
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA proof-reading mechanism for non-proteinogenic amino acid incorporation into glycopeptide antibiotics†
Milda
Kaniusaite
 ab,
Julien
Tailhades
ab,
Edward A.
Marschall
ab,
Robert J. A.
Goode
ac,
Ralf B.
Schittenhelm
ac and
Max J.
Cryle
*ab
ab,
Julien
Tailhades
ab,
Edward A.
Marschall
ab,
Robert J. A.
Goode
ac,
Ralf B.
Schittenhelm
ac and
Max J.
Cryle
*ab
aThe Monash Biomedicine Discovery Institute, Department of Biochemistry and Molecular Biology, Monash University, Clayton, Victoria 3800, Australia. E-mail: max.cryle@monash.edu
bEMBL Australia, Monash University, Clayton, Victoria 3800, Australia
cMonash Proteomics and Metabolomics Facility, Monash University, Clayton, Victoria 3800, Australia
First published on 29th August 2019
Abstract
Non-ribosomal peptide biosynthesis produces highly diverse natural products through a complex cascade of enzymatic reactions that together function with high selectivity to produce bioactive peptides. The modification of non-ribosomal peptide synthetase (NRPS)-bound amino acids can introduce significant structural diversity into these peptides and has exciting potential for biosynthetic redesign. However, the control mechanisms ensuring selective modification of specific residues during NRPS biosynthesis have previously been unclear. Here, we have characterised the incorporation of the non-proteinogenic amino acid 3-chloro-β-hydroxytyrosine during glycopeptide antibiotic (GPA) biosynthesis. Our results demonstrate that the modification of this residue by trans-acting enzymes is controlled by the selectivity of the upstream condensation domain responsible for peptide synthesis. A proofreading thioesterase works together with this process to ensure that effective peptide biosynthesis proceeds even when the selectivity of key amino acid activation domains within the NRPS is low. Furthermore, the exchange of condensation domains with altered amino acid specificities allows the modification of such residues within NRPS biosynthesis to be controlled, which will doubtless prove important for reengineering of these assembly lines. Taken together, our results indicate the importance of the complex interplay of NRPS domains and trans-acting enzymes to ensure effective GPA biosynthesis, and in doing so reveals a process that is mechanistically comparable to the hydrolytic proofreading function of tRNA synthetases in ribosomal protein synthesis.
Introduction
Non-ribosomal peptide biosynthesis plays a major role in the formation of peptide-based natural products, of which many have important medicinal properties.1 The significant diversity of peptide structures produced through non-ribosomal peptide biosynthesis is based on the modular architecture of non-ribosomal peptide synthetase (NRPS) assembly lines. These modules, which are comprised of different catalytic domains, can accept a very wide range of building blocks through the activity of their adenylation (A) domains that perform substrate selection and activation.2 At this point in biosynthesis, the amino acid intermediates are transferred to neighbouring peptidyl carrier protein (PCP) domains, where they are tethered as thioesters via the essential PCP phosphopantetheinyl moiety.3 Formation of peptide bonds is then mediated by the activity of condensation (C) domains, in which the upstream (donor) PCP-bound peptide is transferred onto the aminoacyl-PCP acceptor with concomitant peptide bond formation.4 A-Domains are typically highly selective for their specific amino acid substrates, and hence largely control the amino acid composition of the final non-ribosomal peptides,5 whilst C-domains control the stereochemistry of the upstream (donor) peptide and can also display selectivity for the downstream (acceptor) amino acid.4,6–8 Once the complete peptide has been synthesised, it is typically released from the NRPS through the actions of a terminal thioesterase domain in a process that can generate yet further structural changes to the peptide (through cyclisation/dimerisation etc.).9 Additional diversity in peptide structure can also be installed during biosynthesis by the actions of further catalytic domains found either within the assembly line itself, such as epimerisation performed through (E) domains10 and methylation performed by methyltransferase (MT) domains,11 or through the activity of enzymes acting in trans to the main peptide assembly line such as halogenases or oxidases.12,13 Whilst epimerisation typically occurs on PCP-bound peptides and selectivity is ensured by the actions of the neighbouring C-domain,6,7trans-modifications largely target aminoacyl-PCPs, with the mechanism controlling the modification of specific aminoacyl-PCPs largely unclear.The biosynthesis the glycopeptide antibiotics (GPAs) serves as an important example of a how nature can produce complex, medically-relevant antibiotics through the actions of an equally complex NRPS biosynthesis process (Fig. 1).14 GPAs are a family of highly crosslinked heptapeptides, and include lipid II binding antibiotics exemplified by vancomycin/balhimycin and teicoplanin, as well as other members such as kistamicin and complestatin that have antiviral activity.14 Whilst the biosynthesis of GPA peptides initially appears straightforward, closer inspection reveals significant complexity: in particular, this includes the modification of the structure of amino acids (chlorination and/or hydroxylation of tyrosine residues 2 and 6 within vancomycin and teicoplanin type GPAs) and the peptide itself (through extensive crosslinking of aromatic side chains).15 The crosslinking cascade in GPA biosynthesis has been shown to depend on recruitment of external cytochrome P450 monooxygenases through the conserved X-domain within the final NRPS module, a process unique to GPAs.16,17 The process controlling the modification of amino acid residues within the peptide, however, is less clear. Different mechanisms also exist for incorporation of specific amino acid modifications during NRPS-mediated biosynthesis of vancomycin and teicoplanin-type GPAs. Whilst studies support amino acid modifications as largely occurring on aminoacyl-PCP substrates during GPA biosynthesis, these also indicate that selectivity cannot alone be dictated by selectivity of the trans-modifying enzymes for the correct PCP domains.18–21 Thus, we hypothesised that the selection agent in these modification reactions would be the neighbouring C-domain that would only select the PCP-bound amino acid for peptide elongation once this residue has been appropriately modified. This implies that selectivity in GPA biosynthesis relies on the complex interplay of specificities from multiple enzymatic domains both within and external to the NRPS machinery, which has not yet been investigated.
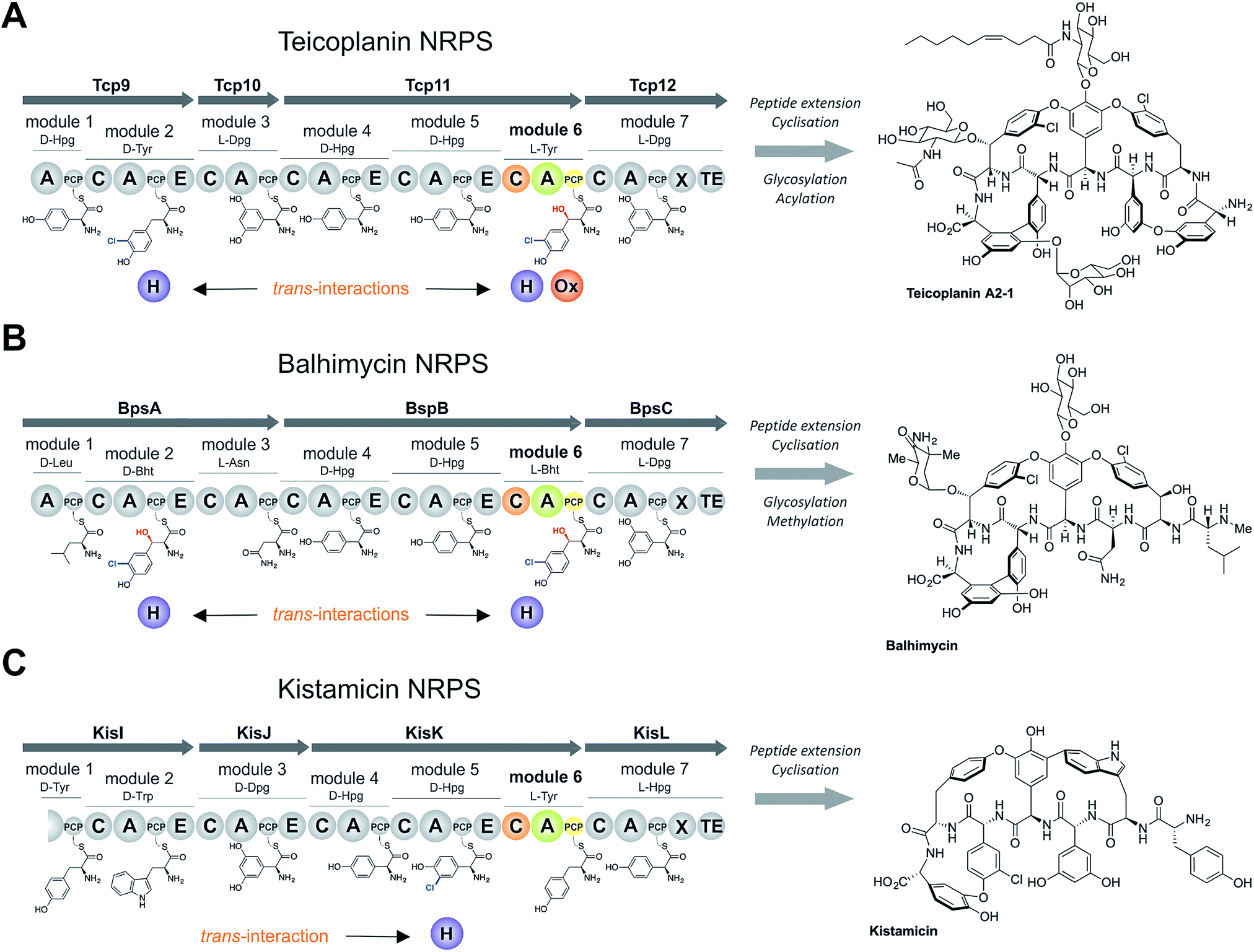 | ||
| Fig. 1 Non-ribosomal peptide biosynthesis of the glycopeptide antibiotics teicoplanin (A), the vancomycin-type GPA balhimycin (B) and kistamicin (C), concentrating on the NRPS proteins and trans-interacting enzymes. Module 6 (M6) of the NRPS machineries are shown in colour, the rest of the NRPS is indicated in grey. A – adenylation domain, C – condensation domain, PCP – peptidyl carrier protein domain, E – epimerisation domain, X – cytochrome P450 recruitment domain, TE – type I thioesterase domain H – flavin-dependent halogenase, Ox – non-heme iron oxygenase, Hpg – 4-hydroxyphenylglycine, Dpg – 3,5-dihydroxyphenylglycine, Tyr – tyrosine (1), Bht – β-hydroxytyrosine (3), Trp – tryptophan, Leu – leucine, Asn – asparagine. | ||
In this work, we describe a complex interplay between low A-domain selectivity, high C-domain selectivity and type-II TE promiscuity, which together constitutes a robust proof-reading system that ensures incorporation of the correct amino acid during NRPS-biosynthesis in a mechanism comparable to that used in tRNA-dependent biosynthesis. We further show that switching C-domains with different aminoacyl-PCP selectivity results in the formation of different peptide products, indicating how amino acid modification in trans can be selectively controlled during non-ribosomal peptide biosynthesis through the exchange of selective C-domains. Our results highlight the importance of a holistic approach to NRPS reconstitution and analysis, and that the selectivity observed in NRPS biosynthesis can stem from the interplay of a number of different mechanisms that need to be understood in concert before effective biosynthetic reengineering can be performed.
