
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of non-ionic carbon superbases: second generation carbodiphosphoranes†
Sebastian
Ullrich
a,
Borislav
Kovačević
b,
Björn
Koch
a,
Klaus
Harms
 a and
Jörg
Sundermeyer
*a
a and
Jörg
Sundermeyer
*a
aFachbereich Chemie, Philipps-University Marburg, Hans-Meerwein-Straße, 35032 Marburg, Germany. E-mail: jsu@staff.uni-marburg.de
bThe Group for Computational Life Sciences, Rudjer Bošković Institute, Bijenička c. 54, HR-10000 Zagreb, Croatia
First published on 16th August 2019
Abstract
A new generation of carbodiphosphoranes (CDPs), incorporating pyrrolidine, tetramethylguanidine, or tris(dimethylamino)phosphazene as substituents is introduced as the most powerful class of non-ionic carbon superbases on the basicity scale to date. The synthetic approach as well as NMR spectroscopic and structural characteristics in the free and protonated form are described. Investigation of basicity in solution and in the gas phase by experimental and theoretical means provides the to our knowledge first reported pKBH+ values for CDPs in the literature and suggest them as upper tier superbases.
Introduction
Much theoretical and synthetical effort has been devoted to lift non-ionic organic bases to the basicity level of common inorganic or metalorganic bases.1,2 With his famous phosphazenes Schwesinger established a widely used and commercially available class of (organo-)superbases.3,4 His homologization concept, the stepwise expansion of the molecular scaffold in order to better delocalize the positive charge formed upon protonation, was also applied to synthesize higher-order N-superbases of guanidines,5,6 imidazolidine amines7 and cyclopropeneimines.8,9 However, such basicity enhancement is accompanied by an unwanted growth of the bases' molecular weight. Therefore, other strategies for augmenting the intrinsic proton affinity have been investigated: in proton sponges, a second nitrogen basicity centre in close proximity to the first one increases the pKBH+ value up to 16 orders of magnitude by intramolecular hydrogen bonding compared to corresponding non chelating bases.10 Additional thermodynamic driving force comes from relief of strain of the aromatic backbone.11 Many derivatives of such proton sponges were designed by combining aforementioned superbasic functionalities with the 1,8-diaminonaphthalene structural motif12 or as proton pincers with different backbones.13Atoms other than nitrogen as basicity centre were also applied, such as phosphorus.14,15 Recently, we demonstrated, that N-phosphazenyl substituted phosphines (PAPs) possess higher pKBH+ values as PIII bases than their corresponding phosphazene PVNtBu counterparts as N bases.16 So far the limit of homologization is reached at the P7 level both in phosphazenyl phosphazenes and phosphazenyl phosphines as both P7 benchmark bases have only been isolated in their protonated form.16,17
Non-ionic carbon is another contender to extend the basicity ladder to unmatched regions.18 In this respect phosphorus (mono-)ylides19,20 as well as bisylidic proton sponges21 were investigated on theoretical and experimental level. Although identified as potential superbases, the application of N-heterocyclic carbenes (NHCs),22 cyclic alkyl amino carbenes (CAACs),23 carbodicarbenes (CDCs),24 and carbodiphosphoranes (CDPs)25 has been exploited predominantly as strong Lewis bases towards transition and main group elements other than the proton.26
The prototypic hexaphenyl carbodiphosphorane ((Ph)6-CDP) was first synthesized 1961 by Ramirez et al.27 Further compounds like the hexamethyl carbodiphosphorane ((Me)6-CDP),28 hexakis(dimethylamino) carbodiphosphorane ((dma)6-CDP),29 and mixed representatives followed.30–32
Herein we promote carbodiphosphoranes with their electron-rich R3P–C–PR3 functionality as exceptionally strong carbon Brønsted bases. As bisylides with a π-symmetric HOMO and σ-symmetric HOMO−1, both mainly located as lone pairs at the carbon, only slightly stabilized by backbonding via negative hyperconjugation,33 they provide outstanding pKBH+ values in particular for the first of two protonation steps. We present a synthesis for hexa(pyrrolidino) carbodiphosphorane ((pyrr)6-CDP) with its calculated first and second proton affinity (PA) of 287.6 and 188.9 kcal mol−1,34 which exceeds the PAs of (Ph)6-CDP (280.0 and 185.6 kcal mol−1)34 and (dma)6-CDP (279.9 and 174.9 kcal mol−1).34 Furthermore we apply the homoligization concept to CDPs by introducing PR2R′ units bearing one intrinsically superbasic substituent R′ to access CDP superbases of second-order.8 We thereby focused on N-tetramethylguanidinyl (tmg) and N-tris(dimethylamino)phosphazenyl (dmaP1) substituents targeting new carbodiphosphoranes sym-(tmg)2(dma)4-CDP and sym-(dmaP1)2(dma)4-CDP.
