
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective carbene insertion into the N–H bond of benzophenone imine†
Jian
Yang
 ,
Peiran
Ruan
,
Wei
Yang
,
Xiaoming
Feng
and
Xiaohua
Liu
*
,
Peiran
Ruan
,
Wei
Yang
,
Xiaoming
Feng
and
Xiaohua
Liu
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn
First published on 18th September 2019
Abstract
Efficient enantioselective insertion of α-diazoesters into the N–H bond of N-sp2-hybridized benzophenone imine was realized by using Rh2(esp)2 and chiral guanidine cooperative catalysis. Both aliphatic and aromatic substituted α-amino esters were obtained in high yields (up to 99%) and good enantioselectivities (up to 95.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.5 er) under mild reaction conditions.
:4.5 er) under mild reaction conditions.
Introduction
Efficient synthesis of enantiomerically enriched α-amino acids and their derivatives remains a hot topic in synthetic organic chemistry and biology.1 The transition-metal-catalyzed enantioselective insertion of an α-diazocarbonyl compound into an N–H bond represents a potentially attractive route to chiral α-amino acid derivatives.2 Significant progress has been made on asymmetric insertion reactions of N-sp3-hybridized N–H sources including primary and secondary anilines (Scheme 1a),3 as well as those of tert-butyl carbamate4 (Scheme 1b). Recently, enantioselective carbene insertion into the N–H bond of C3-substituted indoles and carbazoles was achieved (Scheme 1c).5 With regard to the chiral catalyst variety, different transition metal complexes have proven to be useful, including chiral dirhodium(II) prolinates by McKervey,6 copper complexes with chiral ligands, such as bisoxazoline by Jørgensen,3i spiro bisoxazolines by Zhou,3h bpy* by Fu,4d 2,2′-biimidazole by Zhou,5a a BINOL derivative by our group,3g chiral palladium complexes liganded with guanidine by our group,3a and PyBox by Vranken.5b In addition, cooperative catalytic systems of dirhodium(II) carboxylates with cinchona alkaloids3f or chiral phosphoric acids4c were well applied to the asymmetric N–H bond insertion reaction and others.3b,i,4e,5a In these cases, the coupling of α-diazo compounds and N–H sources is interesting. It seems that insertion of alkyl α-diazoesters into anilines3 and aryl α-diazocarbonyl compounds into tert-butyl carbamate4 and carbazoles5 yielded higher enantioselectivity than the opposite cross combination. This might be due to the nucleophilicity of the nitrogen sources and the stability of the carbene intermediates. This indicates one limitation of these developed catalytic systems.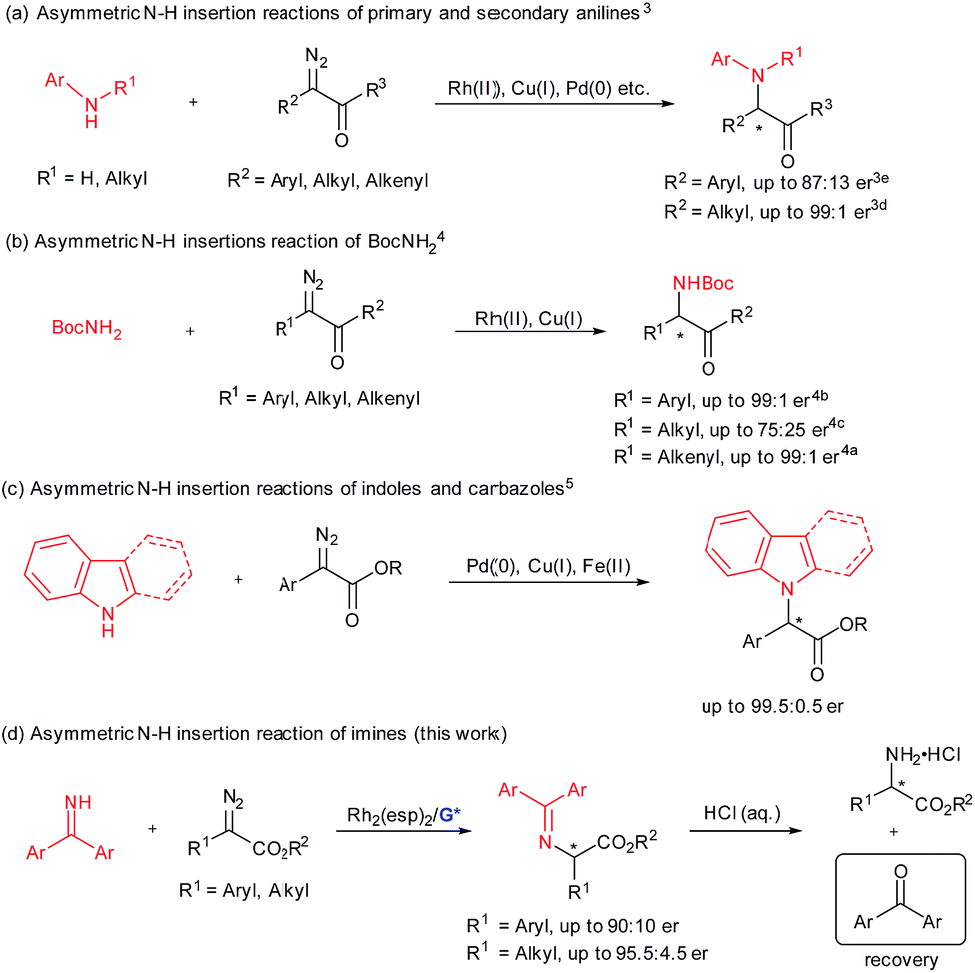 | ||