Experimental
Construct cloning
Protein expression and purification
Tcp11, modules and hybrid modules were co-expressed with the teicoplanin MbtH-like protein Tcp17; KisK module 6 and the A-domain mutant were co-expressed with the kistamicin MbtH-like protein KisM and BpsB module 6 was expressed with the related balhimycin MbtH-like protein. To co-express the proteins, E. coli BL21 (DE3) (NEB) competent cells possessing the plasmid encoding appropriate MbtH-like protein gene were co-transformed together with a plasmid encoding the NRPS module of interest. For selection, two antibiotics (kanamycin and streptomycin) were used; expression of all modules were performed in TB media, supplemented with 50 μg mL−1 kanamycin and 50 μg mL−1 streptomycin, with 10 L of media used for each protein expressed. Cells were incubated with shaking at 37 °C until the OD600nm reached 0.4–0.6. The cultures were then cooled on ice for 15 min and protein expression was induced by the addition of IPTG (0.1 mM); cultures were subsequently grown at 18 °C for 16 h. Tcp39 and Bhp were expressed without an MbtH-like protein according the same procedure using E. coli BL21 (DE3) (NEB) and TB media supplemented with 50 μg mL−1 kanamycin.All NRPS proteins were purified according the same procedure, with the exception of Tcp10 that was purified as described previously.22 Cells were harvested by centrifugation at 4000 rpm for 20 min at 18 °C. Subsequently, the cell pellet was resuspended in Ni-NTA buffer A (50 mM Tris–HCl, pH 8.0; 300 mM NaCl; 10 mM imidazole) supplemented with protease inhibitor cocktail tablets (SIGMAFAST™ Protease Inhibitor Cocktail Tablets, EDTA-Free; Sigma-Aldrich). The cells were lysed by sonication and the lysate cleared by centrifugation at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 40 min at 4 °C. The supernatant was incubated at 4 °C for 1 h with 4 mL of equilibrated (50 mM Tris–HCl, pH 8.0; 300 mM NaCl; 10 mM imidazole) Ni-NTA beads (Macherey-Nagel) with gentle shaking. After incubation, beads were washed twice with 10 column volumes of Ni-NTA buffer A (50 mM Tris–HCl, pH 8.0; 300 mM NaCl; 10 mM imidazole) before the protein was eluted with 5 column volumes of Ni-NTA buffer B (50 mM Tris–HCl, pH 8.0; 300 mM NaCl; 300 mM imidazole). Strep-tag® affinity chromatography connected to ÄKTA PURE system (GE Healthcare) was then used as a second protein purification step: prior to purification, a 5 mL StrepTrap™ HD column (GE Healthcare) was equilibrated with 4 column volumes of StrepTrap buffer A (100 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA). The Ni-NTA eluate was then loaded onto the column, the column washed with 4–6 column volumes of StrepTrap buffer A (100 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA) and bound protein then eluted with StrepTrap buffer B (100 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA; 2.5 mM desthiobiotin). The protein was then further purified by gel-filtration chromatography on a Superose™ 12 prep grade XK 26/70 (320 mL) column (GE Healthcare) or a SRT 10 SEC 300 (105 mL) column (Sepax Technologies) connected to ÄKTA PURE system (GE Healthcare). Initially, the column was equilibrated with 1 column volume of gel-filtration buffer (50 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA). Subsequently, the protein after Strep-tag® purification was loaded onto the column and the flow rate of gel-filtration buffer (50 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA) adjusted to 0.7 mL min−1 for Superose™ 12 column or to 3 mL min−1 for the SRT 10 column (see ESI Fig. S1–S3†). Elution fractions containing monomeric protein were analysed by SDS-PAGE and appropriate fractions combined and concentrated using centrifugal filter units (Amicon® Ultra-15 centrifugal filter units (depending on protein size 30 K and/or 100 K was chosen), Merck Millipore). The protein concentration was determined by measuring protein absorbance at 280 nm using a NanoDrop™ One microvolume UV-Vis spectrophotometer (Thermo Scientific). Concentrated protein was then aliquoted (30–50 μL) into chilled 1.5 mL Eppendorf tubes, flash frozen in liquid nitrogen and stored at −80 °C. For the purification of Tcp39 and Bhp, a simplified, two-step purification procedure (using Ni-NTA and gel filtration) was performed according the same portions of the procedure described above.
000 rpm for 40 min at 4 °C. The supernatant was incubated at 4 °C for 1 h with 4 mL of equilibrated (50 mM Tris–HCl, pH 8.0; 300 mM NaCl; 10 mM imidazole) Ni-NTA beads (Macherey-Nagel) with gentle shaking. After incubation, beads were washed twice with 10 column volumes of Ni-NTA buffer A (50 mM Tris–HCl, pH 8.0; 300 mM NaCl; 10 mM imidazole) before the protein was eluted with 5 column volumes of Ni-NTA buffer B (50 mM Tris–HCl, pH 8.0; 300 mM NaCl; 300 mM imidazole). Strep-tag® affinity chromatography connected to ÄKTA PURE system (GE Healthcare) was then used as a second protein purification step: prior to purification, a 5 mL StrepTrap™ HD column (GE Healthcare) was equilibrated with 4 column volumes of StrepTrap buffer A (100 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA). The Ni-NTA eluate was then loaded onto the column, the column washed with 4–6 column volumes of StrepTrap buffer A (100 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA) and bound protein then eluted with StrepTrap buffer B (100 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA; 2.5 mM desthiobiotin). The protein was then further purified by gel-filtration chromatography on a Superose™ 12 prep grade XK 26/70 (320 mL) column (GE Healthcare) or a SRT 10 SEC 300 (105 mL) column (Sepax Technologies) connected to ÄKTA PURE system (GE Healthcare). Initially, the column was equilibrated with 1 column volume of gel-filtration buffer (50 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA). Subsequently, the protein after Strep-tag® purification was loaded onto the column and the flow rate of gel-filtration buffer (50 mM Tris–HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA) adjusted to 0.7 mL min−1 for Superose™ 12 column or to 3 mL min−1 for the SRT 10 column (see ESI Fig. S1–S3†). Elution fractions containing monomeric protein were analysed by SDS-PAGE and appropriate fractions combined and concentrated using centrifugal filter units (Amicon® Ultra-15 centrifugal filter units (depending on protein size 30 K and/or 100 K was chosen), Merck Millipore). The protein concentration was determined by measuring protein absorbance at 280 nm using a NanoDrop™ One microvolume UV-Vis spectrophotometer (Thermo Scientific). Concentrated protein was then aliquoted (30–50 μL) into chilled 1.5 mL Eppendorf tubes, flash frozen in liquid nitrogen and stored at −80 °C. For the purification of Tcp39 and Bhp, a simplified, two-step purification procedure (using Ni-NTA and gel filtration) was performed according the same portions of the procedure described above.
Enzyme assays
:60 (CoA)/20 (peptidyl-CoA):1 molar ratio of the PCP domain, peptidyl-CoA/CoA and R4-4 Sfp, respectively. The reaction was performed in PCP-loading buffer (50 mM HEPES, pH 7.0; 50 mM NaCl; 10 mM MgCl2) at 30 °C for 1 h. Depending on the number of reactions to be performed, 50–400 μL final volume was used for the loading reaction. Subsequently the excess of peptidyl-CoA or CoA was removed by repeated concentration and dilution (5×) using reconstitution assay buffer (50 mM HEPES, pH 7.0; 50 mM NaCl) and centrifugal concentrators (Amicon® Ultra-0.5 mL centrifugal filters units (depending on protein size 30 K and/or 100 K was chosen), Merck Millipore). Holo-PCP constructs were then immediately used for in vitro reconstitution assays.
Amino acid synthesis
N-Chlorosuccinimide (230 mg, 1.7 mmol), N-bromosuccinimide (306 mg, 1.7 mmol) or N-iodosuccinimide (38 9 mg, 1.7 mmol) in dioxane (2 mL) was added dropwise to a suspension of tyrosine. HCl (250 mg, 1.15 mmol) in dioxane/water/AcOH (8 mL, v/v′/v′′, 1/4) at room temperature. After 15 hours, the reaction was concentrated under reduced pressure to remove the dioxane and the desired products purified using preparative RP-HPLC (gradient of ACN in water of 5–35% over 30 min). After freeze drying, characterisation of the halogenated tyrosine residues 7–11 was performed using LCMS (gradient of 5–35% over 30 min; see ESI Fig. S4–S8†) and by NMR-1H. Nuclear magnetic resonance spectra were recorded on a Bruker Avance III 400 in CD3CN/D2O (v/v′; 20/80). Spectra were consistent with literature reports (Br-Tyr (7),26 I-Tyr (8),27 3,5-dichloro-Tyr (9),28 3,5-dibromo-Tyr (10)28 and 3,5-diiodo-Tyr (11)28).Peptidyl-CoA synthesis
Peptide synthesis for tripeptide 3T-CoA, tetrapeptide 4T-CoA and pentapeptide 5T-CoA CoAs was performed manually by solid phase peptide synthesis (SPPS) (scale 0.05 mmol). 200 mg of 2-chlorotrityl chloride resin was swollen in DCM (4 mL, 30 min), washed with DMF (3×) and incubated with 5% hydrazine solution in DMF (6 mL, 2 × 30 min). The resin was washed with DMF (3x) and a solution of DMF/TEA/MeOH (7:2:1) (4 mL, 15 min) added. The first Fmoc-amino acid (0.065 mmol) was coupled overnight using COMU (0.065 mmol) and 2,6-lutidine (0.13 mmol, 0.12 M). In the second step, unreacted hydrazine moieties were capped with Boc-glycine-OH (0.15 mmol) activated with COMU (0.15 mmol) and 2,6-lutidine (0.15 mmol, 0.12 M) for 1 h. Fmoc removal was performed using 1% DBU solution (3 mL, 3 × 30 s) in DMF followed by Fmoc or Boc-amino acid coupling (0.15 mmol) with COMU (0.15 mmol) and 2,6-lutidine (0.15 mmol, 0.12 M) for 60 min. The last amino acid to be added was always Boc-protected. The cleavage of the hydrazide peptide from resin and removal of side chain protecting groups (tBu and Boc) were accomplished using TFA/TIS/H2O (95:2.5:2.5 v/v′/v′′, 5 mL) with 1.5 h shaking at room temperature. The resin was removed by filtration and washed with TFA (2×). Subsequently, the filtrate was concentrated under a N2 stream to ∼1 mL and the peptide precipitated with ice cold diethyl ether (∼8 mL), followed by centrifugation in a flame-resistant centrifuge (Spintron). The crude peptide hydrazide was then purified by preparative RP-HPLC and characterised by LCMS; these used a gradient of 5–35% ACN in 0.1% aqueous formic acid over 30 min for the tri- and tetrapeptide and 10–40% ACN in 0.1% aqueous formic acid over 30 min for the pentapeptide. The purified hydrazide peptide was dissolved in buffer 1 containing urea (6 M) and NaH2PO4 (0.2 M), pH 3 (obtained via addition of 1 M HCl) to a final concentration of 2–5 mM. The solution was cooled to −15 °C using a salt/ice bath. Subsequently, 0.5 M NaNO2 (0.95 eq.) was added and the mixture stirred for 12 min. Coenzyme A (1.3 eq.) was dissolved in buffer 1 and added to the reaction. After 15 minutes, the pH was adjusted to 6.5 using KH2PO4/K2HPO4 buffer (6:94 v/v 1 M, pH 8.0). The final peptidyl-CoA product was purified using preparative RP-HPLC; this utilised a gradient of 5–35% ACN in 0.1% aqueous formic acid over 30 min for the tri- (3T-CoA) and tetrapeptide (4T-CoA) (ESI Fig. S9 and S10†) and 10–40% ACN in 0.1% aqueous formic acid over 30 min for the pentapeptide (5T-CoA) products (ESI Fig. S11†). Characterisation of peptidyl-CoAs 3T-CoA, 4T-CoA and 5T-CoA was performed by LCMS (ESI Fig. S9–S11†).
HRMS and MS2 measurements
High resolution mass spectrometry measurements were performed on an Orbitrap Fusion mass spectrometer (Thermo) coupled online to a nano-LC (Ultimate 3000 RSLCnano; Thermo) via a nanospray source. Peptides were separated on a 50 cm reverse-phase column (Acclaim PepMap RSLC, 75 μm × 50 cm, nanoViper, C18, 2 μm, 100 Å; Thermo Scientific) after binding to a trap column (Acclaim PepMap 100, 100 μm × 2 cm, nanoViper, C18, 5 μm, 100 Å; Thermo Scientific). Elution was performed on-line with a gradient from 6% MeCN to 30% MeCN in 0.1% formic acid over 30 min at 250 nL min−1. Full scan MS was performed in the Orbitrap at 60000 nominal resolution, with targeted MS2 scans of peptides of interest acquired at 15000 nominal resolution in the Orbitrap using HCD with stepped collision energy (24 ± 5% NCE). QualBrowser (XCalibur 3.0.63, Thermo) was used to view spectra and generate extracted ion chromatograms for the singly charged species at 20 ppm. Predicted MS2 fragments were generated with MS-Product (ProteinProspector v5.22.1, UCSF) and manually assigned to spectra.