Results and discussion
Synthesis
We experienced, that the established synthesis routes to CDPs are inappropriate for phosphines more electron-rich than P(NMe2)3: reactions between such phosphines P(NR2)2R′ and CCl4 did not follow the pattern outlined in ref. 32 and 35 but exclusively led to chlorination of the phosphine, whilst reactions with methylene bromide did not selectively follow the path outlined in ref. 30 and 36, but led to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-mixture of the methylated phosphonium bromide [R′(NR2)2P-Me]Br and the brominated species [R′(NR2)2P-Br]Br. Therefore we further developed an alternative strategy laid out by Appel et al. for the synthesis of (dma)6-CDP.29 The doubly protonated precursors of the second-order carbodiphosphorane superbases, sym-(tmg)2(dma)4-CDP (1) and sym-(dmaP1)2(dma)4-CDP (2), were obtained in an oxidative imination sequence as shown in Scheme 1. Bis[bis(dimethylamino)phosphino]methane (3) was oxidized by CCl4 in presence of tetramethylguanidine (Htmg) or tris(dimethylamino)phosphazene ((dma)P1-H) instead of dimethylamine as nucleophile and auxiliary base. This reaction offers the advantage of preformed C–P-bonds avoiding the preparation of respective PIII nucleophiles.15,20,373 is readily synthesized in two steps on a large scale38 and the selected superbasic building blocks oxidatively introduced as nucleophiles are either commercially available or easily accessible in few steps.4
:1-mixture of the methylated phosphonium bromide [R′(NR2)2P-Me]Br and the brominated species [R′(NR2)2P-Br]Br. Therefore we further developed an alternative strategy laid out by Appel et al. for the synthesis of (dma)6-CDP.29 The doubly protonated precursors of the second-order carbodiphosphorane superbases, sym-(tmg)2(dma)4-CDP (1) and sym-(dmaP1)2(dma)4-CDP (2), were obtained in an oxidative imination sequence as shown in Scheme 1. Bis[bis(dimethylamino)phosphino]methane (3) was oxidized by CCl4 in presence of tetramethylguanidine (Htmg) or tris(dimethylamino)phosphazene ((dma)P1-H) instead of dimethylamine as nucleophile and auxiliary base. This reaction offers the advantage of preformed C–P-bonds avoiding the preparation of respective PIII nucleophiles.15,20,373 is readily synthesized in two steps on a large scale38 and the selected superbasic building blocks oxidatively introduced as nucleophiles are either commercially available or easily accessible in few steps.4