| Scheme 1 Asymmetric N–H insertion reactions. | ||
Benzophenone imine, which is industrially produced by condensation of benzophenone and ammonia, could serve as an ammonia carrier and has been successfully applied to many asymmetric catalytic amination reactions.7 The amination products could be transformed into N-unprotected amino compounds conveniently by one step acidic hydrolysis, and the ammonia carrier precursor benzophenone could be recovered and reused. We became interested in catalytic asymmetric carbene insertion into the N–H bond of N-sp2-hybridized benzophenone imines because the products could be readily transformed into optically active N-unprotected amino acids (Scheme 1d).
Previously, our group demonstrated enantioselective insertion of α-diazocarbonyl compounds into the O–H bond of carboxylic acids by the use of cooperative catalysis of Rh2(OAc)4 and a chiral guanidine.8 In this case, the chiral guanidine salt of the carboxylic acid could act as a proton shuttle9 for the enantioselective proton shift. In view of the strong basicity of guanidine10 and the acidity of the N–H bond of benzophenone imines albeit weak, we plan to extend the cooperative catalysis of the dirhodium(II) complex and chiral guanidine to asymmetric carbene insertion into the N–H bond of benzophenone imine. In this text, we reported the details of such a process which enables the convenient generation of both alkyl and aryl substituted amino acetates in high yield and good enantioselectivities under mild reaction conditions. With regard to reaction partners and the type of proton-transfer carrier, this method complements the studies of Fu4d and Zhou,4a–c using either a chiral copper complex or dirhodium/chiral phosphoric acids for enantioselective N–H insertion of tert-butyl carbamate (Scheme 1b).
Results and discussion
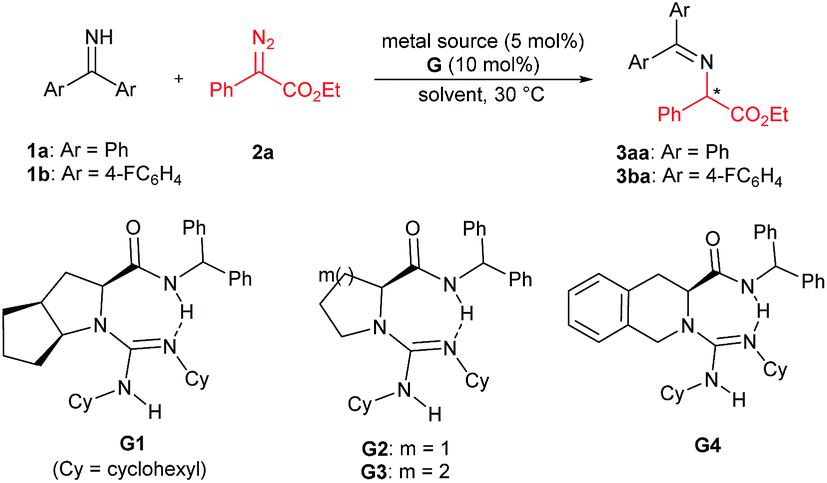
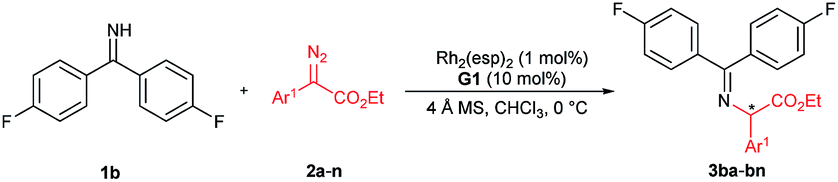
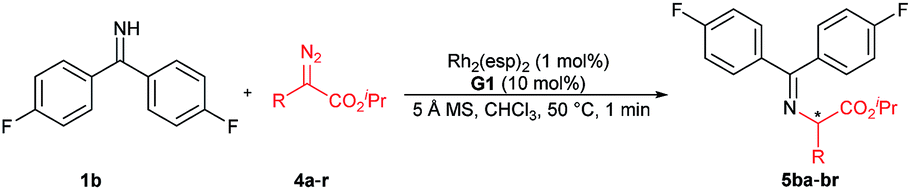
