
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated robotic discovery of type II porous liquids†
Rachel J.
Kearsey
,
Ben M.
Alston
,
Michael E.
Briggs
,
Rebecca L.
Greenaway
 * and
Andrew I.
Cooper
*
* and
Andrew I.
Cooper
*
Department of Chemistry and Materials Innovation Factory, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: aicooper@liverpool.ac.uk; rebecca.greenaway@liverpool.ac.uk
First published on 28th August 2019
Abstract
Porous liquids are an emerging class of materials and to date little is known about how to best design their properties. For example, bulky solvents are required that are size-excluded from the pores in the liquid, along with high concentrations of the porous component, but both of these factors may also contribute to higher viscosities, which are undesirable. Hence, the inherent multivariate nature of porous liquids makes them amenable to high-throughput optimisation strategies. Here we develop a high-throughput robotic workflow, encompassing the synthesis, characterisation and property testing of highly-soluble, vertex-disordered porous organic cages dissolved in a range of cavity-excluded solvents. As a result, we identified 29 cage–solvent combinations that combine both higher cage-cavity concentrations and more acceptable carrier solvents than the best previous examples. The most soluble materials gave three times the pore concentration of the best previously reported scrambled cage porous liquid, as demonstrated by increased gas uptake. We were also able to explore alternative methods for gas capture and release, including liberation of the gas by increasing the temperature. We also found that porous liquids can form gels at higher concentrations, trapping the gas in the pores, which could have potential applications in gas storage and transportation.
Introduction
Porous solids have a plethora of applications, for example in separations and in catalysis, but they do not flow. The development of flowable porous materials could create new opportunities: for example, in separation processes where the porous medium is flowed from a cold zone to a hot zone, thus avoiding more energy-intensive operations such as temperature swing adsorption/desorption.James et al. first proposed the idea of ‘porous liquids’ in 2007.1 These materials were defined as liquids with permanent intrinsic porosity within the constituent molecules, as opposed to the transient extrinsic cavities found in conventional liquids. Three different types of porous liquids were proposed. Type I porous liquids were defined as neat liquids where the molecules themselves contain rigid intrinsic cavities. Type II and III liquids comprise a porous substance either dissolved (Type II) or dispersed (Type III) in a cavity-excluded solvent (Fig. 1a).1 When they were first proposed in 2007, these materials were hypothetical. Even now, there are only a handful of examples of porous liquids,2–6 many of which are based on porous organic cages (POCs).7 POCs are discrete molecules that contain permanent cavities accessible through windows,7 which makes them useful building blocks for porous liquids, either by rendering them low melting (Type I),8,9 or by dissolving them in size-excluded solvents (Type II).10,11 Early efforts here involved decorating the vertices of a POC with alkyl functionalities to promote melting, with the aim of making a Type I porous liquid.8 While the melting point was reduced with increasing alkyl chain length, so was the porosity—probably because of the occupation of cage cavities by the alkyl chains on neighbouring cages.8,9
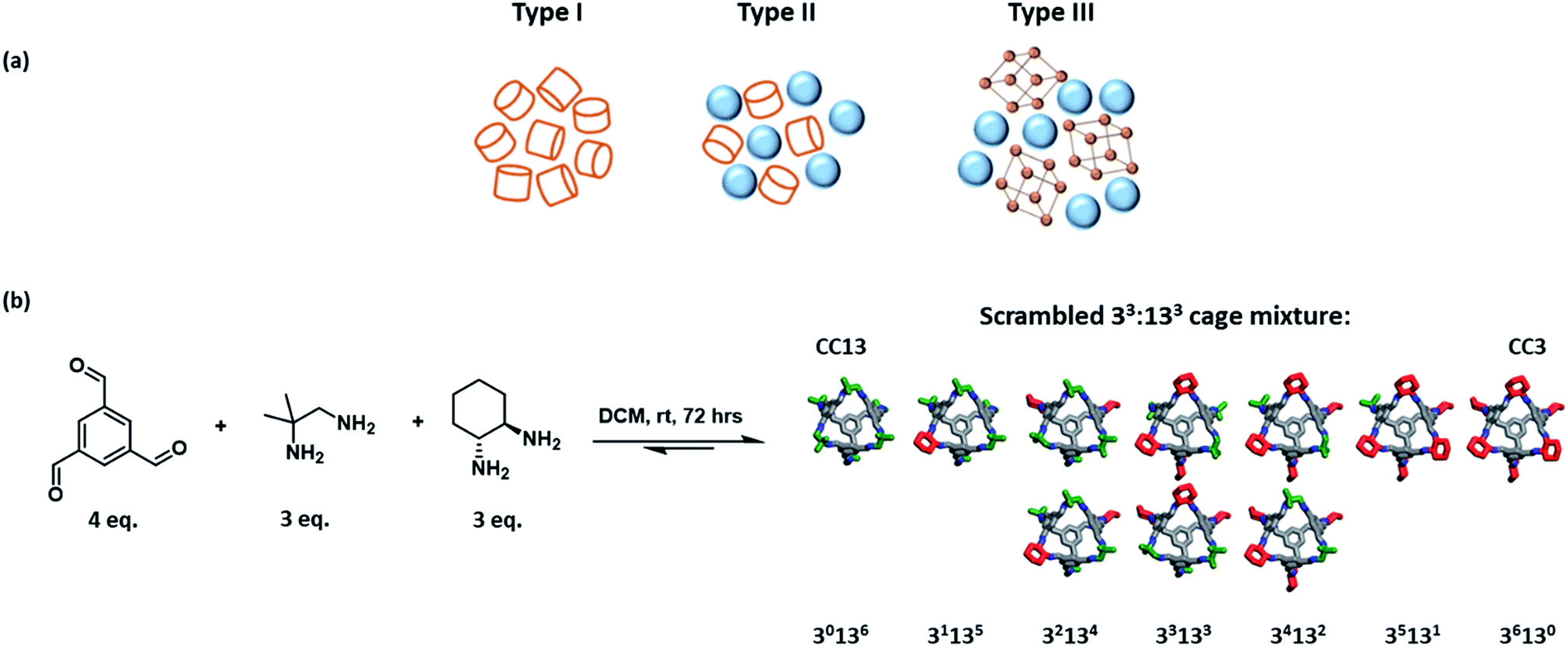 | ||
| Fig. 1 (a) The three different classes of permanently porous liquids: Type I – a neat liquid containing molecules with rigid intrinsic cavities; Type II – a porous material dissolved in a cavity-excluded solvent; Type III – a porous material dispersed in a cavity-excluded solvent; (b) synthesis of a highly-soluble scrambled 33:133 cage mixture: relative positional isomers present in the scrambled mixture are shown, with cyclohexane vertices in red, dimethyl vertices in green, and hydrogens omitted for clarity. | ||
A POC with tethered crown-ether functionality was the basis for the first Type II porous liquid; this molecule was highly soluble in a cavity-excluded solvent, 15-crown-5.10 However, the resulting liquid was somewhat viscous and the cage was prepared by a difficult six-step synthesis, which is hard to scale up. An alternative strategy was to use “dynamic covalent scrambling”. First pioneered in this context by Jiang et al.,12 scrambling greatly improved the solubility of POCs by creating cage mixtures in high-yielding, one-step syntheses that cannot crystallise because of their disorder. Specifically, two porous organic cages, CC3 and CC13, were scrambled in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture (designated as a 33:133 cage; Fig. 1b) and this mixture had good solubility in perchloropropene (PCP), forming a Type II scrambled porous liquid.10,11 While this porous liquid was much more scalable, the use of PCP is problematic. PCP is toxic, has limited commercial availability, and its associated vapour pressure makes the measurement of gas uptakes more challenging. Hence, there is a need for more benign solvents, ideally with lower vapour pressures, that still promote high cage solubilities to create a high density of pore cavities in the liquid. The solvent must, of course, be excluded from the cage cavities. Ideally, the resulting solution should also have relatively low viscosity to facilitate mass transport and handling; for example, if the liquid needs to be pumped around a system for gas separations. This introduces the considerable challenge of identifying cage–solvent combinations that meet multiple criteria, which may be orthogonal: for example, bulky, excluded solvents might also give higher viscosities. In this study, we use automated robotic methods coupled with knowledge-based design to more efficiently explore the large potential materials space.