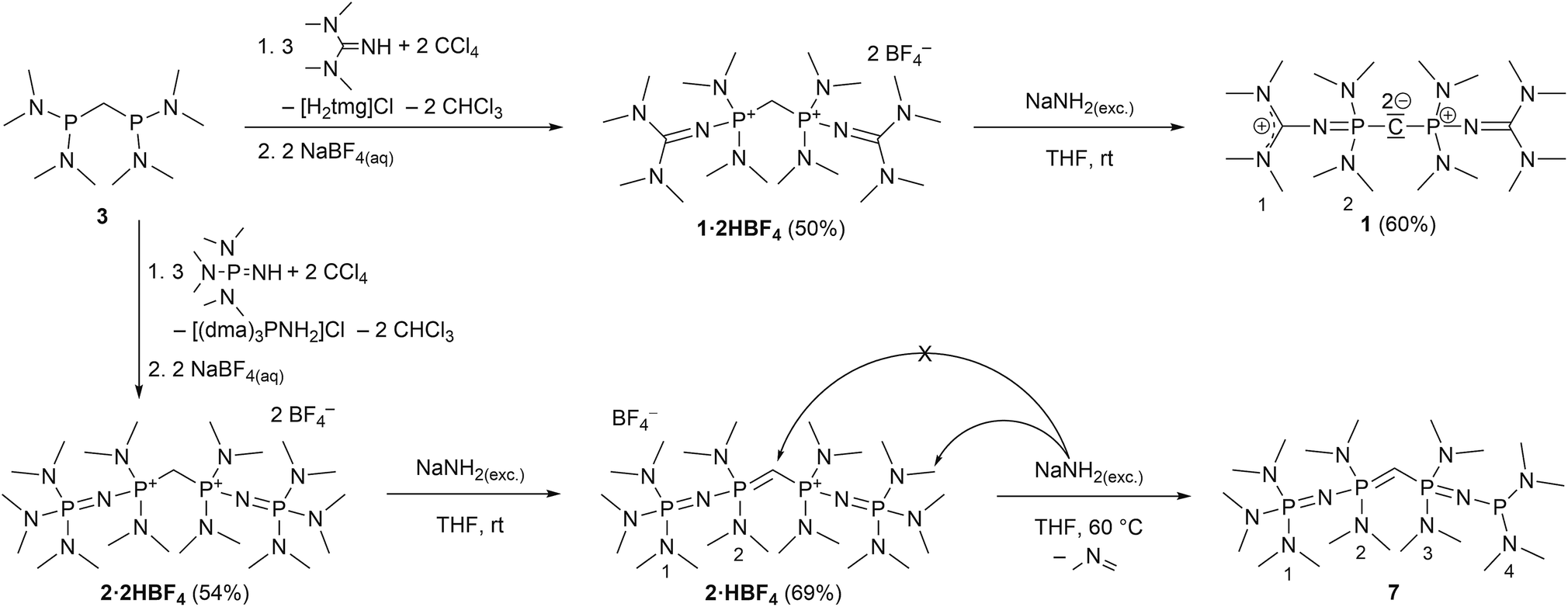 | ||
| Scheme 1 Preparation of CDP precursors 1·2HBF4 and 2·2HBF4 together with subsequent deprotonation to 1 (one exemplary mesomeric structure displayed) and 7, respectively. Numbering schemes refer to assigned NMR signals in the experimental section. | ||
The synthesis of 4·2HBF4, the precursor for (pyrr)6-CDP 4, was accomplished in a one-pot synthesis (Scheme 2), since the intermediate bis[di(pyrrolidino)phosphino]methane (5) turned out to decompose upon vacuum distillation. Starting from bis(dichlorophosphino)methane38 (6), 5 was prepared in situ with an excess of pyrrolidine (Fig. S1 in the ESI†) and directly oxidized with CCl4.
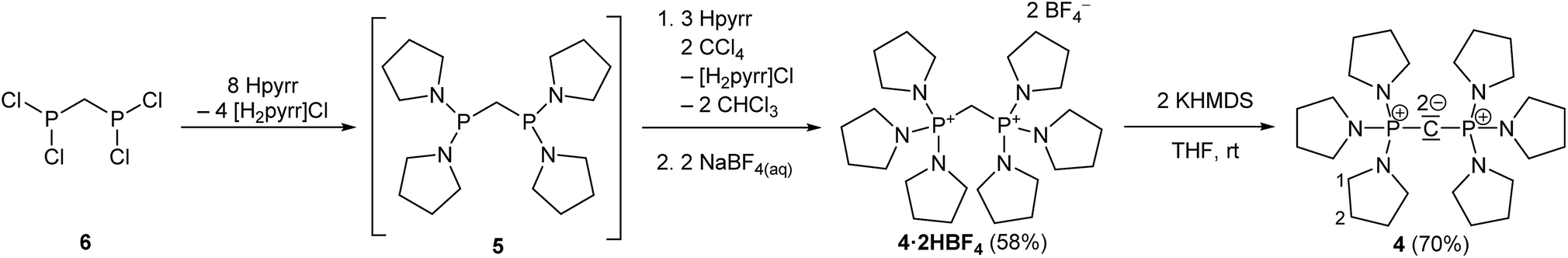 | ||
| Scheme 2 In situ preparation of 5 with subsequent oxidation by CCl4 in presence of excess of pyrrolidine (Hpyrr) to 4·2HBF4. Deprotonation with KHMDS lead to the free CDP 4 (displayed in exemplarily bisylidic notation). The numbering scheme refers to assigned NMR signals in the experimental section. | ||
In all three reactions the respective monoprotonated hydrochloride adducts were identified as products via31P NMR spectroscopy. Therefore the second pKBH+ values in THF of these new CDPs are obviously lower than that of the auxiliary base pyrrolidine (13.5),39 tetramethylguanidine (15.5),40 or tris(dimethylamino)phosphazene 2a (19.7),40 respectively. For purification, the crude products were precipitated with NaBF4 from aqueous solution. These conditions lead to second protonation at the central carbon atom and a strongly alkaline solution. Therefore, even the monoprotonated CDPs can be considered as strong cationic bases in aqueous medium. Similar behaviour was found for (Ph)6-CDP in water, although the latter is slowly hydrolysed under ambient conditions,27 which is not the case for peraminated CDPs 1, 2 and 4 reported here.
