. I. van Kasteren, Y. Kulathu, A. Scholten, I. Berlin, P. P. Geurink, A. de Jong, S. Goerdayal, J. Neefjes, A. J. Heck, D. Komander and H. Ovaa, J. Am. Chem. Soc., 2013, 135, 2867–2870 CrossRef CAS PubMed.
- F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Angew. Chem., Int. Ed. Engl., 2010, 49, 10149–10153 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02226k |
| ‡ Current address: Pepscan, Zuidersluisweg 2, 8243 RC, Lelystad, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2019 |