:3 mixture (designated as a 33:133 cage; Fig. 1b) and this mixture had good solubility in perchloropropene (PCP), forming a Type II scrambled porous liquid.10,11 While this porous liquid was much more scalable, the use of PCP is problematic. PCP is toxic, has limited commercial availability, and its associated vapour pressure makes the measurement of gas uptakes more challenging. Hence, there is a need for more benign solvents, ideally with lower vapour pressures, that still promote high cage solubilities to create a high density of pore cavities in the liquid. The solvent must, of course, be excluded from the cage cavities. Ideally, the resulting solution should also have relatively low viscosity to facilitate mass transport and handling; for example, if the liquid needs to be pumped around a system for gas separations. This introduces the considerable challenge of identifying cage–solvent combinations that meet multiple criteria, which may be orthogonal: for example, bulky, excluded solvents might also give higher viscosities. In this study, we use automated robotic methods coupled with knowledge-based design to more efficiently explore the large potential materials space.
High-throughput synthesis and physiochemical testing are common in areas such as drug discovery13–15 and such methods are increasingly being applied in functional materials chemistry.16–21 For example, we recently reported a high-throughput workflow that enabled a large library of organic imine cages to be synthesised and characterised from binary mixtures of precursors.22 Scrambled cages are typically formed using three different precursors under similar reaction conditions,12 which are translatable onto automated synthesis platforms. Solubility testing has been demonstrated using automated methodology in other areas,14 but this has hitherto not been applied in the discovery of porous liquids. Here, we demonstrate the potential of automation to accelerate the discovery of next-generation Type II porous liquids. This led to the discovery of non-chlorinated replacements for PCP and new Type II porous liquids with even higher levels of porosity.
Results and discussion
Development of high-throughput scrambled cage library
Our first step was to produce a library of structurally diverse scrambled porous organic cage molecules for solubility screening. Typically, the cages are synthesised in a one-pot imine condensation reaction.10–12 Our previous success with high-throughput automation for organic cages derived from two precursors22 allowed us to develop an analogous methodology for three-component scrambled cage mixtures. First, the choice of precursors was considered. The reversibility associated with imine chemistry allows for statistical scrambling, but there is also potential for other outcomes, such as social self-sorting into a new cage species, or narcissistic self-sorting into separate binary ‘parent’ cages.23–26 POCs have also been shown to self-assemble through chiral recognition, with the formation of homochiral cages being possible by using different diastereomeric forms of the diamine precursor.23,27–30 Chiral recognition in cage racemates has also been found to greatly decrease solubility.27,28,31 More generally, having two diamines with unknown relative reactivities means it can be difficult to predict the composition of the final products.To simplify the search space, we selected achiral 1,2-diamino-2-methyl-propane (amine A, Fig. 2) as one of the scrambling partners because it reliably forms CC13; that is, our most soluble unscrambled cage to date.32 Amine A has also been shown to scramble successfully with other vicinal diamines.10 The other scrambling partners (amines B–K, Fig. 2) were selected based on their structural diversity and their ease of access; that is, we focused on diamines that were either commercially available or that were easily synthesised on gram scales, such that we could feed the high-throughput screens with sufficient starting material. POCs are often synthesised under high dilution conditions to avoid the formation of kinetic side-products and impurities, such as polymers or oligomers.7,33 However, there is a limit to the maximum solvent volume (60 mL) that can be accommodated in a single reaction vessel on our automated synthesis platforms. We therefore carried out preliminary studies to identify the highest viable reaction concentration that still avoids formation of polymeric by-products, to maximize the amount of material that we can access (ESI Section 3.1†). We found that reaction concentrations could be increased three-fold compared to those used in our original porous liquid scrambled cage synthesis.12 As a result, this higher concentration was used in subsequent high-throughput screens where possible. Using a robotic synthesis platform (Chemspeed Accelerator SLT-100), 1,3,5-triformylbenzene (TFB), amine A, and the different partner diamines (amines B–K) were combined in varying stoichiometric ratios to give a total of 61 possible scrambled cage mixtures (Fig. 2). These reactions were vortexed at ambient temperature for 3 days to ensure that the reactions had enough time to equilibrate; for full synthetic and purification details, see ESI Section 3.† After a simple purification to remove any insoluble polymer or triethylamine hydrochloride salts by filtration, the isolated materials were analysed to determine which reactions had been successful and, hence, could be used in a solubility screen. 1H NMR spectroscopy and HPLC data were used to identify reactions that resulted in the formation of scrambled cage mixtures. The HPLC spectra showed that the ratio of the cage distributions were governed by the diamine equivalents used in the feed ratio, as observed previously.12 High-resolution mass spectrometry (HRMS) was then used to confirm which scrambled species had formed in each mixture, and powder X-ray diffraction (PXRD) to determine the crystallinity of the isolated material, with the majority being amorphous as desired; for full characterisation data, see Table S5 and Fig. S8–S17.† A reaction was deemed successful if it afforded sufficient material for solubility testing (≥0.2 g) of reasonable purity (≥80%), based on the 1H NMR spectroscopic data. Using this criteria, 44 combinations gave sufficient conversion to both the parent cages and led to scrambled cage mixtures; that is, a 72% overall success rate across the library (Fig. 2).