Results
The sixth tyrosine residue (Tyr-6) present in the GPAs is often highly modified, and given the differences in the modification state of this residue and the alternate pathways through which this is accomplished, we focused on the reconstitution and analysis of the enzymatic steps that generated the natural diversity at this position of the peptide (Fig. 1).14 For this purpose, we concentrated on three different GPA systems – teicoplanin (Fig. 1A), balhimycin (which shares the same peptide core as vancomycin (Fig. 1B)) and kistamicin (Fig. 1C) – with initial experiments performed using the teicoplanin system. These three GPA systems were selected because they differ in M6 residues incorporated in their structure (teicoplanin/balhimycin: 3-chloro-β-hydroxytyrosine (Cl-Bht); kistamicin: tyrosine (Tyr)) or the route in which the 3-chloro-β-hydroxytyrosine residue is biosynthesised. Furthermore, teicoplanin and balhimycin differ in the biosynthetic routes leading to incorporation of the β-hydroxyl moiety. In teicoplanin biosynthesis, two essential modifications (chlorination and hydroxylation) of this tyrosine residue are performed on M6 during the NRPS-mediated peptide biosynthesis, whilst for balhimycin biosynthesis β-hydroxytyrosine (Bht) is selected by M6 and subsequently halogenated during peptide synthesis.19,20,29,30The non-ribosomal peptide synthetase that produces the heptapeptide core of all glycopeptide antibiotics consists of 7 modules that are divided over either three (balhimycin: BpsA-C) or four (teicoplanin: Tcp9-12; kistamicin: KisI-L) separate proteins (Fig. 1).31–34 One of the challenges in assessing the incorporation of the Tyr-6 residue into GPA peptides stems from the nature of the NRPS assembly line in these systems, as the 6th NRPS module responsible for Tyr-6 incorporation is found in a trimodular protein along with the preceding modules 4 and 5 of the peptide assembly line. As the goal of our study was to understand the role of different catalytic domains on the selectivity of the NRPS for the Tyr-6 residue, we first investigated how to divide the teicoplanin M4-6 protein (Tcp11) to obtain single modules of these NRPS assembly lines in a functional state for in vitro characterisation.
Modular division of the NRPS
To study module 6 from the NRPS assembly lines of GPAs, we needed to assay the activity of this module as well as adjacent NRPS modules to determine the selectivity of each reaction occurring during peptide biosynthesis. In particular, we first required access to individual NRPS modules in order to be able to generate specific PCP-bound peptide substrates through the loading of peptidyl-CoA thioesters by promiscuous phosphopantetheinyl transferase enzymes (see Fig. 2D).35 Modules 4–6 of the teicoplanin NRPS are fused within a single polypeptide chain (Tcp11) – as is the case for all GPAs – and thus we first investigated how to divide this large protein (∼450 kDa) into separate modules to allow their individual characterization (Fig. 2). To this end, we attempted two sets of module divisions that either divided the modules between the E/C domains (i.e. C-A-PCP-E architecture, Fig. 2A and B) or C/A domains (A-PCP-E-C architecture, Fig. 2C). Initially, we investigated the use of the C-A-PCP-E module architecture (Fig. 2A and B), and determined that whilst the expression of both modules 4 and 6 was satisfactory, module 5 was extremely aggregation-prone and available only in very low yield even when modified with short N/C-terminal sequences taken from other teicoplanin NRPS proteins (ESI Fig. S1†). In contrast, the behaviour of all three modules using the alternate A-PCP-E-C architecture (Fig. 2C) was significantly improved in terms of protein quality and yield (ESI Fig. S2†). This type of modular architecture is reminiscent of the XU building blocks suggested by Bode and co-workers for the reassembly of NRPS assembly lines in vivo, although as far as we are aware this has not been investigated in vitro.36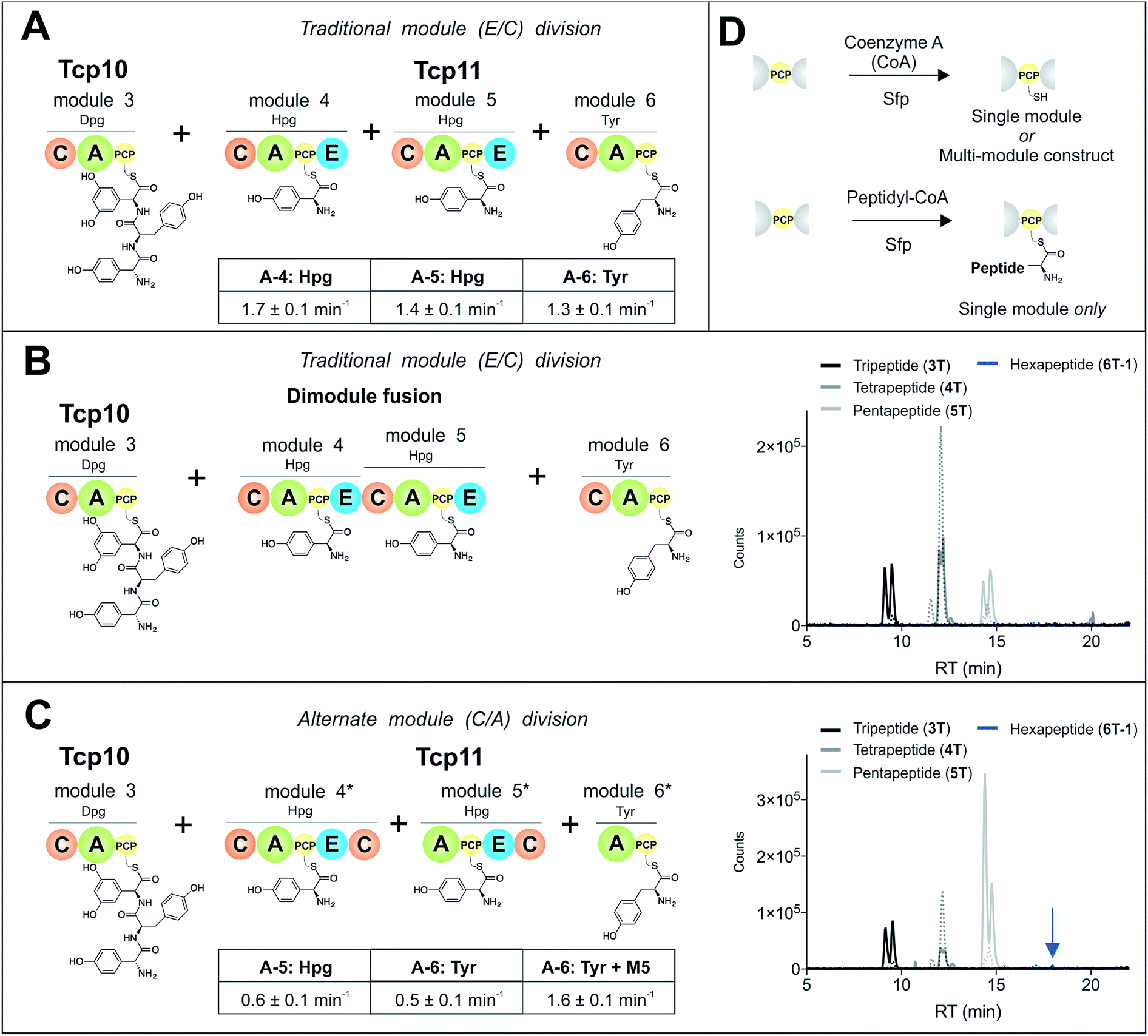 | ||
| Fig. 2 Reconstitution of peptide biosynthesis from the teicoplanin NRPS proteins Tcp10 and Tcp11, utilising two strategies to isolate individual modules 4–6 from Tcp11: either a C-A-PCP-E module architecture (A and B) or A-PCP-E-C architecture (C), together with the rationale behind the need for modularisation of the NRPS – the ability to load individual modules with peptide substrates using phosphopantetheinyl transferases (D). Rate of activation of the natural A-domain substrates for (A) and (C) were determined using a continuous, enzyme-coupled pyrophosphate detection assay; experiments performed in triplicate and standard deviation indicated. Peptide biosynthesis was reconstituted from tripeptide 3T loaded on M3, together with ATP, 4-Hpg and Tyr (1) using both the C-A-PCP-E module architecture and an M4–M5 fusion (B) or the A-PCP-E-C architecture (C). Peptide products were determined by LCMS analysis (ESI, positive mode), with solid lines indicating methylamide peptides (PCP-bound) and dashed lines indicating hydrolysed peptides (tripeptide 3T: black line; tetrapeptide 4T: dark grey line; pentapeptide 5T: light grey line; hexapeptide 6T-1: blue line). A – adenylation domain, C – condensation domain, PCP – peptidyl carrier protein domain, E – epimerisation domain, Hpg – 4-hydroxyphenylglycine, Tyr – tyrosine (1). | ||
Isolated NRPS modules are catalytically competent
Next, we assessed the activity of the adenylation domains present in these isolated modules from teicoplanin biosynthesis (Fig. 2). Here, we assessed their ability to activate and load amino acids in an ATP-dependent manner using the detection of pyrophosphate to determine the activation rate for each amino acid (Fig. 2A and C).22 For teicoplanin modules 4 and 5, these showed that 4-hydroxyphenylglycine (4-Hpg) was the preferred substrate,37 with very low activity for other structurally related amino acids (such as 3,5-Dpg and phenylglycine). The rate of activation by the C-A-PCP-E modules was faster than the A-PCP-E-C constructs (∼1.5 min−1vs. ∼0.6 min−1), which can be explained by the lack of a C-domain adjacent to the A-domain in case of the A-PCP-E-C constructs. Module 6 from the teicoplanin NRPS demonstrated the activation of Tyr (1) as had been anticipated, and here again the C-A-PCP module was ∼3× faster than the A-PCP construct (Fig. 2A and C). However, for this module it was also possible to assay the activity of module 6 in the presence of module 5, which showed that the presence of the adjacent module restored the activity of the M6 A-domain to that observed for other A-domains in this system (∼1.5 min−1). Such interactions between A- and C-domains have been observed previously and can even result in alterations in selectivity of the substrate selected – here, the effect appears restricted to the rate of A-domain substrate activation.38,39Module 6 A-domains can have broad substrate selectivity
Having determined that the teicoplanin M6 A-domain was able to activate the anticipated Tyr-residue, we next investigated the substrate selectivity of this domain (Fig. 3). Here, we utilised a range of modified Tyr/Phe residues (1–11), and showed that this module showed broad substrate selectivity, accepting Phe (5), 3-chlorotyrosine (Cl-Tyr, 2), β-(R)-OH-Tyr (Bht, 3) and even 3-chloro-β-(R)-hydroxytyrosine (Cl-Bht, 4). The alteration of the halogen atom did not have a large effect on the selectivity of this A-domain, which could activate F-Tyr (6), Br-Tyr (7) and I-Tyr (8) at similar rates (∼0.7 min−1, Fig. 3). Perhaps even more surprising was the acceptance of 3,5-di-halogenated Tyr residues (3,5-di-Cl-Tyr (9), 3,5-di-Br-Tyr (10) and 3,5-di-I-Tyr (11)). This ability to accept a broad range of different amino acids – in particular the ability to activate Tyr (1) and Phe (5) that would both be present in the cell – raised a number of intriguing questions regarding how this module could function effectively in vivo to allow the formation of a single peptide product.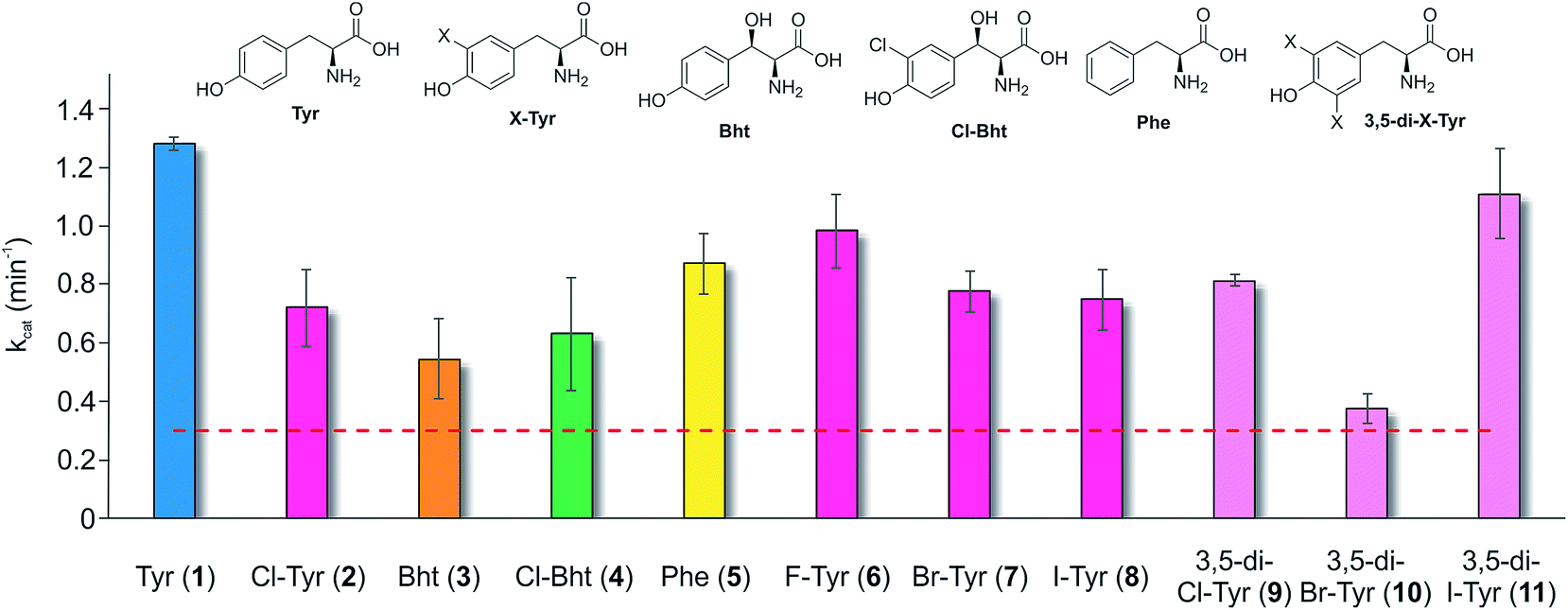 | ||
| Fig. 3 Module 6 A-domain amino acid activation rates determined for the teicoplanin NRPS. Amino acids tested as substrates include tyrosine (1, blue), halogenated tyrosine residues (2 and 6–8, magenta), di-halogenated tyrosine residues (9–11, pale pink), Bht (orange, 3), Cl-Bht (green, 4) and phenylalanine (yellow, 5). Triplicate experiments, standard deviations indicated. Dotted line indicates the method detection limit. | ||
Given the unexpectedly broad substrate tolerance of the teicoplanin M6 A-domain, we next generated the comparable M6 A-PCP constructs from balhimycin and kistamicin biosynthesis to examine the selectivity of these A-domains (Fig. 4). The results obtained for the balhimycin M6 A-domain showed that this domain also possessed a broad substrate tolerance similar to the teicoplanin system (Fig. 4A), whilst in contrast the kistamicin M6 construct was selective for Tyr (1), with no activity towards modified Tyr residues (2–4) or Phe (5) (Fig. 4B). The selectivity pocket for the M6 A-domain from kistamicin matches other Tyr specific pockets, whilst the teicoplanin/balhimycin pockets possess two or three different mutations (ESI Table S1†). Given that two residues appear to be sufficient to distinguish between permissive and selective Tyr-activating domains (ESI Table S1†), we next generated the comparable double mutant (teicoplanin-like pocket) in the kistamicin M6 A-domain. This mutant A-domain was now also able to accept Cl-Bht (4), supporting the hypothesis that these two mutations in the A-domain substrate binding pocket are sufficient to convert a Tyr-selective domain into a permissive Tyr-activating domain (Fig. 4C).