We initially chose benzophenone imine 1a and ethyl 2-diazo-2-phenylacetate 2a as the model substrate to optimize the reaction conditions (Table 1). Several metal sources (see the ESI† for details) were screened in the presence of chiral guanidine G111 in CH2Cl2 at 30 °C, and it was found that Rh2(OAc)4 could promote the reaction to give the N–H insertion product 3aa in good yield with low enantioselectivity (84% yield, 75:25 er, Table 1, entries 1–3). The combination of chiral guanidine with Pd(0), which was efficient in the asymmetric carbene insertion reaction of primary and secondary anilines,3a was completely inefficient in this reaction (entry 1). Next, we used Rh2(OAc)4 to explore different chiral guanidines. After exploring chiral guanidines derived from various amino acids, such as L-proline derived G2, L-pipecolic acid derived G3 and (S)-tetrahydroisoquinoline-3-carboxylic acid derived G4, we obtained enantiomerically enriched products only in the presence of L-ramipril derived G1 (Table 1, entry 3 vs. entries 4–6). The addition of 4 Å molecular sieves slightly benefited the improvement of the yield (Table 1, entry 7). After careful screening of the solvents, CHCl3 was proven to be the best choice, and the N–H insertion product could be obtained in 87% yield and 82:18 er (Table 1, entry 8). Rh2(esp)2 exhibited higher activity, with which a yield of 93% with 82:18 er could be obtained at a reduced reaction temperature (Table 1, entries 9–11). The different substituents on phenyl of benzophenone imines were evaluated (see the ESI† for details), and in the presence of bis(4-fluorophenyl)methanimine 1b as the model substrate and 1 mol% of Rh2(esp)2, the product 3ba could be obtained in 96% yield and 90:10 er (Table 1, entries 12 and 13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal source | G | Solvent | Yieldb (%) | erc |
| a Unless otherwise noted, all reactions were carried out with metal source (5 mol%), G (10 mol%), 1a (0.1 mmol), and 2a (0.12 mmol) in the solvent (0.5 mL) at 30 °C for 5 h. b Isolated yield. c Determined by chiral HPLC analysis. d 4 Å MS (30 mg) was added. e The reaction time was 10 min. f At 0 °C for 8 h. g 1b (0.1 mmol) was used. h Rh2(esp)2 (1 mol%) at 0 °C for 24 h. | |||||
| 1 | Pd2(dba)3 | G1 | CH2Cl2 | N.R. | — |
| 2 | AgNTf2 | G1 | CH2Cl2 | 83 | 50:50 |