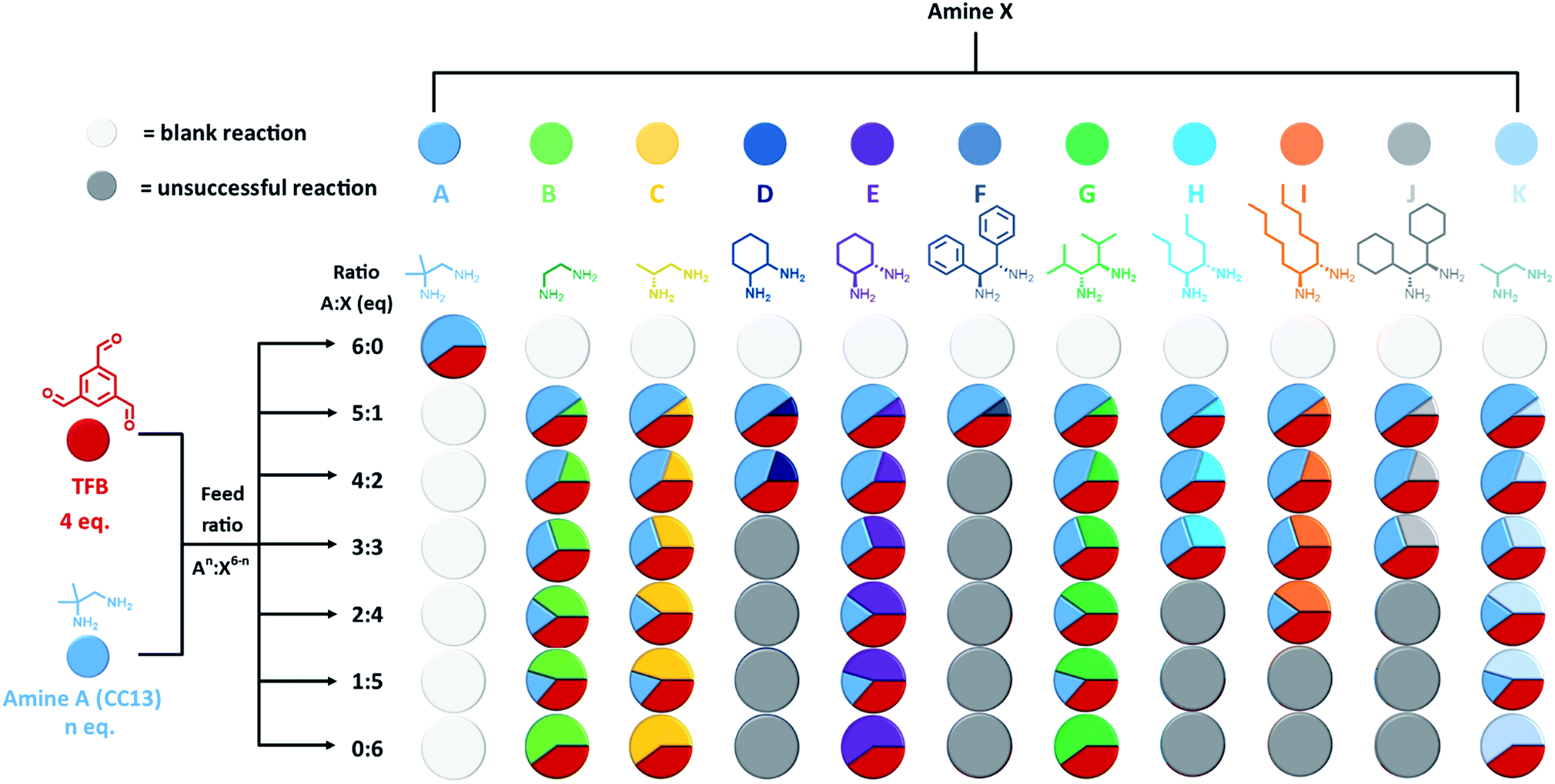 | ||
| Fig. 2 Graphical representation of the scrambled cage mixtures targeted in the high-throughput synthetic screen: the feed ratio of precursors added to each reaction vessel is defined by the diamine combination, An:X6−n (where A is 1,2-diamino-2-methyl-propane, X represents the partner diamine (B–K), and n is the number of equivalents). Unsuccessful reactions are marked with a grey circle. | ||
Choosing suitable size-excluded solvents
The next challenge was to choose appropriate size-excluded solvents that might form Type II porous liquids. Here, knowledge on how to identify appropriate solvent/scrambled cage pairings was limited. The large range of potential solvents available meant that random strategies were undesirable; hence, a more systematic, tiered approach was needed to increase the likelihood of discovering hits. To do this, a preliminary screen was first carried out. We used the known scrambled cage 33:133 (A3:E3 from our high-throughput synthetic screen in this study) in a range of common laboratory solvents to determine if any general solubility trends emerged that might inform the choice of bulkier, size-excluded analogues. This initial test suggested that certain halogenated and aromatic solvents might be suitable to form porous liquids, with 33:133 having high solubility (≥250 mg mL−1) in solvents such as chloroform, trifluoroethanol, and anisole (see Table S6 and Fig. S18† for full data set). These findings were then translated into bulkier, less-common solvent analogues, and the scope of the search was narrowed further: we only included solvents that were liquid at room temperature with a relatively high molecular weight (>100 g mol−1, to favour cavity exclusion) and an elevated boiling point (>150 °C). Our main focus was on finding halogenated and (particularly) non-halogenated aromatic solvents. Applying these selection criteria led to 14 bulky solvents (Fig. 3a); the solubility of 33:133 was tested in these solvents with a minimum threshold loading of 50 mg mL−1: any solvents that failed to meet this solubility threshold were not investigated further (Table S7†). From this screen, five bulky solvents stood out that could dissolve 33:133 at ≥300 mg mL−1 (Fig. 3b); that is, a substantially higher concentration than our first Type II porous liquid that used this scrambled cage mixture (200 mg mL−1 in PCP).10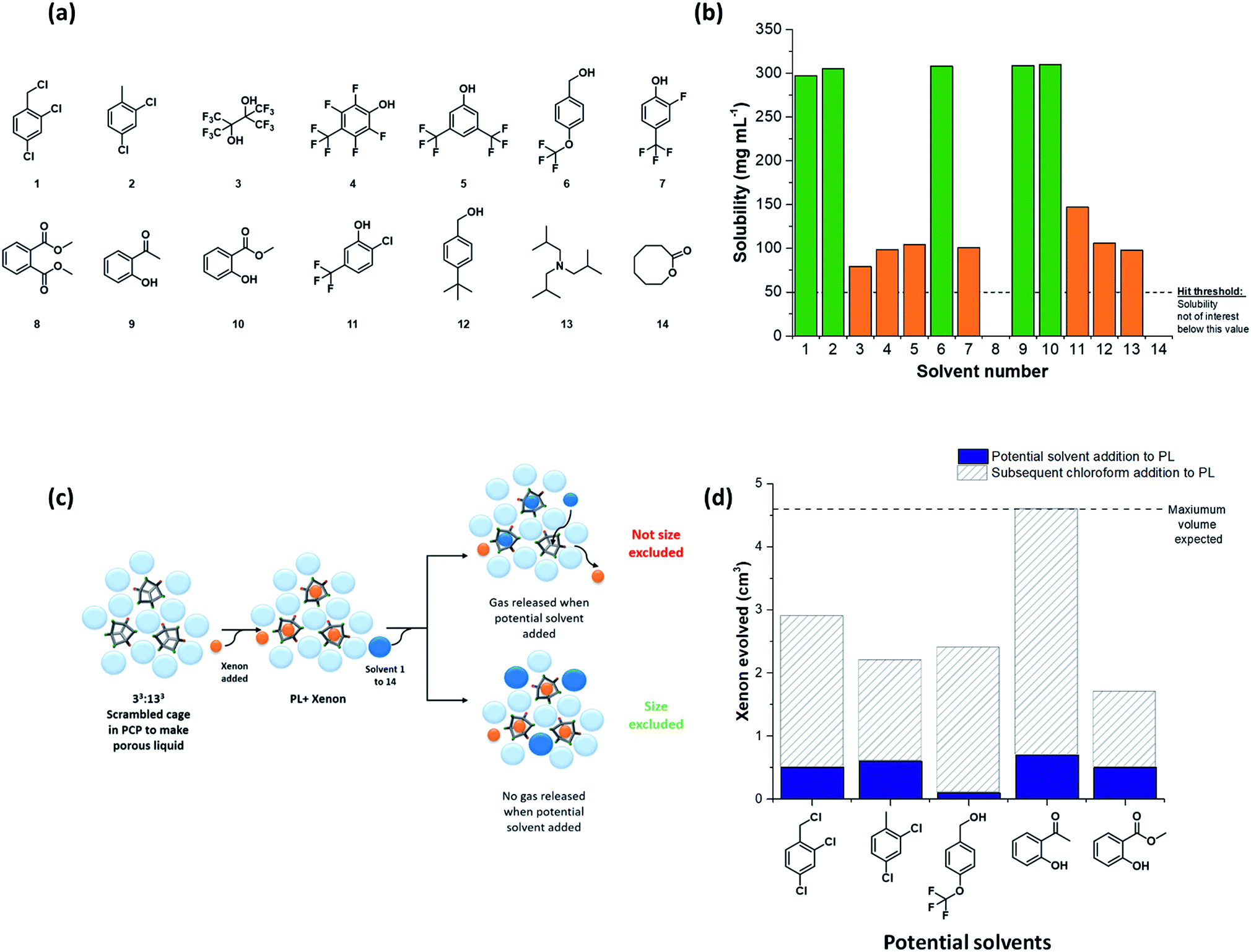 | ||
| Fig. 3 (a) The 14 bulky solvents screened for both cage solubility and size-exclusion from the cage cavity; (b) solubility screen of the scrambled 33:133 cage in these bulky solvents; the highly soluble combinations (∼300 mg mL−1) are highlighted in green; any combinations which fell below a 50 mg mL−1 solubility threshold (i.e., solvents 8 & 14) were not investigated further; (c) scheme illustrating the size-exclusion screen for potential porous liquid solvents – each candidate solvent was added to a known xenon-loaded porous liquid (20% w/v 33:133 in PCP); if little or no gas was evolved upon solvent addition, the solvent was deemed to be size-excluded; (d) the five highly solubilising candidate solvents all displaced small volumes of gas from the known porous liquid (blue bars), but not equating to the total volume in the system, as demonstrated by a subsequent addition of chloroform (grey bars). It could be concluded that the candidate solvents were size-excluded from the cage cavities. | ||
The formation of a Type II porous liquid does not only require high cage solubility: the cage cavities must remain unfilled and accessible to gases in the liquid state. This means that the solvent must not reside in the cage cavities. It was therefore desirable to determine whether or not a solvent was size-excluded before carrying out our high-throughput solubility screen, thus ensuring that only porous liquids were being targeted.
Our previous work showed that gas evolution measurements can give an indication of the gas uptake and the guest selectivity of porous liquids.11 Addition of a small non-excluded liquid guest, such as chloroform, to a gas-loaded porous liquid can displace the gas from the cage pores; by contrast, bulkier size-excluded solvents do not.10,11 We adapted this concept here to create a simple, rapid test for screening solvents for cavity exclusion by investigating whether or not the candidate solvent could displace a gas from a known porous liquid (Fig. 3c). Specifically, we investigated the displacement of xenon from our original 33:133 scrambled porous liquid: a single molecule of xenon can fit inside each cage cavity,34 thus allowing the maximum volume of evolved gas to be calculated, should 100% of the gas be displaced. If a significant volume of xenon was released upon addition of the candidate solvent, then it was assumed that the solvent enters the cage cavities; that is, it is not size-excluded. By contrast, the absence of evolved gas indicates that the candidate solvent is size-excluded and hence a candidate for our high-throughput solubility screen (see Table S8 and Fig. S21†). For the bulkier solvents tested, and in particular those that solubilised large amounts of 33:133, all were deemed to be essentially size-excluded (Fig. 3d).