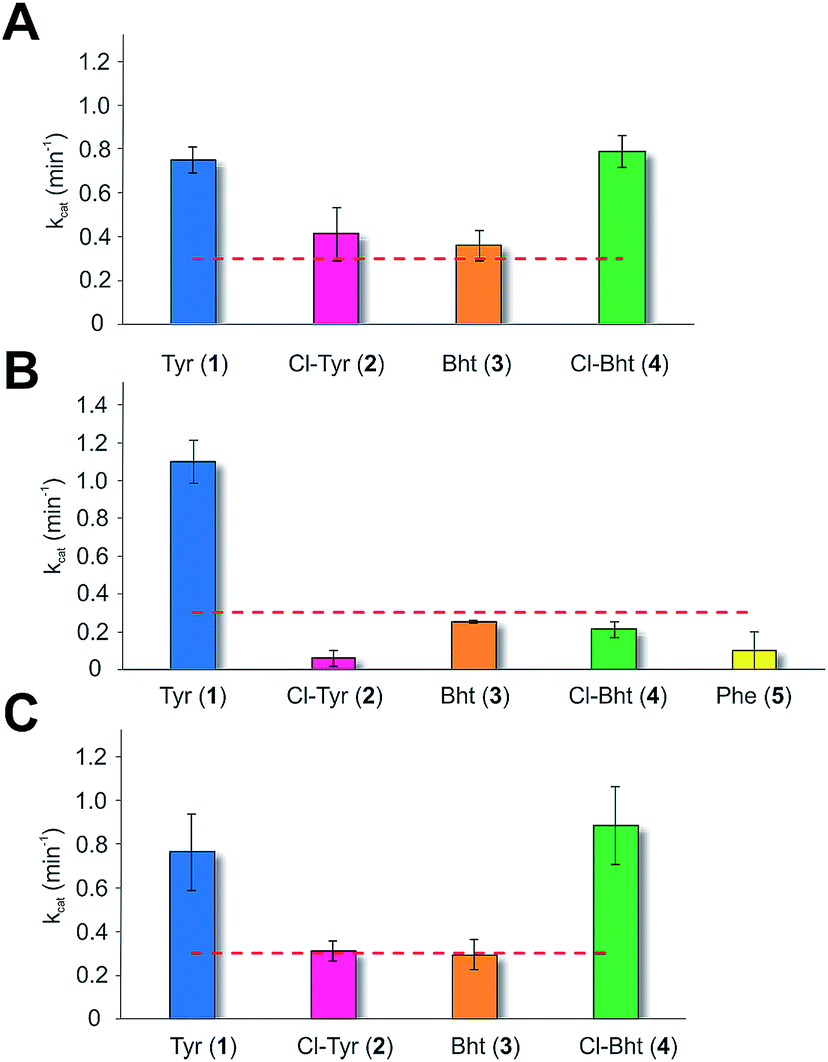 | ||
| Fig. 4 Module 6 A-domain amino acid activation rates determined for the balhimycin (A) and kistamicin (B) NRPS together with the kistamicin double mutant modelled on the teicoplanin A-domain pocket (C). Aromatic amino acids tested include tyrosine (blue, 1), 3-chlorotyrosine (magenta, 2), Bht (orange, 3), Cl-Bht (green, 4) and phenylalanine (yellow, 5). Triplicate experiments, standard deviations indicated. Dotted line indicates the method detection limit. | ||
Hexapeptide forming C-domains are specific for Tyr modification states
Given that the selectivity of the M6 A-domains from teicoplanin and balhimycin biosynthesis was not alone able to explain the specific incorporation of Cl-Bht (4) into the final products of these NRPS assembly lines, we hypothesised that the condensation domain responsible for hexapeptide formation was acting as a selectivity filter to ensure that the M6 bound Tyr (1) residue was properly modified prior to peptide formation. Thus, we next tested the selectivity of the peptide bond forming C-domains responsible for extending the pentapeptide donor substrate onto the M6 PCP-bound Tyr residue acceptor (Fig. 2B and C and 5). As a prequel to these experiments, we first tested the general ability to reconstitute peptide bond formation between the different modules of the teicoplanin NRPS using our purified constructs. To this end, we prepared synthetic peptidyl-CoAs (tripeptide (3T), tetrapeptide (4T) and pentapeptide (5T); ESI Fig. S9–S11†)40,41 and loaded these onto the separated modules 4 and 5 as well as the stand-alone module 3 (Tcp10)22 to examine C-domain activity. Following incubation of modules in an equimolar ratio with the exception of M3 (Tcp10), where we used twice as much to boost potential peptide yield, the cleavage of PCP-bound peptides was affected using methylamine, which allowed NRPS-bound peptides to be identified as their methylamide derivatives as opposed to peptides lost from the assembly line through hydrolysis. Results of these single turnover experiments showed that the C-domain activity of the M3/4 and M4/5 C-domains could be effectively reconstituted using 4-Hpg as the A-domain substrate, with activity of the A-PCP-E-C constructs (Fig. 2C) significantly improved over the C-A-PCP-E type (Fig. 2B). The major problem with the C-A-PCP-E constructs was significant levels of hydrolysis observed from module 4; this was only partially overcome by using a fusion of modules 4 and 5 (Fig. 2B), and was still present even in turnovers using full length Tcp11 (ESI Fig. S12†).Having seen that isolated modules with the A-PCP-E-C construct architecture could be used to reconstitute peptide bond formation, we next turned to the reconstitution of the M5/6 C-domain and different modified Tyr-substrates to examine the effect of different C-domain acceptor substrates on peptide bond formation by this domain (Fig. 5A). In these experiments, we could take advantage of the permissive nature of the teicoplanin M6 A-domain to load various Tyr derivatives (2–4) onto the downstream PCP domain that would normally be generated through the actions of trans-modifying enzymes on the PCP-bound Tyr residue, as neither trans-interacting enzyme could be isolated in a functional form. To this end, we performed peptide reconstitution assays for the M5/6 C-domain using Tyr (1, Fig. 5B) as well as modified Cl-Tyr (2, Fig. 5C), Bht (3, Fig. 5D) and Cl-Bht (4, Fig. 5E) residues as amino acid substrates and quantified the amount of hexapeptide produced in each case. The results of these experiments clearly showed that the activity of the M5/6 C-domain when using Tyr (1) as an acceptor substrate – whilst detectable – was greatly reduced compared to the other modules after overnight incubation (Fig. 5B). In contrast, all of the modified Tyr residues (2–4) led to hexapeptide formation after overnight incubation, showing the importance of these modifications for the catalytic efficiency of this C-domain. Examining the activity of this C-domain using shorter experiments and only M5 and M6 (Fig. 6A) showed that Cl-Bht (4) was the preferred C-domain acceptor substrate, with Bht (3) also a good acceptor substrate and Cl-Tyr (2) significantly poorer than Cl-Bht (4) or Bht (3) (Fig. 6B). These results are in agreement with our hypothesis that the M5/6 C-domain should be selective for the aminoacyl-PCP acceptor substrate: whilst tyrosine is the substrate anticipated to be loaded by the teicoplanin M6 A-domain in vivo,42 the biosynthesis of teicoplanin requires two modifications of this residue prior to peptide elongation by this C-domain. This also indicates that the mechanism of selectivity that lies behind trans-enzyme activity in this case is the selectivity of the M5/6 C-domain for the correctly modified state of the aminoacyl-PCP acceptor substrate, with the C-domain selectivity presumably serving to stall peptide bond formation with incompletely modified aminoacyl-PCP intermediates, allowing sufficient time for the trans-interacting enzymes to act on these residues.