| 3 | Rh2(OAc)4 | G1 | CH2Cl2 | 84 | 75:25 |
| 4 | Rh2(OAc)4 | G2 | CH2Cl2 | 82 | 50:50 |
| 5 | Rh2(OAc)4 | G3 | CH2Cl2 | 88 | 50:50 |
| 6 | Rh2(OAc)4 | G4 | CH2Cl2 | 89 | 50:50 |
| 7d | Rh2(OAc)4 | G1 | CH2Cl2 | 96 | 75:25 |
| 8d | Rh2(OAc)4 | G1 | CHCl3 | 87 | 82:18 |
| 9d | Rh2(oct)4 | G1 | CHCl3 | 88 | 74:26 |
| 10d,e | Rh2(esp)2 | G1 | CHCl3 | 90 | 74:26 |
| 11d,f | Rh2(esp)2 | G1 | CHCl3 | 93 | 82:18 |
| 12d,f,g | Rh2(esp)2 | G1 | CHCl3 | 96 | 90:10 |
| 13d,g,h | Rh2(esp)2 | G1 | CHCl3 | 96 | 90:10 |
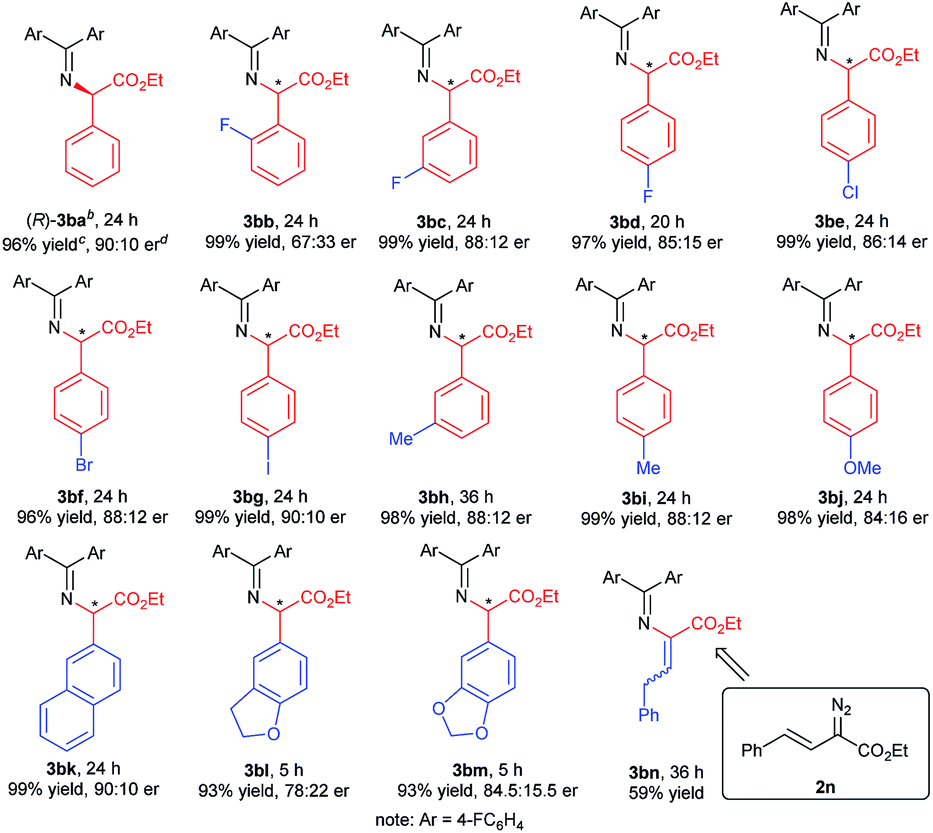
With the optimized reaction conditions in hand, we turned to evaluating the scope of α-diazoesters of this protocol. A variety of α-aryl α-diazoesters 2 were investigated. As shown in Table 2, α-diazoesters bearing halo-substituted phenyl groups at different positions could be smoothly converted to the corresponding products 3bb–3bg in 96%–99% yields with 85:15 to 90:10 er, except for the 2-fluorophenyl substituted one. Generally, substrates with a halogen at the meta- and para-position obviously gave higher enantioselectivity than substrates with a halogen at the ortho-position. Electron-donating groups were tolerated in this reaction, and the desired products 3bh–3bj were generated in 98–99% yields with 84:16 to 88:12 er. 2-Naphthyl or heteroaromatic substituted α-diazoesters were also suitable substrates, affording the related products 3bk–3bm with moderate to good results (93–99% yields, 78:22 to 90:10 er). It is noteworthy that ethyl (E)-2-diazo-4-phenylbut-3-enoate 2n, which contains an alkenyl substituent, underwent the reaction smoothly, but the product 3bnvia [1,4]-H transfer was obtained. This result is different from that of dirhodium(II) carboxylate/chiral spirophosphoric acid cooperatively promoted N–H insertion of vinyl diazoacetates with tert-butyl carbamate in Zhou's work.4a
|
|
|---|
| a Unless otherwise noted, all reactions were carried out with Rh2(esp)2 (1 mol%), G1 (10 mol%), 4 Å MS (30 mg), 1b (0.1 mmol), and 2 (1.2 equiv.) in CHCl3 (0.5 mL) at 0 °C for 24 h. b The absolute configuration of the product 3ba was determined to be R after the transformation into the corresponding N-Boc-protected amine 7ba.12 c Isolated yield. d Determined by chiral HPLC analysis. |
|