High-throughput solubility testing
The five best solvents—that is, those that were both size-excluded and good solvents for the scrambled 33:133 cage (A3:E3)—were next used in a high-throughput solubility screen to target Type II porous liquids. PCP was also included as a benchmark (Fig. 4).10,11 The scrambled cages were first dispensed into vials as stock solutions using a Chemspeed Swing liquid-handling platform (0.1 mL of 300 mg mL−1 concentration in chloroform) to remove the need for dispensing as solids (see Table S10†). After drying, automated liquid dispensing was then used to add the size-excluded solvents in 0.1 mL volumetric increments to each scrambled cage. The samples were then sonicated and inspected visually after each solvent addition to determine if the combination had fully dissolved (Fig. S23†).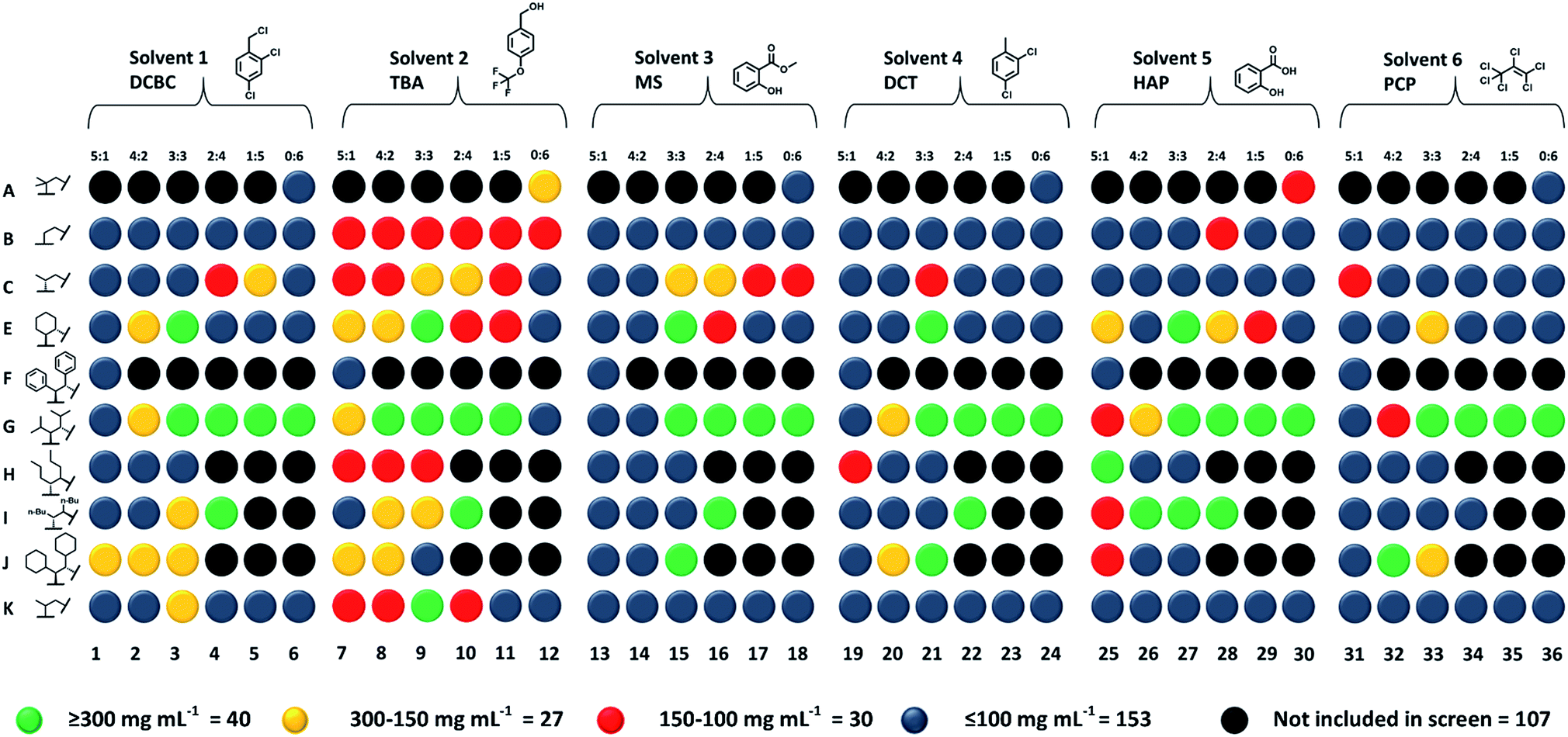 | ||
| Fig. 4 Graphical summary of the results from the high-throughput solubility screen – the solubility of the scrambled cage library was tested in six different solvents (5 new solvents, plus PCP). A ‘hit’ was determined to be a cage/solvent combination with a concentration ≥300 mg mL−1 (green); cage–solvent combinations with a concentration between 150 and 300 mg mL−1 in yellow; 150 to 100 mg mL−1 in red; combinations below the 100 mg mL−1 threshold are grey. All combinations involved diamine A. Each potential porous liquid was assigned a name based on the partner scrambling amine used in the HT synthetic screen (A–K, left), plus a number assigned to each solvent/scrambling ratio combination (1–36, bottom); for example, the 3:3 scrambling ratio of diamine A with diamine B in solvent 2 is B9; the 2:4 scrambling ratio of diamine A with diamine I in solvent 3 is I16, etc. | ||
A solubility of 300 mg mL−1 was set as the upper performance limit since this represents a concentration that is ∼50% greater than our previously reported scrambled cage porous liquid (33:133 in PCP, 200 mg mL−1). Any combinations that were found to hit this limit were scaled-up and investigated in more detail. The lower performance threshold was set at 100 mg mL−1 (3 × 0.1 mL additions); any combinations with solubilities below this were not investigated further. Overall, 40 new scrambled cage/bulky solvent combinations were found with a solubility ≥300 mg mL−1 along with a further 27 that showed similar solubility to our first scrambled porous liquid (Fig. 4 and Table S10†). This illustrates the advantage of using tiered high-throughput solubility screening coupled with pre-screening for size-exclusion for the discovery of new Type II porous liquids. The next step was to scale-up the hits and to study their gas uptake in more detail. However, prior to this, we removed combinations involving PCP (solvent 6, Fig. 4) because of aforementioned problems with toxicity, availability, and purification.
The remaining 36 hits (with a solubility ≥300 mg mL−1) gave us insights into general trends across the scrambled cage families in the five size-excluded solvents. For example, scrambled combinations of A:G gave the most hits above the solubility threshold (sixth row in Fig. 4), suggesting that the addition of isopropyl substituents aids in solubilising the scrambled cage. Also, scrambled cages formed using a 3:3 diamine feed ratio gave the highest number of hits overall, which suggests that this distribution of scrambled cage species gives, in general, more soluble mixtures, perhaps because the 3:3 mixtures are ‘maximally scrambled’ and most effectively lower the lattice energy of these materials.
Scale-up and investigation into porous liquid ‘hits’
Next, the 36 highly-soluble scrambled cage combinations were scaled up, and the reaction conditions used for their formation optimised. This was done because the generalised reaction conditions used for the robotic screen did not necessarily result in the optimal conversion for each specific cage. For example, in the synthesis of A3:K3, the use of dichloromethane rather than chloroform afforded better conversion, whereas A2:I4 benefitted from heating at reflux. Some of the scrambled cages were found to scale well, with A3:E3 (33:133) being made on a 30 g scale in 74% yield, and A3:K3 on an 18 g scale in 89% yield. Other combinations that utilised novel synthesised diamines, such as A3:J3, were lower yielding and contained impurities even after multiple synthesis and purification attempts. Generally speaking, small molecular or oligomeric impurities in both the cage and solvent10,11 have been shown to compete with other guests for occupation of the cavities in the porous liquids. It was important to ensure the porosity of a system was not significantly reduced by impurities, and therefore only scrambled cages with ≥95% purity, as determined by 1H NMR spectroscopy were used. Likewise, distilled size-excluded solvents stored over activated molecular sieves were used in subsequent studies, to remove any possible smaller-sized impurities that might penetrate the cage pores, including water. This afforded 30 potential new porous liquid combinations for further study (Table S12†).Initially, each of the potential porous liquid combinations was made up using 200 mg of the scrambled cage in 1 mL of the size-excluded solvent. However, for certain combinations (such as K9) the mixture formed a gel rather than a free-flowing solution at this concentration (Fig. S67†). This turned out to be a common occurrence at higher concentrations, making some systems unsuitable as porous liquids. POCs are known to self-assemble in the solid state in an ordered fashion to form interconnected pore networks.35–39 At higher solution concentrations we believe that these cages start forming ordered structures, which could explain the observed gel formation. After removal of the systems that gelled, this left 29 porous liquid samples for further study. Previously, gas uptake measurements on Type II porous liquids have relied on NMR experiments and gas evolution studies to indicate whether a system has improved porosity over the neat parent solvent.10,11 Here, we used similar methods to determine the gas uptake properties of the 29 potential porous liquids. Our earlier scrambled porous liquid (A3:E3 (200 mg) in PCP (1 mL), E33) provided a benchmark value for xenon evolution (4.4 ± 0.2 cm3, equating to 95.6 μmol gPL−1) enabling direct comparison with new materials, which all contained the same mass of cage (200 mg). The 29 prepared samples were loaded with xenon gas, and the volume evolved after displacement with chloroform was measured (Table S12†). Using this method, three new combinations were found to have xenon uptakes that were comparable to E33 (Fig. 5a). One of these hits (G16, A2:G4 in solvent 3), however, showed some gelation and was not investigated further, leaving two final PLs: E9 – A3:E3 in solvent 2, and E27 – A3:E3 in solvent 5. These systems both contain the same 33:133 scrambled cage (A3:E3) that we employed in our first scrambled porous liquid, but in different solvents: 2-hydroxyacetophenone (HAP) and 4-(trifluoromethoxy)benzyl alcohol (TBA), respectively. These combinations are referred to as 33:133HAP, 33:133TBA, and 33:133PCP (Fig. 5b).