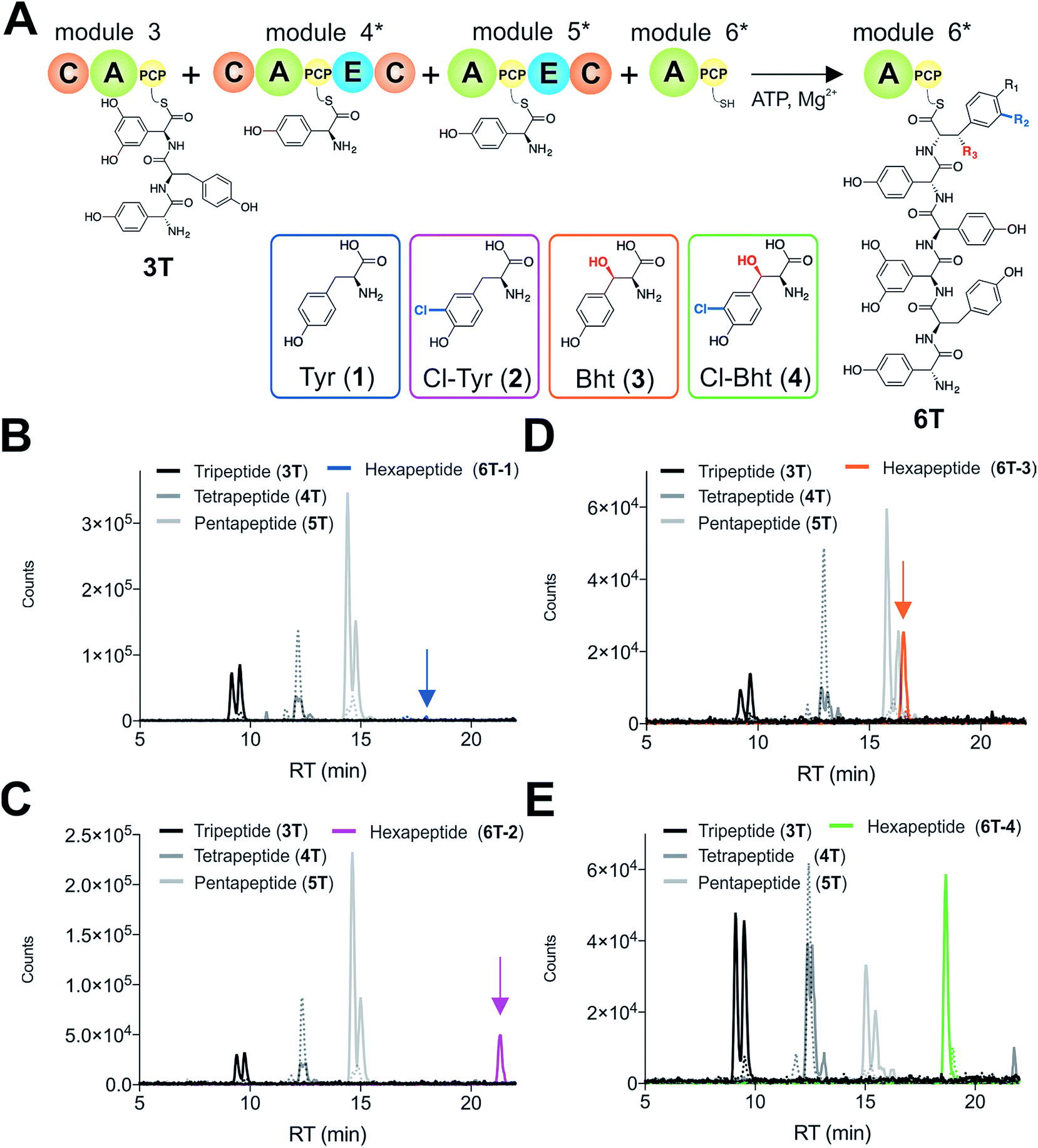 | ||
| Fig. 5 Reconstitution of tripeptide extension using the separated modules (A-PCP-E-C architecture) from teicoplanin biosynthesis M3–M6 (Tcp10/Tcp11) using different substrates for module 6 (A). Peptide biosynthesis reconstituted using ATP, tripeptide (3T)-loaded M3, 4-Hpg, plus tyrosine (1) (B), Cl-Tyr (2) (C), Bht (3) (D) and Cl-Bht (4) (E) as M6 substrates, and determined by LCMS analysis (ESI, positive mode) with solid lines indicating methylamide peptides (PCP-bound) and dashed lines indicating hydrolysed peptides (tripeptide 3T: black line; tetrapeptide 4T: dark grey line; pentapeptide 5T: light grey line; hexapeptides 6T: blue line (Tyr, 6T-1), magenta line (Cl-Tyr, 6T-2), orange line (Bht, 6T-3) or green line (Cl-Bht, 6T-4)). A – adenylation domain, C – condensation domain, PCP – peptidyl carrier protein domain, E – epimerisation domain, Tyr – tyrosine (1), Cl-Tyr – 3-chlorotyrosine (2), Bht – β-hydroxytyrosine (3), Cl-Bht – 3-chloro-β-hydroxytyrosine (4). | ||
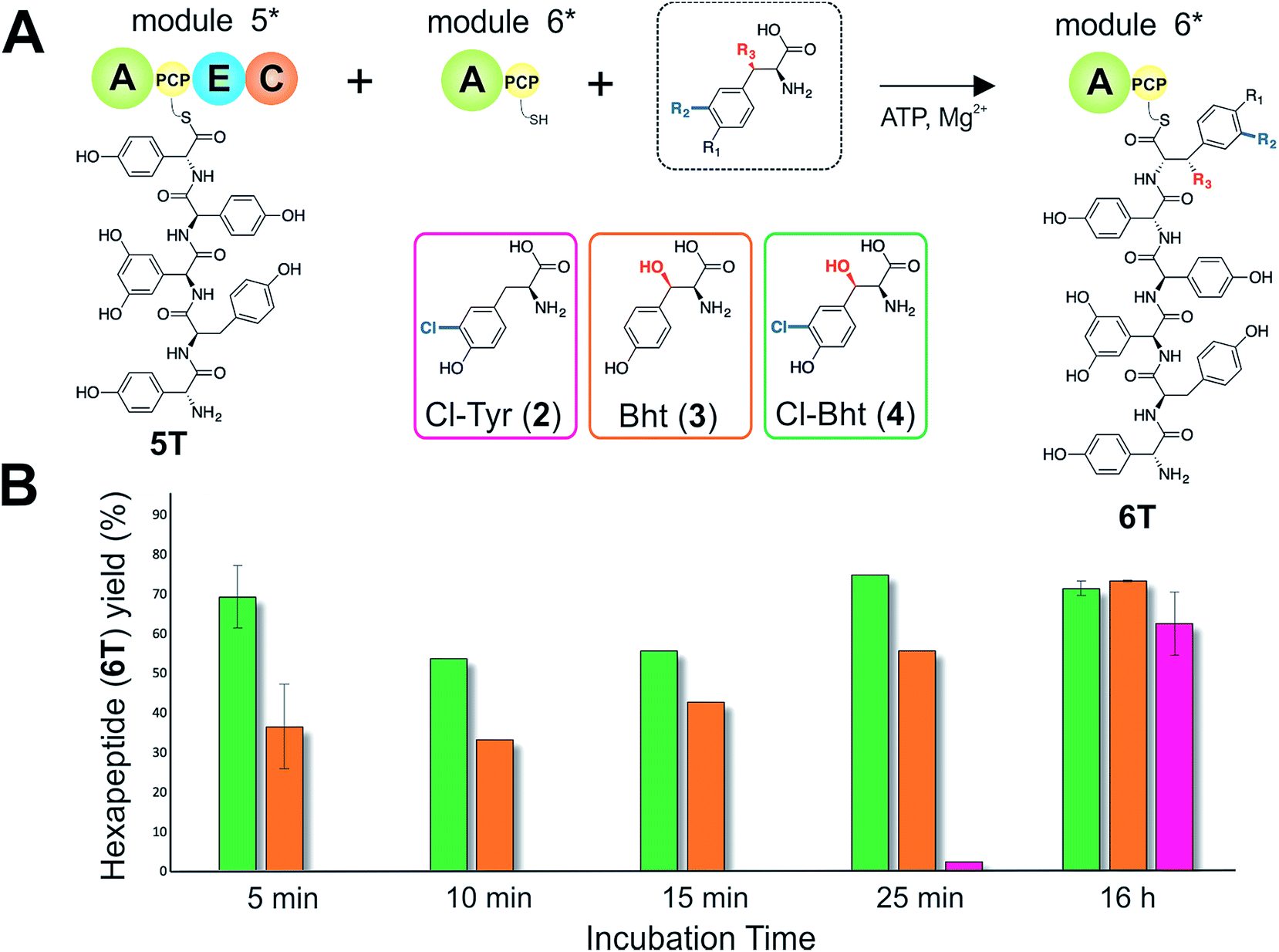 | ||
| Fig. 6 The rate of pentapeptide extension by teicoplanin module 6 using differently modified tyrosine residues as substrates (A). Hexapeptide biosynthesis reconstituted using ATP, pentapeptide (5T)-loaded M5, plus Cl-Tyr (2), Bht (3) and Cl-Bht (4) as M6 substrates, analysed by LCMS analysis (ESI, positive mode) at various time intervals (B). 5 min and 16 h experiments were performed in triplicate with the standard deviation indicated; other time points are the result of single experiments. A – adenylation domain, C – condensation domain, PCP – peptidyl carrier protein domain, E – epimerisation domain, Cl-Tyr – 3-chlorotyrosine (2), Bht – β-hydroxytyrosine (3), Cl-Bht – 3-chloro-β-hydroxytyrosine (4). | ||
NRPS assembly lines can be reprogrammed by C-domain exchange
With evidence that the teicoplanin M5/6 C-domain acts as a logic gate to ensure Tyr-6 modification in this system, we now turned our attention to the comparable C-domains from the balhimycin and kistamicin systems.32,34 Whilst we were unable to access soluble M5 constructs for either the balhimycin or kistamicin NRPS assembly lines, we were able to express a modified A-PCP-E-C teicoplanin module 5 in which the C-domain was exchanged with the equivalent kistamicin C-domain (Fig. 7A). Whilst it was disappointing to be unable to analyse the activity of the balhimycin M5/6 C-domain, the kistamicin M5/6 C-domain provides a perfect vehicle to examine C-domain selectivity for tyrosine as a substrate, and also the potential viability of altering amino acid modification in trans through C-domain exchange within the NRPS machinery. Reconstitution of this hybrid module 5 together with kistamicin module 6 clearly demonstrated the direct incorporation of Tyr (1) into the product hexapeptide, supporting the ability to control peptide extension by the exchange of C-domains exhibiting different aminoacyl-PCP specificities (Fig. 7C). In comparison, the use of teicoplanin module 6 did not lead to efficient reconstitution of hexapeptide formation, which we attribute to the importance of conserving C/A interfaces for effective NRPS reconstitution using our module division strategy. To examine the acceptance of Cl-Bht (4) by the kistamicin M5/6 C-domain, we made use of the double mutant kistamicin M6 construct in which we had altered the amino acid selectivity pocket in the adenylation domain that we had shown to be able to activate Cl-Bht (4). Using this construct, we could show that the kistamicin M5/6 C-domain was now no longer permissive for this larger, modified tyrosine residue – tyrosine was now the preferred substrate (Fig. 7B). These experiments show that Cl-Bht (4) is not accepted for peptide extension by the kistamicin M5/6 C-domain, and hence that the teicoplanin and kistamicin C-domains show altered specificities for modified Tyr residues as their acceptor substrates during GPA biosynthesis. However, these results did yet not explain how the permissive M6 A-domains present in the NRPS assembly line of both balhimycin and teicoplanin could lead to effective rates of peptide formation in the presence of amino acid substrates that were competitive for the A-domain but that would not be accepted by the C-domain.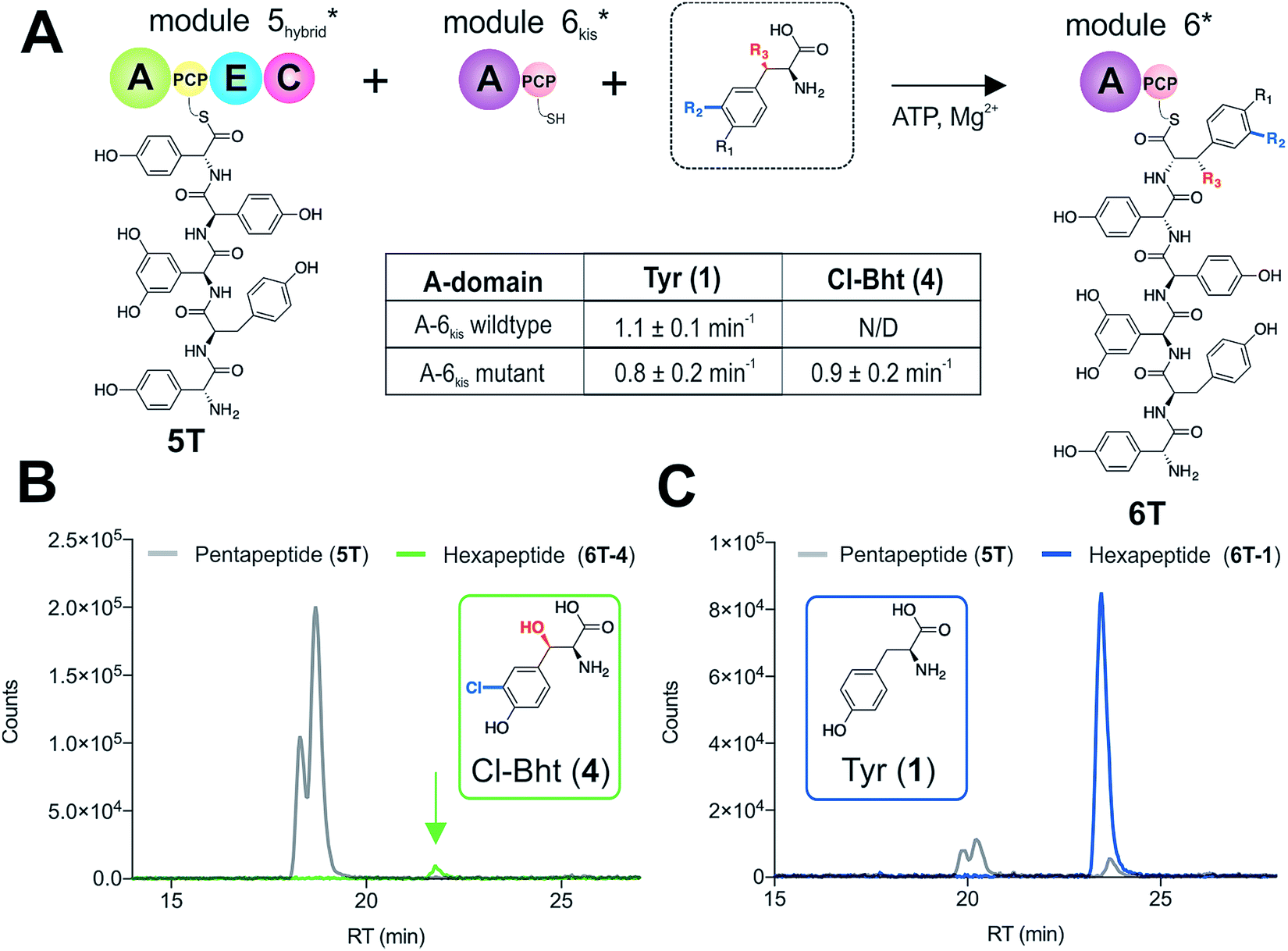 | ||
| Fig. 7 Reconstitution of pentapeptide extension by exchanging the teicoplanin Cl-Bht specific M5/6 C-domain with the Tyr-specific M5/6 C-domain from kistamicin biosynthesis (A). Peptide biosynthesis reconstituted using ATP, pentapeptide (5T)-loaded M5 hybrid, plus Cl-Bht (4) (B) and Tyr (1) (C) as M6kis substrates, and determined by LCMS analysis (ESI, positive mode) with solid lines indicating methylamide peptides (PCP-bound) peptides (pentapeptide 5T: light grey line; hexapeptides 6T: blue line (Tyr, 6T-1) or green line (Cl-Bht, 6T-4)). A – adenylation domain, C – condensation domain, PCP – peptidyl carrier protein domain, E – epimerisation domain, Tyr – tyrosine (1), Cl-Tyr – 3-chlorotyrosine (2), Bht – β-hydroxytyrosine (3), Cl-Bht – 3-chloro-β-hydroxytyrosine (4). | ||
Proofreading by the type-II thioesterase
To explore how the NRPS assembly line could function effectively in the presence of competing M6 A-domain substrates, we next turned to competitive assays (Fig. 8A) in which two different amino acid substrates that are both accepted by the M6 A-domain were included in peptide extension assays (Tyr/Cl-Bht (1/4), Fig. 8B–D; and Phe/Cl-Bht (5/4), Fig. 8F). In these competitive assays, the overall production of hexapeptide 6T-4 was reduced, which is in line with the activation of both substrates by the A-domain but with the product peptide only being formed from PCP6-bound Cl-Bht (4) (Fig. 8D and F) due to the selectivity of the M5/6 C-domain for the acceptor aminoacyl-PCP substrate. To stimulate the removal of the Phe/Tyr (5/1) residues from PCP6 we then included the Type-II TE enzyme from the teicoplanin system (Tcp39)31,43 into these assays, which we hypothesised should be active against all PCP-bound amino acid intermediates and that should be effective in removing long-lived PCP-bound amino acids that were not accepted by the M5/6 C-domain. Given that type-II TE enzymes often appear interchangeable amongst different biosynthetic pathways and lack of knowledge concerning the stoichiometry of expression within the teicoplanin system, we tested a range of different TE ratios relative to M6. At relatively low concentrations (2.5 mol%) the inclusion of this TE enzyme enabled the biosynthesis of the hexapeptide 6T-4 to proceed with the same efficiency for the Tyr/Cl-Bht (1/4) and Phe/Cl-Bht (5/4) mixtures as if only Cl-Bht (4) was present (Fig. 8E and G). Inclusion of higher ratios of the TE domain beyond 30% of the levels of M6 led to a loss of peptide extension efficiency, presumably due to excessive aminoacyl-PCP hydrolysis in these cases. However, it is important to consider that in the natural system the TE/PCP ratio would be expected to be <15 mol% at a 1:1 NRPS/TE stoichiometry, and that the need for low levels of TE in these in vitro assays stems from the fact that the majority of the NRPS machinery is absent (ESI Fig. S13†). These experiments show how the selectivity of the M5/6 C-domain for the acceptor substrate works together with a type-II TE domain to ensure that non-ribosomal peptide biosynthesis still produces a highly specific peptide product even when A-domain selectivity is limited. This is particularly relevant with the high activity of the teicoplanin M6 A-domain for Phe (5), which is naturally present in cells and would compete with Tyr (1) for incorporation into GPAs if no C-domain gating mechanism was present. We also tested whether the same effect could be observed when using the separate TE domain (Bhp) responsible for cleavage of PCP-Bht from the stand alone BpsD (A-PCP) NRPS module that produces Bht (β-hydroxytyrosine, 3) in balhimycin biosynthesis.19,30,44 In these GPA systems, Bht (3) is produced by Cytochrome P450 mediated hydroxylation of PCP-Tyr before being hydrolysed specifically through the actions of Bhp.29,44 Here, our experiments showed that Bhp was not able to replicate the proofreading function of the Tcp39 enzyme (data not shown), which fits with the selectivity of this enzyme for one specific PCP (that found in BpsD) as opposed to the PCP domains from the main peptide producing NRPS.