Excited by the expected reaction profile, we next examined the attractive scenario of preparing alkyl substituted amino acids which are less enantioselective in the insertion reaction of tert-butyl carbamate.4c We reoptimized the reaction conditions between fluoro-containing benzophenone imine 1b and 2-diazopropanoate (see the ESI† for details). To our delight, the cooperative catalysis of Rh2(esp)2 and chiral guanidine G1 occurred much more efficiently for alkyl substituted diazoesters than for aryl substituted ones. All the reactions were completed within one minute at increased reaction temperatures. Thus, we examined the reaction of benzophenone imine 1b with various α-alkyl α-diazoesters 4, with the results listed in Table 3. It was found that the length of the α-alkyl chain attached to α-diazoesters had a limited influence on the enantioselectivity (93:7 to 95.5:4.5 er) but the yield dramatically varied (30–96% yields) due to the formation of β-H elimination byproducts.3d,15 The reaction was also tolerable to α-alkyl α-diazoesters with important functional groups, including phenyl, halogen, nitrile, alkenyl, propargyl, ester, silyl ester, and ether, and the corresponding products 5bf–5bp were obtained in moderate to good yields (35–99% yields) with high enantioselectivities (85:15 to 95:5 er). Generally, the use of substrates with electron-withdrawing groups gave obviously higher yields than the use of those with electron-donating groups (5bf–5bnvs.5bo–5bp) because α-alkyl α-diazoesters containing electron-donating groups easily underwent β-H eliminate to form the corresponding olefins. Notably, the protected isatin and oxindole derived substrates could undergo the reaction smoothly and afforded the desired products 5bq and 5br with excellent results (99% yield, 95:5 and 95.5:4.5 er, respectively).
|
|
|---|
| a Unless otherwise noted, all reactions were carried out with Rh2(esp)2 (1 mol%), G1 (10 mol%), 5 Å MS (30 mg), 1b (0.1 mmol), and 4 (2.0 equiv.) in CHCl3 (0.5 mL) at 50 °C for 1 min. b The absolute configuration of the products 5ba and 5bf was determined to be S after further transformation into known compounds13,14 (see the ESI for details). c Isolated yield. d Determined by chiral HPLC analysis. e 4p (4.0 equiv.) was added. |
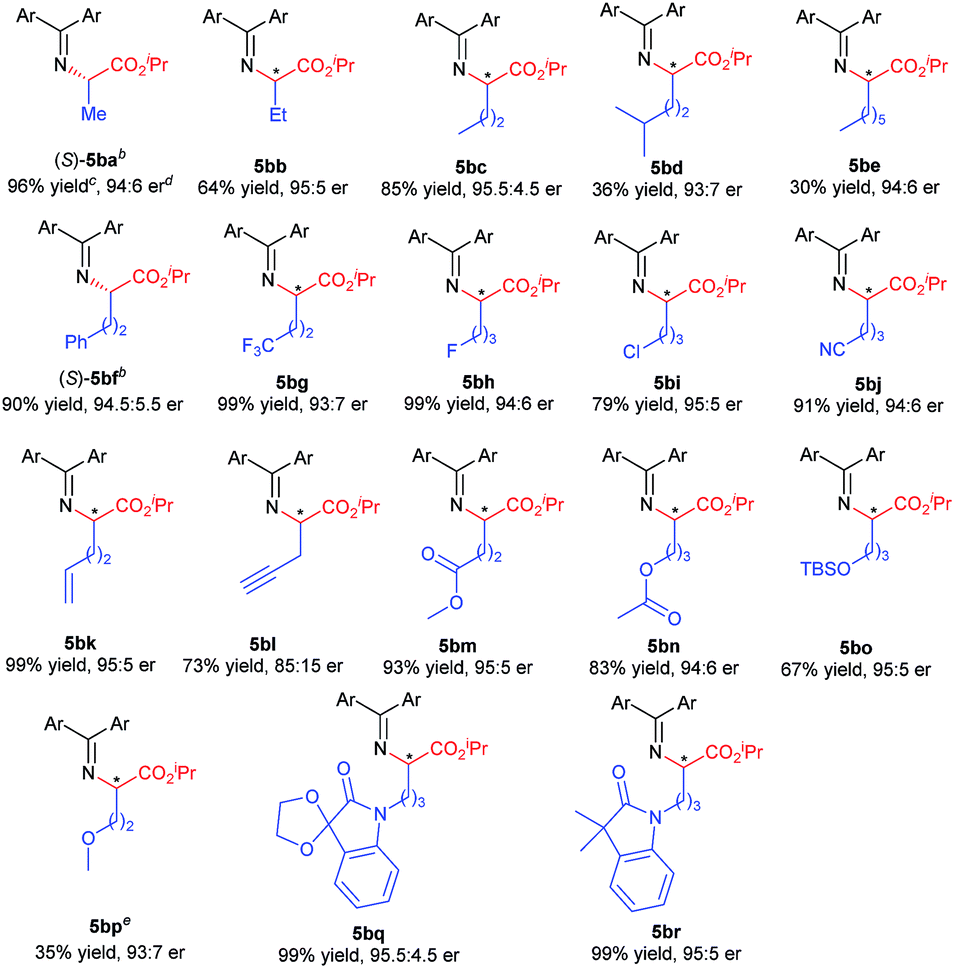
|