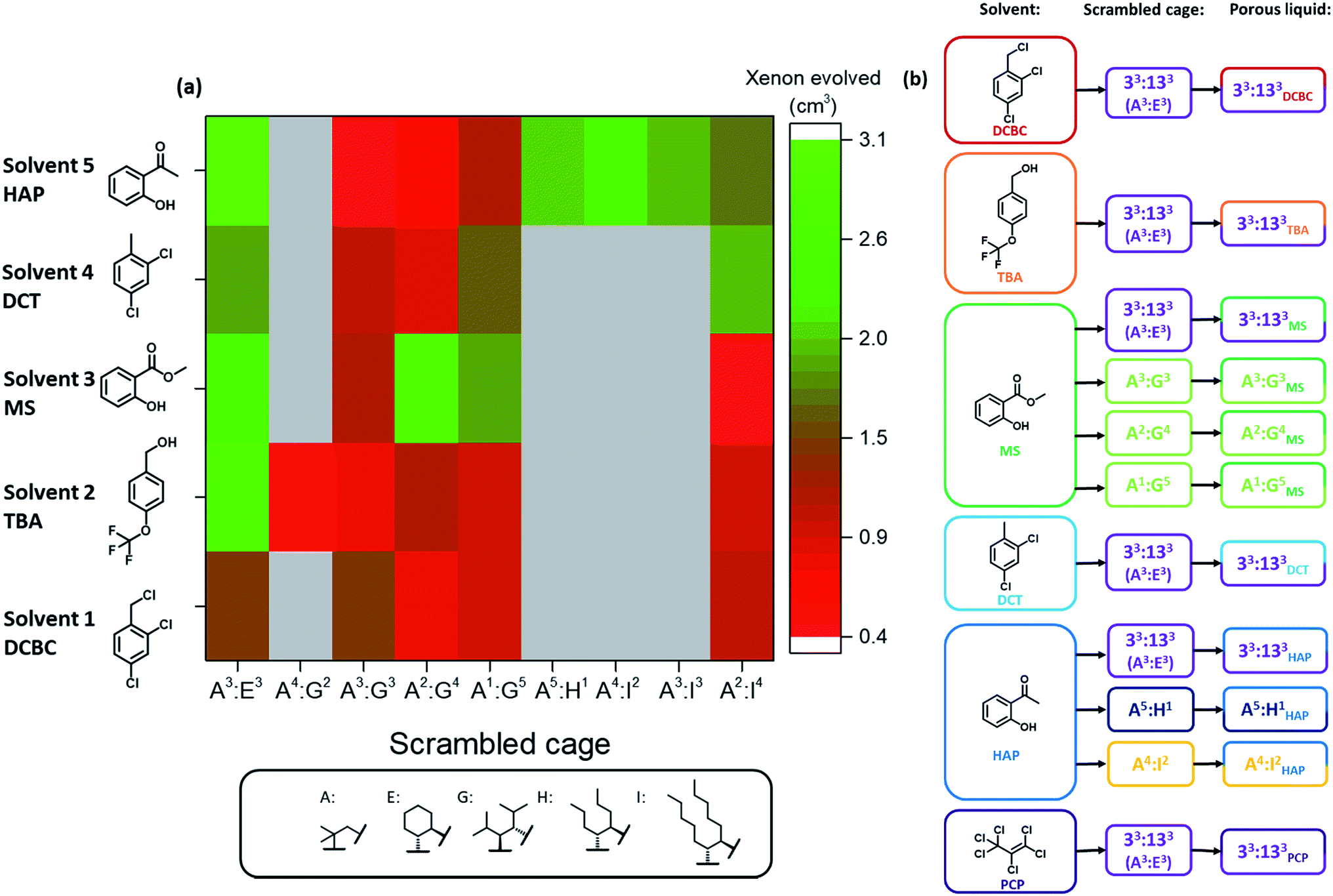 | ||
| Fig. 5 (a) Plot summarising the results from the porosity screen using xenon evolution as a measure for porosity; (b) naming system for the porous liquid families arising from the tiered screens, for example, the scrambled cage 33:133 (A3:E3) in 2,4-dichlorobenzyl chloride (DCBC) is referred to as 33:133DCBC. The colours show the parent cages and corresponding solvents. | ||
Overall, 26 of the new combinations were found to have lower xenon evolutions than our previously reported scrambled porous liquid, despite the fact that the cage solubility in these combinations was in all cases much higher than for our original PCP/33:133 liquid, 33:133PCP. This could be due to several reasons including the viscosity of the liquids, poorer xenon solubility in the solvents, or peripheral alkyl chains occupying neighboring cage cavities (e.g., for diamines H and I).
Investigation into type II porous liquid properties
To probe this further, we carried out separate studies on the effect of changing each component in the porous liquid on the gas uptake. For example, A3:E3 was soluble in all six solvents used in the screen, thus allowing us to study the effect of changing the bulky solvent. Several scrambled cage mixtures containing different diamine feed ratios (A3:G3, A2:G4, A1:G5), as well as different alkyl groups (A5:H1, A4:I2), were also soluble in solvents 3 and 5, respectively, allowing us to study the effect of changing the cage.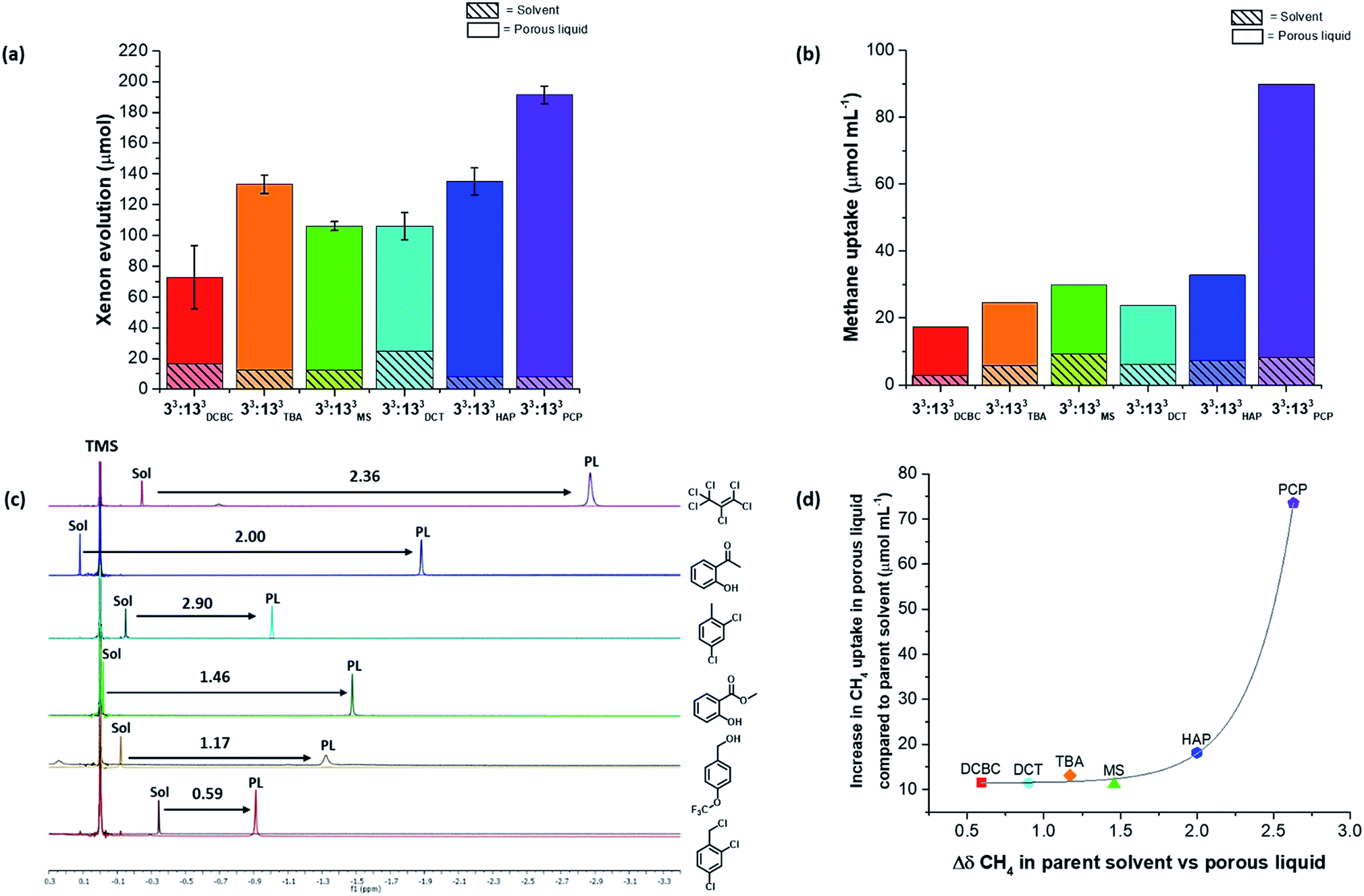 | ||
| Fig. 6 Effect on gas uptake of changing the solvent in Type II porous liquids. (a) Average xenon uptake measured by displacement from the neat solvents (1 mL, dashed lines) and from each porous liquid (200 mg 33:133 with 1 mL solvent, 20% w/v) using chloroform (1.0 molar equivalent relative to cage) – average of 3 measurements, with the standard deviation shown as error bars; (b) methane uptake in neat solvents and porous liquids (20% w/v) measured using 1H NMR spectroscopy; (c) comparison of the methane upfield shifts in the neat solvents and the porous liquids (Sol – indicates CH4 in neat solvent, PL – indicates CH4 in porous liquid); (d) comparison of the difference in upfield shift of methane between the neat solvent and the porous liquid, plotted against the increase in methane uptake in each porous liquid with respect to the neat solvent. | ||
Although gas displacement measurements can give an indication of porosity levels, they cannot confirm directly whether a liquid is permanently porous, nor the extent to which the gas molecules occupy the cage cavities. NMR experiments allowed us to study the host-guest behaviour of these porous liquids—we used 1H NMR spectroscopy to study the methane uptake. Although the new porous liquid combinations exhibited lower CH4 uptakes compared to 33:133PCP at 200 mg mL−1, they all showed a marked improvement over the respective neat parent solvents (Fig. 6b). In agreement with previous studies,10,11 an upfield shift of the methane signal was apparent in comparison to the neat parent solvents (Fig. 6c). This indicates that the gas molecules experience a shielding effect and enter the cage cavities on the NMR timescale. The degree to which the methane protons were shifted varied significantly, and the most pronounced shifts seem to correlate with the liquids that exhibited the highest overall methane uptakes (Fig. 6d). There also appears to be a correlation between the methane solubility in the parent solvent and the resulting methane uptake in the corresponding porous liquid. For example, neat PCP dissolves the most methane of all the solvents (7.1 μmol mL−1), and the porous liquid, 33:133PCP, exhibits the highest overall methane uptake (81.7 μmol mL−1), whereas 33:133DCBC, and its solvent; 2,4-dichlorobenzyl chloride, gave the lowest values (14.2 and 2.8 μmol mL−1, respectively) – see Fig. S70.† In previous work,11 a correlation between the heats of absorption in the solid state and the uptake in the porous liquid was observed, but these additional findings also show that the solvent influence is just as important.