The bis(tetrafluoridoborate) salts of 1, 2 and 4 were obtained in 50–60% yield as water and air stable, colourless solids, indefinitely storable. They are well soluble in polar organic solvents like methanol, acetonitrile or DMSO but insoluble in less polar solvents such as ethers and hydrocarbons.
For the liberation of the free CDPs different suitable bases were identified: for 4 potassium bis(trimethylsilyl)amide (KHMDS) is of sufficient basicity, whilst for 1 the more basic sodium amide (NaNH2) is necessary for full deprotonation. Both new bases 1 and 4 could be isolated in 70% and 60% yield, respectively, from n-hexane as pure colourless crystalline solids, indefinitely storable at room temperature under inert conditions. Contrastingly we were not able to isolate 2 as free CDP base form. Sodium amide in liquid ammonia or suspended in THF at room temperature selectively abstracts the first proton under formation of 2·HBF4 as colourless solid in 69% yield. At elevated temperature the central carbon atom is not further deprotonated, even though it is the thermodynamically most acidic site (see Theoretical Calculations). Instead NaNH2 deprotonates selectively one of the dimethylamino groups at the terminal phosphazene moiety which results in the irreversible elimination of N-methylmethanimine and reduction of the phosphazene to a phosphine (Scheme 1). A related deprotonation and reduction of tetrakis(dimethylamino)phosphonium bromide under the action of NaNH2 was described by Pinchuk et al.41 In case of 2 this reaction is slow but highly selective and 7 could be obtained as sole product as pale yellow highly viscous oil. The proposed configuration was confirmed via1H, 13C, and 31P NMR spectroscopy and by HR mass spectrometry. 7 can be considered as a hybrid between mixed valence phosphazenyl phosphines15,16 and ylidic PIII/PV compounds of the type (Me2N)3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H)–PR2 (ref. 42) or other ylide-functionalized phosphines.43 Further attempts to deprotonate 2·2HBF4 with other bases or reducing agents resulted either in only single deprotonation (benzyl potassium in THF), in an unselective disintegration (nBuLi) or in the same deprotonation of the P-NMe2 group (potassium in liquid ammonia, ethylene diamine, THF, or DME or an excess of benzyl potassium in THF). The reaction of potassium hydride in THF gave a mixture of 7 as minor component and presumably free CDP 2 as major product by means of 31P NMR spectroscopy (Fig. S29 in the ESI†). Clearly the acidity of PV-attached NMe2 groups limits the accessibility of 2. Under the action of excess of strong inorganic bases at elevated temperatures the stability limit of these phosphazene moieties seems to have been reached.
C(H)–PR2 (ref. 42) or other ylide-functionalized phosphines.43 Further attempts to deprotonate 2·2HBF4 with other bases or reducing agents resulted either in only single deprotonation (benzyl potassium in THF), in an unselective disintegration (nBuLi) or in the same deprotonation of the P-NMe2 group (potassium in liquid ammonia, ethylene diamine, THF, or DME or an excess of benzyl potassium in THF). The reaction of potassium hydride in THF gave a mixture of 7 as minor component and presumably free CDP 2 as major product by means of 31P NMR spectroscopy (Fig. S29 in the ESI†). Clearly the acidity of PV-attached NMe2 groups limits the accessibility of 2. Under the action of excess of strong inorganic bases at elevated temperatures the stability limit of these phosphazene moieties seems to have been reached.