Furthermore, the synthetic utility of this reaction was investigated. A gram-scale synthesis of the products 3ba and 5br was carried out as shown in Scheme 2. Under the standard reaction conditions, the corresponding product 3ba was generated in 99% yield and 90:10 er (Scheme 2a). The optically active N-unprotected 2-amino-2-phenylacetate 6ba could be obtained easily by one step acidic hydrolysis in excellent yield and enantioselectivity after recrystallization. Meanwhile, the benzophenone could be recovered in high yield from the organic phase. The absolute configuration of the product 3ba was determined to be R after the transformation into the corresponding N-Boc-protected amine 7ba, whose structure was determined by X-ray crystal analysis.12 It is noteworthy that the absolute configuration of the products 5ba and 5bf was determined to be mainly S in comparison with the known chiral compounds after transformation (see the ESI† for details). An enantiodivergent phenomenon16 was observed from the substrate variation in these cases (3ba/5ba/5bf). Similarly, the alkyl substituted amino acetate 6br was afforded at a gram-scale with maintained enantioselectivity (Scheme 2b, 93% total yield, 94:6 er). This N–H insertion reaction of benzophenone imine provides a new convenient route to various chiral α-amino acids.
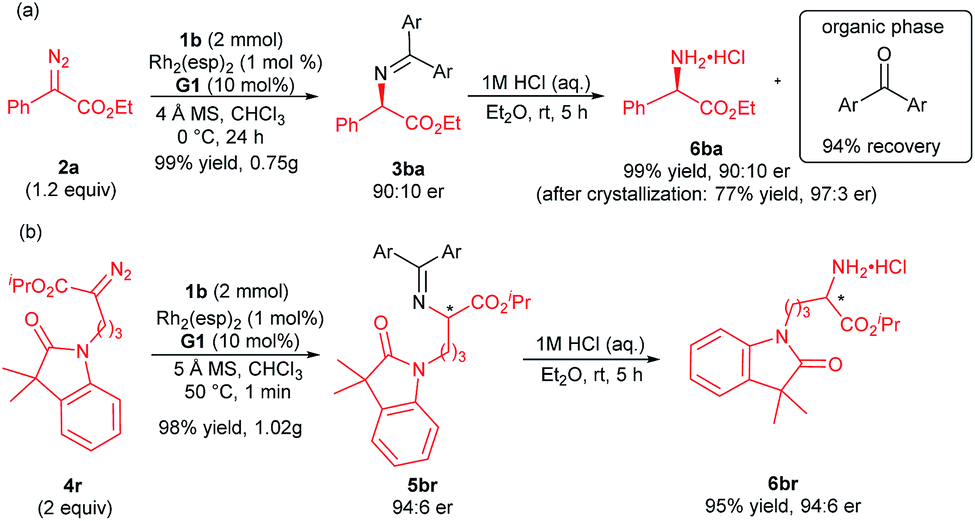 | ||
| Scheme 2 Scaled-up version and further transformation. | ||
Conclusions
We have developed the first catalytic asymmetric N–H insertion of α-diazoesters with benzophenone imines. A cooperative catalytic system of a dirhodium(II) complex and chiral guanidine showed high efficiency. Both aryl and alkyl substituted α-amino esters were prepared in high yields and with good enantioselectivities under mild reaction conditions. In particular, this method complements the substrate limitation in N–H insertion of tert-butyl carbamate. We are currently extensively studying the mechanism and the use of chiral guanidine for other asymmetric reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21625205) for financial support.Notes and references
- For selected reviews, see: (a) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef; (b) M. Calmes and J. Daunis, Amino Acids, 1999, 16, 215 CrossRef CAS PubMed; (c) V. Constantinou-Kokotou and G. Kokotos, Amino Acids, 1999, 16, 273 CrossRef CAS PubMed.