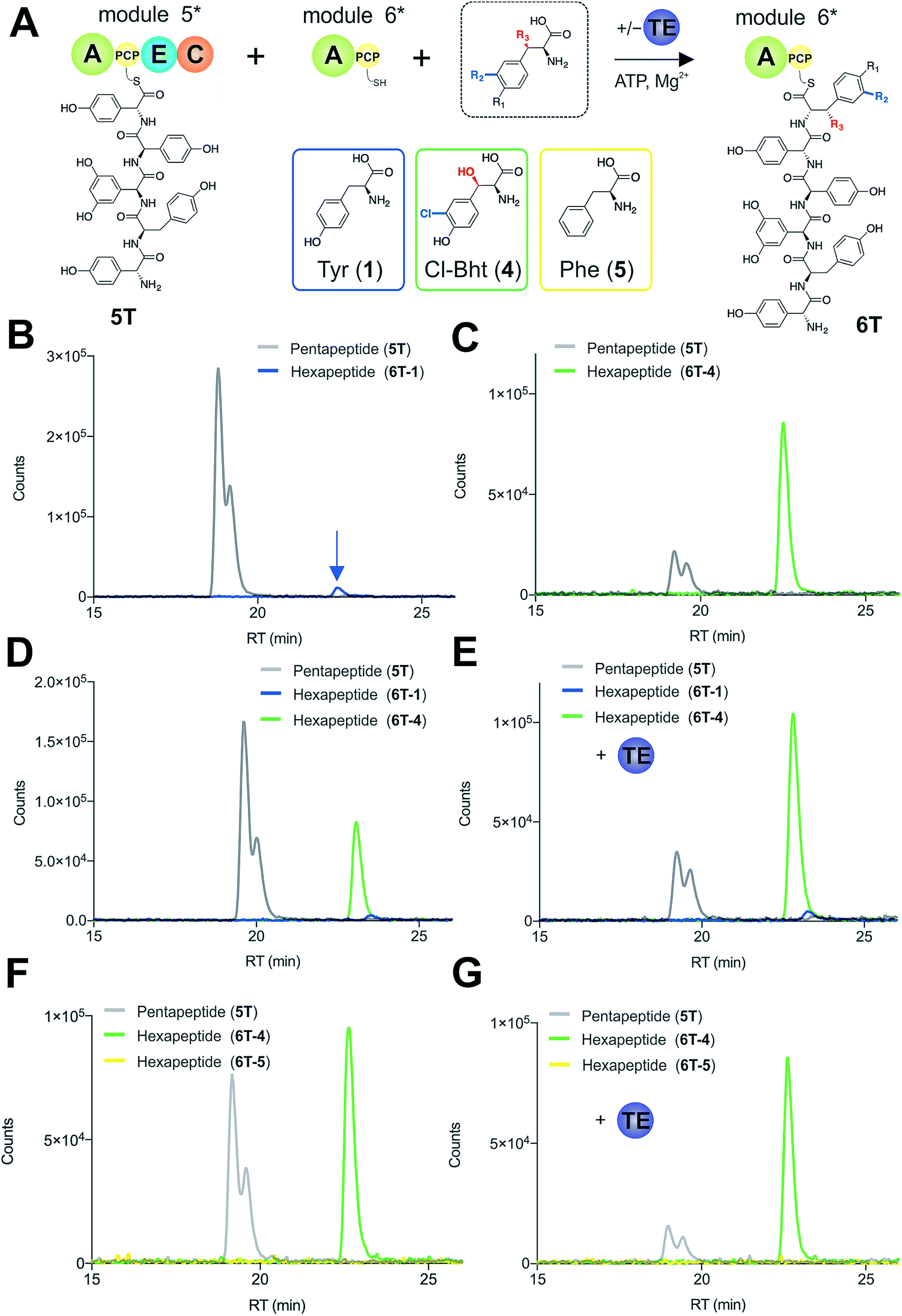 | ||
| Fig. 8 Reconstitution of pentapeptide extension coupled with the actions of a type-II TE enzyme (A). Peptide biosynthesis reconstituted using ATP, pentapeptide (5T)-loaded M5, plus different combinations of possible amino acids as M6 substrates, as determined by LCMS analysis (ESI, positive mode) with solid lines indicating methylamide peptides (PCP-bound) peptides (pentapeptide 5T: light grey line; hexapeptides 6T: blue line (Tyr, 6T-1), green line (Cl-Bht, 6T-4) or yellow line (Phe, 6T-5)). Results of peptide extension using tyrosine (1) alone (B), Cl-Bht (4) alone (C), an equimolar ratio of Tyr (1) and Cl-Bht (4) (D) as well as an equimolar ratio of Tyr (1) and Cl-Bht (4) together with the incorporation of the type-II TE enzyme Tcp39 (E). Results of peptide extension using an equimolar ratio of Phe (5) and Cl-Bht (4) (F) as well as an equimolar ratio of Phe (5) and Cl-Bht (4) together with the incorporation of the type-II TE enzyme Tcp39 shown (G). A – adenylation domain, C – condensation domain, PCP – peptidyl carrier protein domain, E – epimerisation domain, TE – type-II thioesterase, Tyr – tyrosine (1), Cl-Bht – 3-chloro-β-hydroxytyrosine (4), Phe – phenylalanine (5). | ||
Discussion
Within non-ribosomal peptide biosynthesis, one of the major issues that slows the reengineering of these systems is the complexity of the peptide biosynthesis process, including the large number of different catalytic domains that can be active in both cis and trans on PCP-bound substrates. Whilst the typical functions of the different NRPS domains and ancillary proteins are known, the effects that the specificities of multiple catalytic domains on a single step in NRPS-mediated peptide extension have not been widely investigated. This is particularly the case for the activity of condensation domains, where the classical role of these domains as stereochemical gatekeepers4,6–8 has been recently been supplemented with a number of extended catalytic functions in a variety of systems.45–48 Beyond extending the activity of C-domains away from peptide bond formation, a major question remains as to what extent these domains act as a selectivity filter for their acceptor aminoacyl-PCP substrates with regards to modification of aminoacyl-PCP domains in trans, given that the mechanism leading to the specific targeting of desired PCP-bound substrates has not previously been resolved and can lead to unexpected products after biosynthetic redesign.49 In this work, we have concentrated on understanding the pentapeptide extension step during the non-ribosomal biosynthesis of GPAs, given that there is not only significant diversity within the biosynthesis of GPAs for the modification state of the Tyr residue present on module 6 of the NRPS, but also several different mechanisms by which modification of this residue is performed.14,19,42 Most GPAs demonstrate both chlorination and hydroxylation of the M6 Tyr residue, and within teicoplanin biosynthesis this is unarguably a complex process, requiring two distinct modifications of this PCP-bound tyrosine residue. Despite the requirement for two trans-interacting enzymes to modify this PCP-bound Tyr residue, this process is also very efficient at ensuring the product GPA bears both substituents at this position. Now, through the reconstitution of pentapeptide extension in vitro, we have determined by proxy that the actions of these trans-modifying enzymes are gated by the acceptor selectivity of the M5/6 C-domain. Whilst C-domains are known to be able to enforce selectivity for their acceptor substrates,4 this is the first time to our knowledge that this selectivity has been demonstrated to be present and to control the modification of PCP-bound amino acids via the actions of trans-acting enzymes. The low acceptance of partially modified Tyr residues as well as very low levels of Tyr incorporation into hexapeptide also provides an explanation why only small amounts of such peptide products can be observed from modified strains in which the trans-modifying enzymes have been disrupted.20,21,50 Previous work has shown that the halogenation of amino acids during GPA biosynthesis occurs on aminoacyl-PCP substrates, but that the halogenase itself could not distinguish between the PCP domains whose substrates either should (PCP2, PCP6) or should not (PCP1) be chlorinated.18 The preference of the M5/6 C-domain for Bht (3) over Cl-Tyr (2) suggests that the halogenase would be expected to act before the hydroxylase to ensure effective incorporation of the Cl-Bht (4) residue in teicoplanin biosynthesis. This would further suggest that the halogenase should have higher affinity for the PCP-bound substrate than the hydroxylase, and as the selectivity is unable to be reconstituted using an isolated PCP this further suggests that the interaction interface utilised by the halogenase lies elsewhere in module 6.18 The need for C-domain scanning of the acceptor-PCP state as well as the interaction of this PCP with trans-interacting enzymes and type-II TE enzymes supports a flexible structural model of NRPS systems, and possibly one with limited higher order structure as recently reported from EM data.51 The selectivity of the teicoplanin M5/6 C-domain for Cl-Bht (4) stands in contrast to the selectivity of the comparable C-domain from the related kistamicin NRPS, which we could show is specific for PCP-bound Tyr (1) as an acceptor substrate and rejects large, modified Tyr residues. These results show that C-domains can play important roles in ensuring the correct modification of PCP-bound amino acids during NRPS biosynthesis, and that this selectivity must be taken into account when attempting to alter the production of such NRPS-produced peptides via the addition/deletion of trans-modifying enzymes (Fig. 9).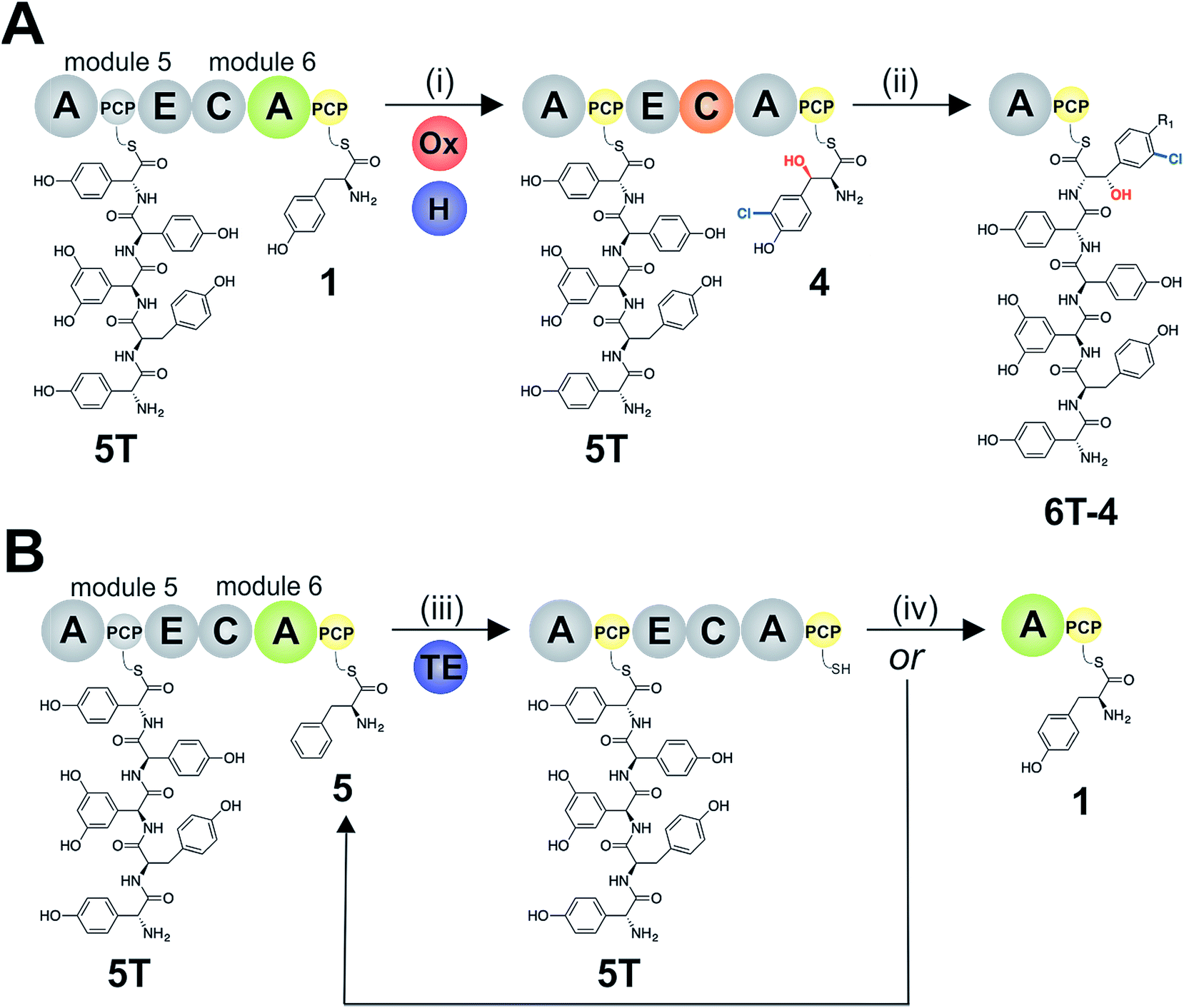 | ||
| Fig. 9 Schematic representation of A-domain and C-domain selectivity interfacing with the activity of trans-modifying enzymes and the type-II TE enzyme during teicoplanin biosynthesis. (A) Pentapeptide 5T extension commences by the activation of Tyr (1) by the M6 A-domain, which is in turn modified by trans enzymes (i) to generate PCP-bound Cl-Bht (4), the recognised M5/6 C-domain acceptor substrate; this domain then catalyses peptide extension to generate the desired PCP-bound hexapeptide 6T-4 product (ii). (B) Incorrect amino acid activation (e.g. of Phe (5)) by the M6 A-domain leads to a PCP-bound intermediate that is not accepted by the halogenase or the M5/6 C-domain, which leads to a long-lived aminoacyl-PCP intermediate that is eventually cleaved by the type-II TE enzyme (iii). At this point, another round of amino acid activation catalysed by the M5/6 A-domain can lead to the loading of the correct amino acid (Tyr, iv) and entry into the productive pathway (A, i + ii), or the loading of an incorrect substrate that then will proceed to another round of TE-mediated substrate cleavage (iii). A – adenylation domain, C – condensation domain, PCP – peptidyl carrier protein domain, E – epimerisation domain, H – flavin-dependent halogenase, Ox – non-heme iron oxygenase, TE – type-II thioesterase. | ||