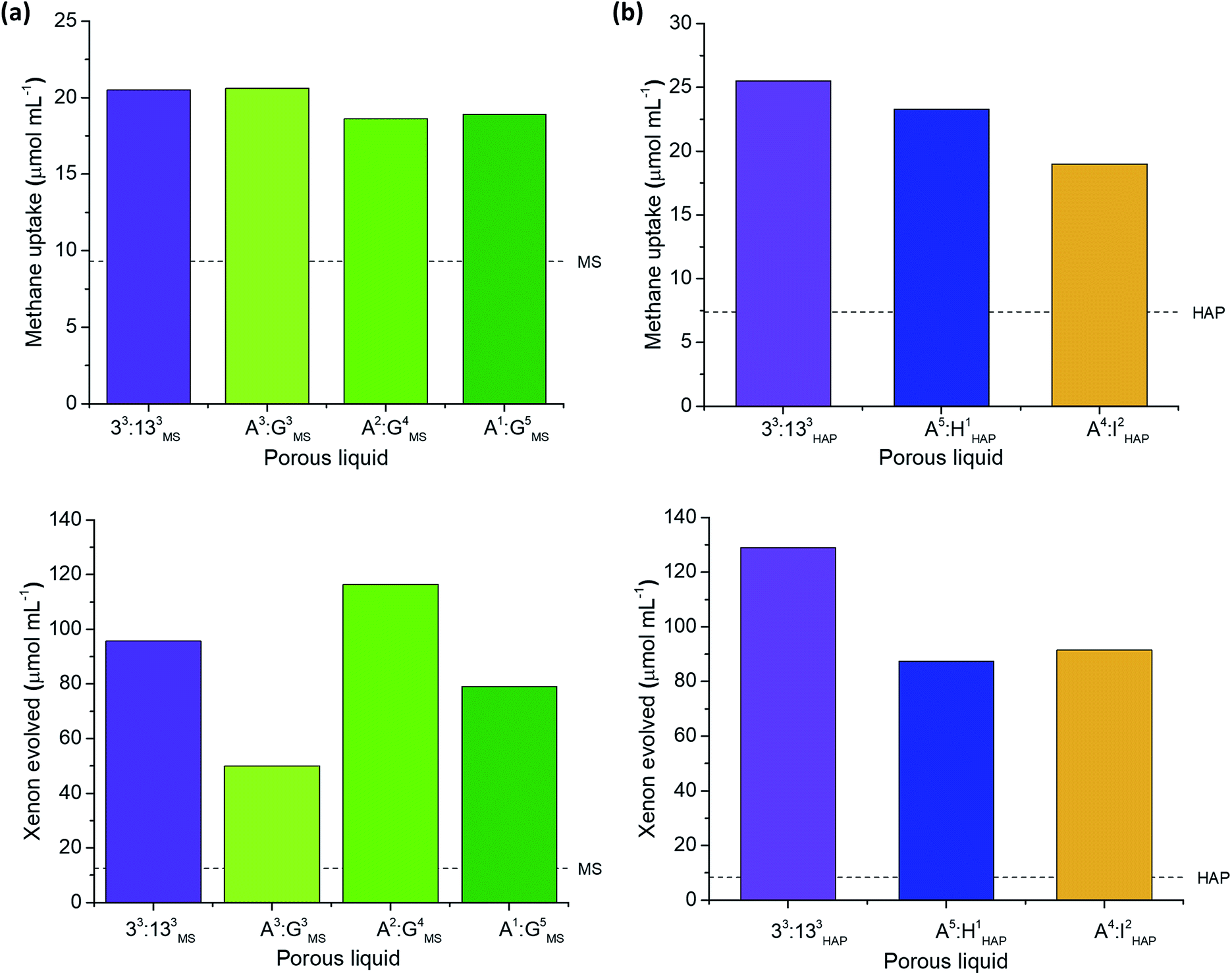 | ||
| Fig. 7 Effect of changing the scrambled cage components in Type II porous liquids on gas uptake. (a) Effect on methane (upper) and xenon (lower) uptake of changing the scrambled diamine feed ratio; (b) effect on methane (upper) and xenon (lower) uptake of changing the peripheral functionality on the scrambled cage. | ||
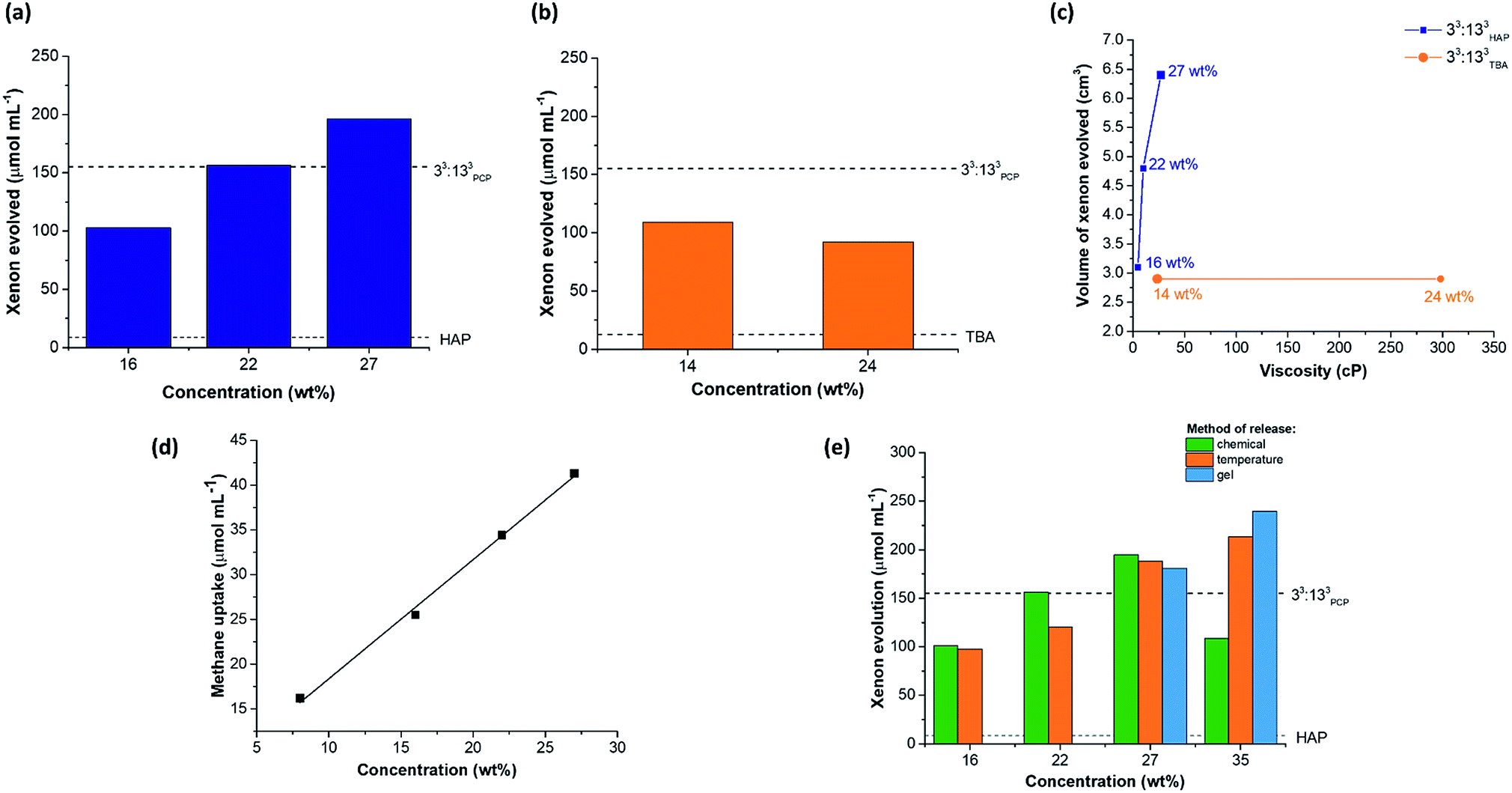 | ||
| Fig. 8 Changing the concentration of scrambled Type II porous liquids allows us to exceed the porosity levels in our previous scrambled porous liquids. (a) Xenon uptake in 33:133HAP at different concentrations (200, 300, and 400 mg in 1 mL solvent, equating to 16, 22 and 27 wt% solutions) measured by displacement with chloroform (1.0 molar equiv. per cage), compared to the uptake in the neat solvent (HAP – dashed line); the maximum Xe uptake attained in our earlier 33:133PCP porous liquid is also marked for reference; (b) xenon uptake in 33:133TBA at different concentrations (200 and 400 mg in 1 mL solvent, equating to 14 and 24 wt% solutions) measured by displacement with chloroform (1.0 molar equiv. per cage), compared to the uptake in the neat solvent (TBA – dashed line); (c) comparison of the amount of xenon evolved from the two porous liquids at different concentrations compared to their viscosities; (d) methane uptake in 33:133HAP at different concentrations measured by 1H NMR spectroscopy; (e) comparison of the amount of xenon evolved from 33:133HAP at different concentrations and using different release mechanisms (chemical displacement with chloroform – green; increased temperature – orange; formation of gel and release on heating – blue). | ||
Investigation into release mechanisms and stability
Throughout this study, small liquid additives were used to actively displace gaseous guests from the cavities within the porous liquids. However, this is unlikely to be a scalable or practical method for gas capture and release. Previously, we showed that sonication could be used as a method to release gas from a porous liquid and this allowed for repeated gas addition and release cycles.10,11 While an improvement on chemical displacement, it still is not an attractive procedure for larger scale processes. Temperature and pressure swings are more practically relevant methods for removing gases from liquids, but with porous liquids, lowering the pressure is challenging due to the solvent's associated vapour pressure. We therefore decided to investigate temperature as a way to liberate xenon from 33:133HAP. At a concentration of 16 wt%, xenon-loaded 33:133HAP released its guest when the temperature was increased to 40 °C and 60 °C. During the first cycle, 97.8 ± 2.3 μmol mL−1 of xenon was evolved from the porous liquid, which was comparable to the volume released using chemical displacement (Fig. 8e). Although the cage appeared to suffer slight decomposition (Fig. S78 and S79†), the process could be cycled a further time on the same sample evolving a similar volume of xenon (78.1 ± 11.4 μmol mL−1), showing the temperature displacement can be cycled twice. The volume of xenon released from more concentrated samples of 33:133HAP (22 and 27 wt%) on heating was also comparable to chemical displacement. However, at 35 wt%, temperature release afforded a significantly higher xenon evolution (213.3 ± 9.6 μmol mL−1) than observed when using chemical displacement with chloroform, in proportion with the increased cage concentration (Fig. 8e). Unfortunately, this process could not be cycled because the cage appeared to decompose upon heating at this higher concentration, perhaps because of polymerisation. The physical state and appearance of the samples also changed during the process, with the viscosity increasing and with foaming occurring while xenon was released (Fig. S79†).Next, the gas retention of 33:133HAP was studied, since the ability to capture and store a gas in a porous liquid might be useful in certain applications. A sample of 33:133HAP at 27 wt% was loaded with xenon and left standing in a sealed vial for 48 hours, after which chloroform was used to chemically displace the remaining gas from the porous liquid. After 2 days, the sample had retained 121.6 μmol mL−1 of xenon, compared to an immediate uptake of 196.3 μmol mL−1, indicating that gas is slowly lost from the porous liquid over time.