- For selected reviews of N–H insertion, see: (a) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (b) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC; (c) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed; (d) Z.-H. Zhang and J.-B. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS; (e) C. J. Moody, Angew. Chem., Int. Ed., 2007, 46, 9148 CrossRef CAS PubMed; (f) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS.
- For asymmetric N–H insertion of primary and secondary anilines, see: (a) Y. Zhu, X. H. Liu, S. X. Dong, Y. H. Zhou, W. Li, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2014, 53, 1636 CrossRef CAS PubMed; (b) G. Liu, J. Li, L. Qiu, L. Liu, G. Y. Xu, B. Ma and J. T. Sun, Org. Biomol. Chem., 2013, 11, 5998 RSC; (c) X. F. Xu, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2012, 51, 9829 CrossRef CAS PubMed; (d) S.-F. Zhu, B. Xu, G.-P. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2012, 134, 436 CrossRef CAS PubMed; (e) H. Saito, D. Morita, T. Uchiyama, M. Miyake and S. Miyairi, Tetrahedron Lett., 2012, 53, 6662 CrossRef CAS; (f) H. Saito, T. Uchiyama, M. Miyake, M. Anada, S. Hashimoto, T. Takabatake and S. Miyairi, Heterocycles, 2010, 81, 1149 CrossRef CAS; (g) Z. R. Hou, J. Wang, P. He, J. Wang, B. Qin, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2010, 49, 4763 CrossRef CAS PubMed; (h) B. Liu, S.-F. Zhu, W. Zhang, C. Chen and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834 CrossRef CAS PubMed; (i) S. Bachmann, D. Fielenbach and K. A. Jørgensen, Org. Biomol. Chem., 2004, 2, 3044 RSC.
- For asymmetric N–H insertion of tert-butyl carbamate, see: (a) J.-X. Guo, T. Zhou, B. Xu, S.-F. Zhu and Q.-L. Zhou, Chem. Sci., 2016, 7, 1104 RSC; (b) B. Xu, S.-F. Zhu, X.-D. Zuo, Z.-C. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 3913 CrossRef CAS PubMed; (c) B. Xu, S.-F. Zhu, X.-L. Xie, J.-J. Shen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 11483 CrossRef CAS PubMed; (d) E. C. Lee and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 12066 CrossRef CAS PubMed; (e) Y. Ni, X. Guo, W. H. Hu and S. Y. Liu, Chin. J. Org. Chem., 2014, 34, 107 CrossRef CAS.
- For asymmetric N–H insertion of C3-substituted indoles and carbazoles, see: (a) H.-Q. Shen, B. Wu, H.-P. Xie and Y.-G. Zhou, Org. Lett., 2019, 21, 2712 CrossRef CAS PubMed; (b) V. Arredondo, S. C. Hiew, E. S. Gutman, I. D. U. A. Premachandra and D. L. Van Vranken, Angew. Chem., Int. Ed., 2017, 56, 4156 CrossRef CAS PubMed.
- C. F. García, M. A. McKervey and T. Ye, Chem. Commun., 1996, 1465 RSC.
- For selected examples, see:
(a) L. Lykke, D. Monge, M. Nielsen and K. A. Jgensen, Chem.–Eur. J., 2010, 16, 13330 CrossRef CAS PubMed;
(b) A. J. Lin, H. B. Mao, X. Zhu, H. M. Ge, R. X. Tan, C. J. Zhu and Y. X. Cheng, Chem.–Eur.J., 2011, 17, 13676 CrossRef CAS PubMed;
(c) H. Choubane, A. F. Garrido-Castro, C. Alvarado, A. Martín-Somer, A. Guerrero-Corella, M. Daaou, S. Díaz-Tendero, M. C. Maestro, A. Fraile and J. Alemá, Chem. Commun., 2018, 54, 3399 RSC;
(d) M. Y. Li, S. Berritt, L. Matuszewski, G. G. Deng, A. Pascual-Escudero, G. B. Panetti, M. Poznik, X. D. Yang, J. J. Chruma and P. J. Walsh, J. Am. Chem. Soc., 2017, 139, 16327 CrossRef CAS PubMed;
(e) M. J. O'Donnell, J. T. Cooper and M. M. Mader, J. Am. Chem. Soc., 2003, 125, 2370 CrossRef PubMed;
(f) B. M. Trost and Y. L. Wang, Angew. Chem., Int. Ed., 2018, 57, 11025 CrossRef CAS PubMed.