For analytical reasons the monoprotonated forms of 1 and 4 were prepared on NMR scale either via commutation between the free CDP and its bisprotonated form or by protonating the free CDPs with one equivalent triflimidic acid (HTFSI).
Structural features
For X-ray structure determination suitable single crystals were obtained from n-hexane for both presented CDPs 4 and 1. They crystallize solvent-free in space group P21/c or Pbca, respectively, with one complete molecule per asymmetric unit (Fig. 1). Contrary to the parent compound (dma)6-CDP, one of the hitherto two reported linear CDPs,29,44 a bent structure with P–C–P angles of 155.9(2)° and 147.30(9)°, respectively is found. Since the potential for bending at the central P–C–P carbon atom in polymorphic (Ph)6-CDP is very flat44 and reveals high dependence of the crystallization method,45 the obtained crystals of (dma)6-CDP from the melt are maybe the reason for its linearity.29 The P–Ccentral distances are with 1.606 Å (4) and 1.618 Å (1) in the for CDPs reported range: (dma)6-CDP: 1.584(1) Å,29 (Me)6-CDP: 1.594(3) Å,46 (Ph)6-CDP: 1.601–1.635 Å.44,47 On average, pyrrolidine N–P distances in 4 are 1.68 Å while those of dma and tmg groups in 1 are 1.70 Å and 1.66 Å respectively. | ||
| Fig. 1 Molecular structure of 4 (top) and 1 (bottom). Hydrogen atoms omitted for clarity, ellipsoids at 50% probability. Selected bond length/Å and angles/°: 4 P1–C1 1.605(2), P1–N1 1.672(2), P1–N2 1.678(2), P1–N3 1.694(2), P2–C1 1.606(2), P2–N4 1.699(2), P2–N5 1.669(2), P2–N6 1.671(2), P1–C1–P2 155.9(2), C1–P1–N1 110.2(1), C1–P1–N2 115.1(1), C1–P1–N3 121.8(1), C1–P2–N4 118.4(1), C1–P2–N5 111.3(1), C1–P2–N6 117.1(1), N1–P1–C1–P2 168.0(4), N4–P2–C1–P1 130.6(4). 1 P1–C1 1.619(1), P1–N4 1.680(1), P1–N5 1.714(1), P1–N1 1.665(1), N1–C2 1.298(2), N2–C2 1.377(2), N3–C2 1.382(2), P2–C1 1.617(1), P2–N9 1.719(1), P2–N10 1.680(1), P2–N6 1.664(1), N6–C11 1.299(2), N7–C11 1.376(2), N8–C11 1.379(2), P2–C1–P1 147.30(9), C1–P1–N4 109.52(6), C1–P1–N5 121.56(6), C1–P1–N1 119.85(6), C2–N1–P1 128.1(1), C1–P2–N9 120.76(6), C1–P2–N10 110.08(6), C1–P2–N6 119.47(6), C11–N6–P2 127.3(1), N4–P1–C1–P2 162.2(2), N10–P2–C1–P1 155.8(2). | ||