While studying the stability of these porous liquids, 33:133HAP was observed to form a gel at high cage concentrations if left standing for prolonged time periods (≥72 hours) (Fig. S85†). This gel could be switched back to a liquid by heating and stirring the sample at 80 °C, and the process of gelation could be prevented entirely by continuously agitating the solution (Fig. S86†). Previous reports have shown that supramolecular assemblies can possess thermo-responsive sol–gel behaviour, with cooling promoting gel formation, and heating reforming the solution.40 We investigated this here as a capture–release mechanism for gases by studying the sol–gel behaviour of 33:133HAP at 27 and 35 wt%. Samples of these porous liquids were saturated with xenon and cooled to between 0–6 °C until gelation occurred – even lower temperatures caused the cage to precipitate out of solution, rather than promoting gel formation. Subsequent heating of the gels to 60–80 °C led to the liquid being reformed along with the release of xenon. Both samples showed gas evolutions comparable to those obtained by chemical displacement or by heating the corresponding liquid: gelled samples – 181 μmol mL−1 for 27 wt%, and 240 μmol mL−1 for 35 wt% (Fig. 8e). The process could not be cycled at either concentration, however, with the 35 wt% sample showing signs of decomposition, alongside a change in the physical state, preventing the addition of further xenon (Fig. S82†). While the 27 wt% sample appeared unchanged based on the 1H NMR spectra, the liquid did not appear to maintain its porosity to xenon after a single gelation-thermal release cycle, as determined by chemical release on addition of a small liquid additive to the re-saturated sample (Fig. S83, Table S61†). Even though the sol–gel capture–release process was not cyclable, we did find that the xenon could be retained within the 33:133HAP gel at 35 wt% for over 28 days. The xenon-loaded gel retained 68% of the gas after 7 days, and 50% after 28 days, based on temperature release. This could lead to interesting capture–release applications, where the gaseous guest could be loaded into the flowable porous liquid, then stored as a gel, before being released after heating to reform the liquid, particularly if the cage stability problem can be overcome (for example, by reducing the imine bonds and tying them with formaldehyde to form stable aminals).41
Conclusions
Prior to this study, there was limited knowledge on the design considerations for Type II porous liquids. We expanded this field by using high-throughput automation to aid in the synthesis, characterisation and solubility testing for a library of 29 scrambled cages in six cavity-excluded solvents. This involved the design of a tiered high-throughput workflow that includes informed decisions, for example to focus efforts on combinations where the solvent is size excluded.This approach led to new porous liquids that comprise solvents that are significantly easier to source and to handle than our first solvent choice, PCP, and which moreover can in some cases be much more porous, storing up to 55% more xenon than 33:133PCP. This is because the cage solubility in these new solvents is much higher, although we also found that this does not always translate into porosity improvements for all combinations (Fig. 7), probably because peripheral functionalities on certain cages can compromise the cage pores.