- F. Tan, X. H. Liu, X. Y. Hao, Y. Tang, L. L. Lin and X. M. Feng, ACS Catal., 2016, 6, 6930 CrossRef CAS.
- (a) Y. Liang, H. L. Zhou and Z.-X. Yu, J. Am. Chem. Soc., 2009, 131, 17783 CrossRef CAS PubMed; (b) Y.-Y. Ren, S.-F. Zhu and Q.-L. Zhou, Org. Biomol. Chem., 2018, 16, 3087 RSC; (c) M.-L. Li, M.-Q. Chen, B. Xu, S.-F. Zhu and Q.-L. Zhou, Acta Chim. Sin., 2018, 76, 883 CrossRef CAS.
- For selected reviews, see: (a) S. X. Dong, X. M. Feng and X. H. Liu, Chem. Soc. Rev., 2018, 47, 8525 RSC; (b) W. D. Cao, X. H. Liu and X. M. Feng, Chin. Chem. Lett., 2018, 29, 1201 CrossRef CAS; (c) X. Fu and C.-H. Tan, Chem. Commun., 2011, 47, 8210 RSC; (d) X.-Y. Cui, C.-H. Tan and D. Leow, Org. Biomol. Chem., 2019, 17, 4689 RSC. For selected examples, see: (e) A. Hjelmencrantz and U. Berg, J. Org. Chem., 2002, 67, 3585 CrossRef PubMed; (f) Z. P. Yu, X. H. Liu, L. Zhou, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2009, 48, 5195 CrossRef CAS PubMed; (g) S. X. Dong, X. H. Liu, X. H. Chen, F. Mei, Y. L. Zhang, B. Gao, L. L. Lin and X. M. Feng, J. Am. Chem. Soc., 2010, 132, 10650 CrossRef CAS PubMed; (h) S. X. Dong, X. H. Liu, Y. Zhu, P. He, L. L. Lin and X. M. Feng, J. Am. Chem. Soc., 2013, 135, 10026 CrossRef CAS PubMed; (i) T. F. Kang, P. Zhao, J. Yang, L. L. Lin, X. M. Feng and X. H. Liu, Chem.–Eur. J., 2018, 24, 3703 CrossRef CAS PubMed; (j) Y. Tang, Q. G. Chen, X. H. Liu, G. Wang, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 9512 CrossRef CAS PubMed; (k) Q. G. Chen, Y. Tang, T. Y. Huang, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2016, 55, 5286 CrossRef CAS PubMed; (l) Y. Tang, J. Xu, J. Yang, L. L. Lin, X. M. Feng and X. H. Liu, Chem, 2018, 4, 1658 CrossRef CAS; (m) S. S. Guo, P. Dong, Y. S. Chen, X. M. Feng and X. H Liu, Angew. Chem., Int. Ed., 2018, 57, 16852 CrossRef CAS PubMed.
- CCDC 1935053 (G1). (See the ESI† for details) contains the supplementary crystallographic data for this paper..
- CCDC 1915990 (7ba). (See the ESI† for details) contains the supplementary crystallographic data for this paper..
- F. Ulatowski and J. Jurczak, J. Org. Chem., 2015, 80, 4235 CrossRef CAS PubMed.
- P. A. Evans, J. E. Robinson and J. D. Nelson, J. Am. Chem. Soc., 1999, 121, 6761 CrossRef CAS.
- For selected examples of β-H elimination of metal carbenes, see: (a) D. F. Taber, R. J. Herr, S. K. Pack and J. M. Geremia, J. Org. Chem., 1996, 61, 2908 CrossRef CAS PubMed; (b) Y. Yasutomi, H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 4510 CrossRef CAS PubMed; (c) T. Goto, K. Takeda, N. Shimada, H. Nambu, M. Anada, M. Shiro, K. Ando and S. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 6803 CrossRef CAS PubMed.
- (a) G. Zanoni, F. Castronovo, M. Franzini, G. Vidari and E. Giannini, Chem. Soc. Rev., 2003, 32, 115 RSC; (b) T. Tanaka and M. Hayashi, Synthesis, 2008, 3361 CAS; (c) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675 RSC; (d) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080 CrossRef CAS PubMed; (e) W. D. Cao, X. M. Feng and X. H. Liu, Org. Biomol. Chem., 2019, 17, 4689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1915990 and 1935053. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03354h |
| This journal is © The Royal Society of Chemistry 2019 |