Single crystals obtained from reaction control samples during the synthesis of 4·2HBF4 turned out to be a cocrystallizate of 4·2HCl and pyrrolidinium chloride (Fig. 2). Cations and anions form a C–H⋯Cl⋯H–N hydrogen bond network with C⋯Cl distances of 3.600(2) Å and N⋯Cl distances of 3.018(2) Å and 3.048(2) Å, the slightly longer distance involving the bridging chlorine atom. Similar weak hydrogen bonds were described for (Ph)6-CDP·2H+ with [InCl4]− (3.60 Å and 4.03 Å),48 [BeCl4]2− (3.55 Å and 3.58 Å),49 I− (3.80 Å and 3.81 Å)50 and Cl− (3.38 Å)49 anions. The difference between the latter and 4·2HCl probably arise from a less polarized C–H-bond due to the stronger electron pair donor 4. Single crystals of the isolated 4·2HBF4 were additionally obtained from chloroform and exhibits no significant differences in the structural properties (displayed in the ESI†). Fig. 2 shows the molecular structures of 1·2HBF4 and 2·2HBF4 as well. All three bisprotonated CDPs exhibit a strong influence of charge delocalization as the reason for their extraordinary basicity: upon protonation the P–C bonds elongate from 1.606 Å (4) and 1.618 Å (1) to 1.799 Å in 4·2HCl and 1.821 Å in 1·2HBF4 and 2·2HBF4, whilst the P–N bonds become shorter to average 1.62 Å for pyrrolidine and 1.64 Å for dimethylamine substituents. This complies with distances found in protonated phosphazenes51 and phosphorus ylids52 and proves the electron donating effect of the amino substituents. The P–N bonds to the tmg groups in 1·2HBF4 exhibits with 1.58 Å (1.66 Å in 1) clearly double-bond character. The P–NC angles are expanded from 127° and 128° to 132° and 136°. A diminishing difference of formal N–C single and double bonds in the tmg group indicates the conjugation within the CN3 moiety. The formal P–N single and double bonds of the phosphazene substituents in 2·2HBF4 equalize at 1.57–1.59 Å with P–N–P angles between 134° and 142°. Similar influence of negative hyperconjugation for charge delocalization was found in superbasic PAPs16 and protonated diphosphazenes.53 The P–C–P angles in the bisprotonated forms (4: 120°, 1: 113°, 2: 121°) are more acute than in the free CDPs (4: 156°, 1: 147°). The difference to ideal tetrahedral geometry presumably arise from the bulkiness of the PR3 moieties.
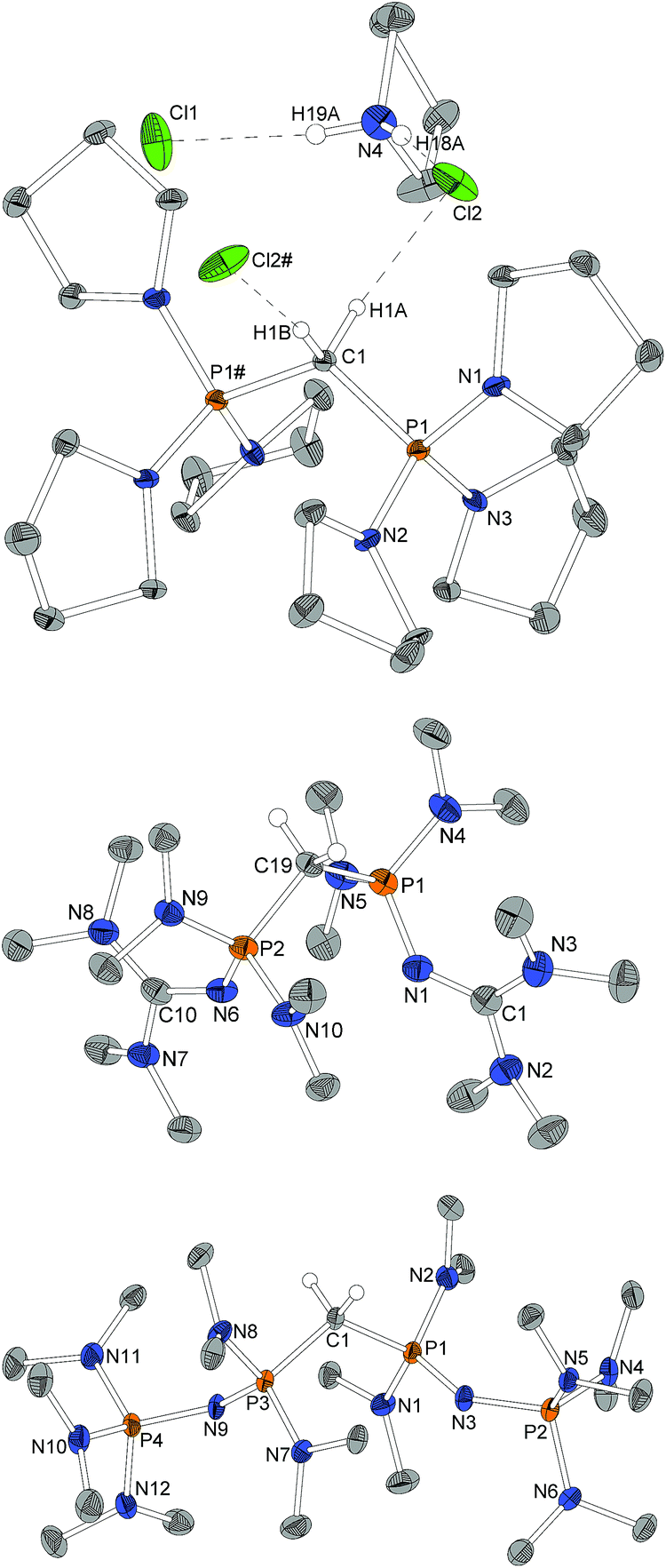 | ||
| Fig. 2 Molecular structure of 4·2HCl with pyrrolidinium chloride as cocrystallizate as well as of 1·2HBF4 and 2·2HBF4 (only one of the two independent molecules depicted, structure factors given for both). Peripheral hydrogen atoms and BF4-anions omitted for clarity, ellipsoids at 50% probability. # marked atoms generated via a 2-fold axes through C1. Selected bond length/Å and angles/°: 4·2HCl P1–C1 1.799(1), P1–N1 1.612(2), P1–N2 1.630(2), P1–N3 1.616(2), P1–C1–P1# 119.5(1), N1–P1–C1 103.26(9), N2–P1–C1 109.07(7), N3–P1–C1 115.21(8), N1–P1–C1–P1# 177.03(7), C1–H1A⋯Cl2 3.600(2), 173.8; C1–H1B⋯Cl2# 3.600(2), 173.8; N4–H18A⋯Cl2 3.048(2), 174(3); N4–H19A⋯Cl1 3.018(2), 172(3). 