Whilst the role of the C-domain in gating the modification of M6 Tyr residues via acceptor selectivity could be seen as an extension of known domain selectivity, the activity of the A-domain responsible for the loading of the Tyr-residue on M6 in the teicoplanin NRPS displays unexpectedly low selectivity for Tyr (1). As A-domains are typically considered to be the main source of amino acid selectivity during NRPS-mediated peptide biosynthesis, this result was unexpected. A-domains have been engineered to possess wide substrate acceptance,52 however this often involves substrates that are not present in the producer strains, and hence this does not pose a problem for the NRPS machinery to maintain the selectivity of the product peptide. One well known example of an A-domain with wide substrate tolerance is found in gramicidin biosynthesis, although here the tolerance of the A-domain is not limited by C-domain editing due to the similar activity of the peptide products produced in this case.53 Whilst for balhimycin type GPAs, where the low selectivity of the M6 A-domain would allow the direct activation of Bht (3) formed by an NRPS module outside of the main NRPS, in the case of teicoplanin this lack of selectivity is somewhat puzzling. Whilst the ability to switch the strictly selective Tyr A-domain from kistamicin biosynthesis into a permissive Tyr domain via two pocket mutations indicates the source of this low selectivity in the case of the teicoplanin M6 A-domain, it is harder to explain from a biosynthetic perspective. One possible explanation could be the ability of such a domain to recycle modified Tyr residues that are lost from the main NRPS via hydrolysis, although in doing so these permissive A-domains then open up the problem of NRPS stalling due to the incorporation of Phe (5). To overcome the rapid inactivation of the NRPS by activation of amino acids that are not processed by the C-domain, a type-II TE enzyme is required to remove these PCP-bound amino acids and to regenerate the NRPS to engage in another round of aminoacylation (Fig. 9).54,55 This process is reminiscent of the hydrolytic proofreading performed by tRNA synthetases, in which incorrectly loaded aminoacyl tRNAs that have been formed by are hydrolysed by editing domains, contained either within the tRNA synthetase or an external enzyme.56–59 In the case of the NRPS machinery, the low selectivity A-domain and trans-enzymes forms a range of different aminoacyl-PCP states in a manner akin to the tRNA synthetase activation site, whilst the selectivity of the C-domain combined with non-selective TE activity towards long-lived (i.e. stalled) PCP-intermediates functions as an equivalent of the tRNA editing site. Thus, the commonality of the selectivity problem faced in both ribosomal and non-ribosomal peptide biosynthesis has led to a similar solution being adopted by both biosynthetic machineries. This example of type-II TE activity shows the importance these enzymes for maintaining the selectivity of the NRPS during peptide biosynthesis, and not merely to activate “blocked” PCP domains, for example caused by the loading of acetyl-CoA during phosphopantetheinylation of apo-PCP domains.60 Indeed, this editing function of type-II TE enzymes has long been recognised in modular polyketide synthase (PKS) systems,61 and more recently also in iterative PKS systems.62 Whilst the combination of A-domain/C-domain/type-II TE activity to generate selectivity would initially appear to be a somewhat inefficient process, most of the residues found in GPAs (and indeed all residues in the case of teicoplanin and kistamicin) are derived from the shikimate pathway,15 which during the biosynthesis of GPAs would be expected to rapidly deplete the pool of non-cognate amino acids in favour of conversion into appropriate monomers for the NRPS. This process would also explain how the balhimycin NRPS would be able to effectively incorporate Bht (3) in the presence of Tyr (1), as the importance of the hydroxyl group for C-domain acceptance would cause PCP-bound Tyr residues to be long lived, and thus substrates for type-II TE enzyme mediated cleavage. During this time, the conversion of free Tyr (1) into Bht (3) via the 8th NRPS module present in this system would be expected to occur,42 which would lead to Bht (3) formation that could then be accepted by the main NRPS. This process might also explain why such GPAs utilise two Bht residues (at positions 2 and 6 of the peptide), due to the inability to maintain both Tyr (1) and Bht (3) concentrations at the same time as performing effective GPA biosynthesis.
Beyond the identification of C-domain selectivity as a mechanism to ensure effective trans-enzyme modification of aminoacyl-PCPs, in this work we could show that exchange of this M5/6 C-domain for the comparable domain from the kistamicin system (that contains an unmodified Tyr-6)34 allows this selectivity filter in NRPS biosynthesis to be controlled by switching C-domains that display different acceptor specificities. In order to assay the acceptance of Cl-Bht (4) by the kistamicin M5/6 C-domain we first had to reduce the selectivity of the kistamicin M6 A-domain for tyrosine by converting this domain into a teicoplanin-like M6 A-domain, as the protein interface between M5/6 C-domain and the teicoplanin M6 A-domain was not conducive to effective peptide biosynthesis. This highlights the importance of maintaining the C/A domain interface in order to allow the modules to reassemble when adopting the A-PCP-E-C module architecture, and is supported by structures of complete NRPS modules indicating a large C/A yet smaller A/C interface.51,63,64 Altering the A-domain selectivity of the kistamicin M6 A-domain was successfully accomplished by incorporating two mutations in the A-domain pocket (I5 to V; G7 to A), which supports the role of these mutations in the reduced substrate selectivity observed for some Tyr-activating A-domains found in GPA biosynthesis (see ESI Table S1†). These results demonstrate that the selectivity of trans-acting NRPS enzymes need not be strictly tied to PCP-recognition, but can be controlled through the gating of the modification process by the upstream C-domain. This indicates that the modification of NRPS assembly lines in vivo through the alteration of trans-interacting enzymes first requires an understanding of the inherent specificities of the C-domains within these NRPS assembly lines. Once characterised, such exchange reactions via NRPS hybridisation approaches could be used to generate the specific, desired modification of non-ribosomally produced peptides in vivo. Taken together, our results show how A-domains, C-domains and housekeeping type-II TE enzymes can act together to ensure that NRPS-mediated peptide biosynthesis retains high selectivity even when A-domains – typically thought of as highly selective domains responsible for controlling peptide sequence – display reduced specificities for monomer selection. This implies that a combination of A-and C-domain redesign should be included in reengineering paradigms for NRPS biosynthesis pathways, and once more underpins the importance of a holistic approach for studying these fascinating molecular machines that produce many of our most important medicinal agents.