A serendipitous discovery of gelation at higher cage concentrations suggests a new way to store gases in gelled porous liquids, though this will require us to overcome the chemical instability of these systems to heating when dissolved at such high concentrations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) under the Grants EP/R005710/1 and EP/N004884/1, and the European Research Council under FP7, RobOT, ERC Grant Agreement No. 321156, for financial support. B. M. A. thanks the Leverhulme Research Centre for Functional Materials Design for supporting this research. We acknowledge the MicroBioRefinery for assistance with QTOF-MS measurements. We thank Prof. Stuart James (Queen's University Belfast) for helpful discussions, Rob Clowes for technical support with equipment, and Benjamin Egleston for providing purified hexachloropropene.Notes and references
- N. O'Reilly, N. Giri and S. L. James, Chem.–Eur. J., 2007, 13, 3020–3025 CrossRef PubMed.
- P. Li, H. Chen, J. A. Schott, B. Li, Y. Zheng, S. M. Mahurin, D. Jiang, G. Cui, X. Hu, Y. Wang, L. Li and S. Dai, Nanoscale, 2019, 11, 1515–1519 RSC.
- J. Zhang, S. H. Chai, Z. A. Qiao, S. M. Mahurin, J. Chen, Y. Fang, S. Wan, K. Nelson, P. Zhang and S. Dai, Angew. Chem., Int. Ed., 2015, 54, 932–936 CrossRef CAS PubMed.
- W. Shan, P. F. Fulvio, L. Kong, J. A. Schott, C.-L. Do-Thanh, T. Tian, X. Hu, S. M. Mahurin, H. Xing and S. Dai, ACS Appl. Mater. Interfaces, 2018, 10, 32–36 CrossRef CAS PubMed.
- R. Gaillac, P. Pullumbi, K. A. Beyer, K. W. Chapman, D. A. Keen, T. D. Bennett and F.-X. Coudert, Nat. Mater., 2017, 16, 1149–1154 CrossRef CAS PubMed.
- S. Horike and S. Kitagawa, Nat. Mater., 2017, 16, 1054–1055 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- N. Giri, C. E. Davidson, G. Melaugh, M. G. Del Pópolo, J. T. A. Jones, T. Hasell, A. I. Cooper, P. N. Horton, M. B. Hursthouse and S. L. James, Chem. Sci., 2012, 3, 2153 RSC.
- G. Melaugh, N. Giri, C. E. Davidson, S. L. James and M. G. Del Pópolo, Phys. Chem. Chem. Phys., 2014, 16, 9422–9431 RSC.
- N. Giri, M. G. Del Pópolo, G. Melaugh, R. L. Greenaway, K. Rätzke, T. Koschine, L. Pison, M. F. C. Gomes, A. I. Cooper and S. L. James, Nature, 2015, 527, 216–220 CrossRef CAS PubMed.
- R. L. Greenaway, D. Holden, E. G. B. Eden, A. Stephenson, C. W. Yong, M. J. Bennison, T. Hasell, M. E. Briggs, S. L. James and A. I. Cooper, Chem. Sci., 2017, 8, 2640–2651 RSC.
- S. Jiang, J. T. A. Jones, T. Hasell, C. E. Blythe, D. J. Adams, A. Trewin and A. I. Cooper, Nat. Commun., 2011, 2, 207 CrossRef PubMed.
- E. H. Kerns, J. Pharm. Sci., 2001, 90, 1838–1858 CrossRef CAS PubMed.
- J. Alsenz and M. Kansy, Adv. Drug Delivery Rev., 2007, 59, 546–567 CrossRef CAS PubMed.
- K. Kwon and S. N. Peterson, Methods Mol. Biol., 2014, 1140, 75–88 CrossRef CAS PubMed.
- F. Blanc, S. Y. Chong, T. O. McDonald, D. J. Adams, S. Pawsey, M. A. Caporini and A. I. Cooper, J. Am. Chem. Soc., 2013, 135, 15290–15293 CrossRef CAS PubMed.
- T. Willhammar, J. Su, Y. Yun, X. Zou, M. Afeworki, S. C. Weston, H. B. Vroman, W. W. Lonergan and K. G. Strohmaier, Inorg. Chem., 2017, 56, 8856–8864 CrossRef CAS PubMed.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- R. Lai, B. S. Kang and G. R. Gavalas, Angew. Chem., Int. Ed., 2001, 40, 408–411 CrossRef CAS PubMed.
- P. Wollmann, M. Leistner, U. Stoeck, R. Grünker, K. Gedrich, N. Klein, O. Throl, W. Grählert, I. Senkovska, F. Dreisbach and S. Kaskel, Chem. Commun., 2011, 47, 5151 RSC.
- S. Han, Y. Huang, T. Watanabe, Y. Dai, K. S. Walton, S. Nair, D. S. Sholl and J. C. Meredith, ACS Comb. Sci., 2012, 14, 263–267 CrossRef CAS PubMed.
- R. L. Greenaway, V. Santolini, M. J. Bennison, B. M. Alston, C. J. Pugh, M. A. Little, M. Miklitz, E. G. B. Eden-Rump, R. Clowes, A. Shakil, H. J. Cuthbertson, H. Armstrong, M. E. Briggs, K. E. Jelfs and A. I. Cooper, Nat. Commun., 2018, 9, 2849 CrossRef CAS PubMed.
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244–1248 CrossRef CAS PubMed.
- K. Acharyya, S. Mukherjee and P. S. Mukherjee, J. Am. Chem. Soc., 2013, 135, 554–557 CrossRef CAS PubMed.
- S. Klotzbach and F. Beuerle, Angew. Chem., Int. Ed., 2015, 54, 10356–10360 CrossRef CAS PubMed.
- M. Kołodziejski, A. R. Stefankiewicz and J.-M. Lehn, Chem. Sci., 2019, 10, 1836–1843 RSC.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS PubMed.
- T. Hasell, M. A. Little, S. Y. Chong, M. Schmidtmann, M. E. Briggs, V. Santolini, K. E. Jelfs and A. I. Cooper, Nanoscale, 2017, 9, 6783–6790 RSC.
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, M. Schmidtmann, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 7154–7157 CrossRef CAS PubMed.
- T. Hasell, J. L. Culshaw, S. Y. Chong, M. Schmidtmann, M. A. Little, K. E. Jelfs, E. O. Pyzer-Knapp, H. Shepherd, D. J. Adams, G. M. Day and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 1438–1448 CrossRef CAS PubMed.
- M. E. Briggs and A. I. Cooper, Chem. Mater., 2017, 29, 149–157 CrossRef CAS PubMed.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- J. Tian, P. K. Thallapally and B. P. McGrail, CrystEngComm, 2012, 14, 1909 RSC.
- A. Avellaneda, P. Valente, A. Burgun, J. D. Evans, A. W. Markwell-Heys, D. Rankine, D. J. Nielsen, M. R. Hill, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2013, 52, 3746–3749 CrossRef CAS PubMed.
- M. Mastalerz, M. W. Schneider, I. M. Oppel and O. Presly, Angew. Chem., Int. Ed., 2011, 50, 1046–1051 CrossRef CAS PubMed.
- E. O. Pyzer-Knapp, H. P. G. Thompson, F. Schiffmann, K. E. Jelfs, S. Y. Chong, M. A. Little, A. I. Cooper and G. M. Day, Chem. Sci., 2014, 5, 2235–2245 RSC.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- M. Liu, M. A. Little, K. E. Jelfs, J. T. A. Jones, M. Schmidtmann, S. Y. Chong, T. Hasell and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 7583–7586 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, experimental details and measurements (PDF). See DOI: 10.1039/c9sc03316e. |
| This journal is © The Royal Society of Chemistry 2019 |