1·2HBF4 P1–C19 1.820(2), P1–N4 1.644(2), P1–N5 1.639(2), P1–N1 1.580(2), N1–C1 1.330(3), N2–C1 1.351(3), N3–C1 1.346(3), P2–C19 1.822(2), P2–N9 1.640(2), P2–N10 1.643(2), P2–N6 1.586(2), N6–C10 1.335(3), N7–C10 1.332(3), N8–C10 1.349(3), P1–C19–P2 113.4(1), N4–P1–C19 104.3(1), N5–P1–C19 109.7(1), N1–P1–C19 110.8(1), C1–N1–P1 136.1(2), N9–P2–C19 105.4(1), N10–P2–C19 108.8(1), N6–P2–C19 111.8(1), C10–N6–P2 132.6(2), N4–P1–C19–P2 169.1(1), N9–P2–C19–P1 165.2(1). 2·2HBF4 P1–C1/P5–C22 1.820(4)/1.822(4), P1–N1/P5–N19 1.626(4)/1.630(4), P1–N2/P5–N20 1.642(4)/1.650(4), P1–N3/P5–N21 1.573(4)/1.571(4), P2–N3/P6–N21 1.582(4)/1.589(4), P2–N4/P6–N22 1.648(4)/1.639(4), P2–N5/P6–N23 1.639(4)/1.639(4), P2–N6/P6–N24 1.650(4)/1.655(4), P3–C1/P7–C22 1.819(5)/1.817(5), P3–N7/P7–N13 1.636(4)/1.645(4), P3–N8/P7–N14 1.647(4)/1.635(4), P3–N9/P7–N15 1.575(4)/1.567(4), P4–N9/P8–N15 1.577(4)/1.579(4), P4–N10/P8–N16 1.644(4)/1.652(4), P4–N11/P8–N17 1.636(4)/1.648(4), P4–N12/P8–N18 1.655(4)/1.637(4), P3–C1–P1/P5–C22–P7 120.9(2)/121.7(2), N1–P1–C1/N19–P5–C22 110.8(2)/109.8(2), N2–P1–C1/N20–P5–C22 103.8(2)/104.0(2), N3–P1–C1/N21–P5–C22 107.9(2)/108.4(2), P1–N3–P2/P5–N21–P6 138.2(3)/135.7(3), N7–P3–C1/N14–P7–C22 111.2(2)/112.4(2), N8–P3–C1/N13–P7–C22 105.0(2)/103.2(2), N9–P3–C1/N15–P7–C22 107.9(2)/107.4(2), P3–N9–P4/P7–N15–P8 133.6(3)/141.6(3), N2–P1–C1–P3/N20–P5–C22–P7 164.8(3)/166.8(3), N8–P3–C1–P1/N13–P7–C22–P5 165.6(3)/164.4(3). | ||
NMR spectroscopic features
All six presented compounds were characterized by 1H, 13C, and 31P NMR spectroscopy. Selected chemical shifts and couplings are collected in Table 1. Proton shifts of bis- and monoprotonated CDPs lie around 3 ppm for CH2 and below 1 ppm for CH groups, both decreasing with increasing basicity of the parent CDP indicating less polarized C–H bonds. This shielding trend is not observed in the 13C NMR shifts of the carbon nuclei: the most basic CDP 1 exhibits a triplet at 9.5 ppm compared to −1.6 ppm (4) and −6.8 ppm ((dma)6-CDP).29 Surprisingly the 13C chemical shift for 1 is even higher than for its monoprotonated form (1·HTFSI: 9.3 ppm) contrasting the typical trend observed for other CDPs.31,54 The 1JPC couplings drastically increase with step by step deprotonation indicating larger s-character of the ylidic P–C bonds. In the 31P NMR spectra signals for the