Author contributions
M. J. C. designed the study. M. K. cloned constructs, expressed proteins and performed all activity assays. J. T. synthesised and purified peptidyl-CoA substrates; E. A. M. synthesised and purified halogenated amino acids. R. J. A. G and R. B. S. performed and analysed HRMS and MS2 experiments. M. J. C. wrote the manuscript and prepared the figures with input from all co-authors.Conflicts of interest
The author(s) declare no competing interests.Acknowledgements
G. Stier (BZH-Heidelberg) for fusion protein vectors and J. Yin (University of Chicago) for the R4-4 Sfp expression plasmid; M. H. Hansen for assistance with sequence alignments; G. L. Challis (University of Warwick/Monash University), J. J. De Voss (University of Queensland), E. Stegmann and N. Ziemert (University of Tübingen) for helpful discussions. This work was supported by Monash University, EMBL Australia and the National Health and Medical Research Council (APP1140619 to (M. J. C.)) and further supported under the Australian Research Council's Discovery Projects funding scheme (project number DP170102220 and DP190101272 to M. J. C.).References
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef CAS PubMed.
- T. Izoré and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 1120–1139 RSC.
- P. J. Belshaw, C. T. Walsh and T. Stachelhaus, Science, 1999, 284, 486–489 CrossRef CAS PubMed.
- A. Stanišić and H. Kries, ChemBioChem, 2019, 20, 1347–1356 CrossRef PubMed.
- L. Luo, R. M. Kohli, M. Onishi, U. Linne, M. A. Marahiel and C. T. Walsh, Biochemistry, 2002, 41, 9184–9196 CrossRef CAS PubMed.
- S. L. Clugston, S. A. Sieber, M. A. Marahiel and C. T. Walsh, Biochemistry, 2003, 42, 12095–12104 CrossRef CAS PubMed.
- K. Bloudoff, C. D. Fage, M. A. Marahiel and T. M. Schmeing, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 95–100 CrossRef CAS PubMed.
- M. E. Horsman, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- D. B. Stein, U. Linne and M. A. Marahiel, FEBS J., 2005, 272, 4506–4520 CrossRef CAS PubMed.
- S. Mori, A. H. Pang, T. A. Lundy, A. Garzan, O. V. Tsodikov and S. Garneau-Tsodikova, Nat. Chem. Biol., 2018, 14, 428–430 CrossRef CAS PubMed.
- A. Greule, J. E. Stok, J. J. De Voss and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 757–791 RSC.
- V. Agarwal, Z. D. Miles, J. M. Winter, A. S. Eustáquio, A. A. El Gamal and B. S. Moore, Chem. Rev., 2017, 117, 5619–5674 CrossRef CAS PubMed.
- G. Yim, M. N. Thaker, K. Koteva and G. Wright, J. Antibiot., 2014, 67, 31–41 CrossRef CAS PubMed.
- E. Stegmann, H.-J. Frasch and W. Wohlleben, Curr. Opin. Microbiol., 2010, 13, 595–602 CrossRef CAS PubMed.
- M. Peschke, M. Gonsior, R. D. Süssmuth and M. J. Cryle, Curr. Opin. Struct. Biol., 2016, 41, 46–53 CrossRef CAS PubMed.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed.
- T. Kittilä, C. Kittel, J. Tailhades, D. Butz, M. Schoppet, A. Büttner, R. J. A. Goode, R. B. Schittenhelm, K.-H. van Pee, R. D. Süssmuth, W. Wohlleben, M. J. Cryle and E. Stegmann, Chem. Sci., 2017, 8, 5992–6004 RSC.
- O. Puk, D. Bischoff, C. Kittel, S. Pelzer, S. Weist, E. Stegmann, R. D. Süssmuth and W. Wohlleben, J. Bacteriol., 2004, 186, 6093–6100 CrossRef CAS PubMed.
- S. Stinchi, L. Carrano, A. Lazzarini, M. Feroggio, A. Grigoletto, M. Sosio and S. Donadio, FEMS Microbiol. Lett., 2006, 256, 229–235 CrossRef CAS PubMed.
- F. Beltrametti, A. Lazzarini, C. Brunati, A. Marazzi, S. Jovetic, E. Selva and F. Marinelli, J. Antibiot., 2003, 56, 773–782 CrossRef CAS PubMed.
- T. Kittilä, M. Schoppet and M. J. Cryle, ChemBioChem, 2016, 17, 576–584 CrossRef PubMed.
- J. Bogomolovas, B. Simon, M. Sattler and G. Stier, Protein Expression Purif., 2009, 64, 16–23 CrossRef CAS PubMed.
- M. Ralser, R. Querfurth, H.-J. Warnatz, H. Lehrach, M.-L. Yaspo and S. Krobitsch, Biochem. Biophys. Res. Commun., 2006, 347, 747–751 CrossRef CAS PubMed.
- M. Sunbul, N. J. Marshall, Y. Zou, K. Zhang and J. Yin, J. Mol. Biol., 2009, 387, 883–898 CrossRef CAS PubMed.
- Y. Yokoyama, T. Yamaguchi, M. Sato, E. Kobayashi, Y. Murakami and H. Okuno, Chem. Pharm. Bull., 2006, 54, 1715–1719 CrossRef CAS PubMed.
- J. R. Cochrane, J. M. White, U. Wille and C. A. Hutton, Org. Lett., 2012, 14, 2402–2405 CrossRef CAS PubMed.
- X. Xu, R. Ge, L. Li, J. Wang, X. Lu, S. Xue, X. Chen, Z. Li and J. Bian, Eur. J. Med. Chem., 2018, 143, 1325–1344 CrossRef CAS PubMed.
- M. J. Cryle, A. Meinhart and I. Schlichting, J. Biol. Chem., 2010, 285, 24562–24574 CrossRef CAS PubMed.
- O. Puk, P. Huber, D. Bischoff, J. Recktenwald, G. Jung, R. D. Süssmuth, K.-H. van Pée, W. Wohlleben and S. Pelzer, Chem. Biol., 2002, 9, 225–235 CrossRef CAS PubMed.
- M. Sosio, H. Kloosterman, A. Bianchi, P. de Vreugd, L. Dijkhuizen and S. Donadio, Microbiology, 2004, 150, 95–102 CrossRef CAS PubMed.
- S. Pelzer, R. Sussmuth, D. Heckmann, J. Recktenwald, P. Huber, G. Jung and W. Wohlleben, Antimicrob. Agents Chemother., 1999, 43, 1565–1573 CrossRef CAS PubMed.
- B. Nazari, C. C. Forneris, M. I. Gibson, K. Moon, K. R. Schramma and M. R. Seyedsayamdost, MedChemComm, 2017, 8, 780–788 RSC.
- A. Greule, T. Izoré, D. Iftime, J. Tailhades, M. Schoppet, Y. Zhao, M. Peschke, I. Ahmed, A. Kulik, M. Adamek, R. J. A. Goode, R. B. Schittenhelm, J. A. Kaczmarski, C. J. Jackson, N. Ziemert, E. H. Krenske, J. J. De Voss, E. Stegmann and M. J. Cryle, Nat. Commun., 2019, 10, 2613 CrossRef PubMed.
- J. Yin, A. J. Lin, D. E. Golan and C. T. Walsh, Nat. Protoc., 2006, 1, 280 CrossRef CAS PubMed.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C.-P. Niesert and H. B. Bode, Nat. Chem., 2017, 10, 275 CrossRef PubMed.
- R. S. Al Toma, C. Brieke, M. J. Cryle and R. D. Suessmuth, Nat. Prod. Rep., 2015, 32, 1207–1235 RSC.
- S. Meyer, J.-C. Kehr, A. Mainz, D. Dehm, D. Petras, R. D. Süssmuth and E. Dittmann, Cell Chem. Biol., 2016, 23, 462–471 CrossRef CAS PubMed.
- R. Li, R. A. Oliver and C. A. Townsend, Cell Chem. Biol., 2017, 24, 24–34 CrossRef CAS PubMed.
- J. Tailhades, M. Schoppet, A. Greule, M. Peschke, C. Brieke and M. J. Cryle, Chem. Commun., 2018, 54, 2146–2149 RSC.
- C. Brieke, V. Kratzig, K. Haslinger, A. Winkler and M. J. Cryle, Org. Biomol. Chem., 2015, 13, 2012–2021 RSC.
- J. Recktenwald, R. Shawky, O. Puk, F. Pfennig, U. Keller, W. Wohlleben and S. Pelzer, Microbiology, 2002, 148, 1105–1118 CrossRef CAS PubMed.
- O. Yushchuk, B. Ostash, T. H. Pham, A. Luzhetskyy, V. Fedorenko, A. W. Truman and L. Horbal, ACS Chem. Biol., 2016, 11, 2254–2264 CrossRef CAS PubMed.
- S. Mulyani, E. Egel, C. Kittel, S. Turkanovic, W. Wohlleben, R. D. Süssmuth and K.-H. van Pée, ChemBioChem, 2010, 11, 266–271 CrossRef CAS PubMed.
- N. M. Gaudelli, D. H. Long and C. A. Townsend, Nature, 2015, 520, 383–387 CrossRef CAS PubMed.
- J. B. Patteson, Z. D. Dunn and B. Li, Angew. Chem., Int. Ed., 2018, 57, 6780–6785 CrossRef CAS PubMed.
- T. Izoré, J. Tailhades, M. H. Hansen, J. A. Kaczmarski, C. J. Jackson and M. J. Cryle, Proc. Natl. Acad. Sci., 2019, 116, 2913–2918 CrossRef PubMed.
- B. J. C. Law, Y. Zhuo, M. Winn, D. Francis, Y. Zhang, M. Samborskyy, A. Murphy, L. Ren, P. F. Leadlay and J. Micklefield, Nat. Catal., 2018, 1, 977–984 CrossRef CAS.
- X. Yin, Y. Chen, L. Zhang, Y. Wang and T. M. Zabriskie, J. Nat. Prod., 2010, 73, 583–589 CrossRef CAS PubMed.
- M. Schoppet, M. Peschke, A. Kirchberg, V. Wiebach, R. D. Süssmuth, E. Stegmann and M. J. Cryle, Chem. Sci., 2019, 10, 118–133 RSC.
- M. J. Tarry, A. S. Haque, K. H. Bui and T. M. Schmeing, Structure, 2017, 25, 783–793 CrossRef CAS PubMed.
- F. Ishikawa, A. Miyanaga, H. Kitayama, S. Nakamura, I. Nakanishi, F. Kudo, T. Eguchi and G. Tanabe, Angew. Chem., Int. Ed., 2019, 58, 6906–6910 CrossRef CAS PubMed.
- N. Kessler, H. Schuhmann, S. Morneweg, U. Linne and M. A. Marahiel, J. Biol. Chem., 2004, 279, 7413–7419 CrossRef CAS PubMed.
- E. Yeh, R. M. Kohli, S. D. Bruner and C. T. Walsh, ChemBioChem, 2004, 5, 1290–1293 CrossRef CAS PubMed.
- D. Schwarzer, H. D. Mootz, U. Linne and M. A. Marahiel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14083–14088 CrossRef CAS PubMed.
- O. Nureki, D. G. Vassylyev, M. Tateno, A. Shimada, T. Nakama, S. Fukai, M. Konno, T. L. Hendrickson, P. Schimmel and S. Yokoyama, Science, 1998, 280, 578–582 CrossRef CAS PubMed.
- E. Schmidt and P. Schimmel, Science, 1994, 264, 265–267 CrossRef CAS PubMed.
- T. Hussain, V. Kamarthapu, S. P. Kruparani, M. V. Deshmukh and R. Sankaranarayanan, Proc. Natl. Acad. Sci., 2010, 107, 22117–22121 CrossRef CAS PubMed.
- J. Ling, N. Reynolds and M. Ibba, Annu. Rev. Microbiol., 2009, 63, 61–78 CrossRef CAS PubMed.
- M. Kotowska and K. Pawlik, Appl. Microbiol. Biotechnol., 2014, 98, 7735–7746 CrossRef CAS PubMed.
- M. L. Heathcote, J. Staunton and P. F. Leadlay, Chem. Biol., 2001, 8, 207–220 CrossRef CAS PubMed.
- P. A. Storm and C. A. Townsend, Chem. Commun., 2018, 54, 50–53 RSC.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. Leigh Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef CAS PubMed.
- A. Tanovic, S. A. Samel, L.-O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03678d |
| This journal is © The Royal Society of Chemistry 2019 |