monoprotonated forms lie between the bisprotonated at higher and the free CDPs at lower values and correlate with the group electronegativity of the phosphines ((dma)6-CDP: 27.72 ppm; (dma)6-CDP·HCl: 54.16 ppm).29 This is not exactly the case for the bisprotonated and free CDPs. The 31P NMR signals of all three forms of 2 are multiplets corresponding to an AA′XX′ spin system with 2JPP and 4JPP coupling (Fig. S22, S25, and S29 in the ESI†). 7 exhibits four individual signals in shape of two doublets of doublets for bridging phosphorus atoms and two doublets for terminal phosphorus atoms with the PIII atom being characteristically deshielded.15,161H and 13C NMR signals are slightly shifted to higher frequencies in comparison with 2·HBF4, indicating that the mixed valent PIII/PV phosphanylphosphazene substituent is a poorer donor than corresponding P2 bisphosphazene.| δ H (2JPH/4JPH) | δ C (1JPC/3JPC) | δ P | |
|---|---|---|---|
| a In CD3CN. b In THF-d8. c In C6D6. d In C6D6, assigned from the isolated mixture of the reaction between 2·2HBF4 and KH in THF (Fig. S29 in the ESI). | |||
| 4·2HBF4 | 3.43 (19) | 26.4 (110) | 32.7 |
| 4·HTFSI | 0.93 (7) | 10.3 (192) | 40.1 |
| 4 | — | −1.6 (280) | 11.5 |
| 1·2HBF4 | 3.16 (17) | 25.2 (112) | 20.8 |
| 1·HTFSI | 0.55 (4) | 9.3 (185) | 37.1 |
| 1 | — | 9.5 (209) | 18.2 |
| 2·2HBF4 | 2.87 (19) | 25.6 (122/7) | 23.2–22.7, 20.6–20.3 |
| 2·HBF4 | 0.25 (6/3) | 12.6 (194/4) | 34.3–33.6, 16.5–15.8 |
| 2 | — | 7.7–7.0, 6.2–5.6 | |
| 7 | 0.42 (3/2) | 13.0 (187/186/2) | 109.9, 39.9, 37.0, 15.1 |
NMR titration experiments were conducted for 4 against (tmg)P1-tBu (pKBH+ in THF: 29.1)6 and (dma)P4-tBu (pKBH+ in THF: 33.9).20 The pKBH+ value for 4 therefore has to be in between 30.1 and 32.9, since only free (tmg)P1-tBu and protonated 4 or protonated (dma)P4-tBu and free 4 were detected, respectively. Basicity of 1 was determined via titration against (pyrr)P4-tBu (pKBH+ in THF: 35.3)20 as reference. Protonated and base forms of both species were quantified by 31P NMR integration and a pKBH+ value of 35.8 in THF was determined for 1. To our knowledge this is the first report of an experimental pKBH+ value for a carbodiphosphorane. It approves 1 to be an exceptional strong non-ionic carbon base, 0.5 orders of magnitude more basic than the strongest uncharged Schwesinger-type nitrogen superbase measured in THF20 and 2.3 orders of magnitude more basic than the so far strongest uncharged carbon superbase H2CP(2,4,6-(MeO)3–C6H2)2Ph (pKBH+ in THF: 33.5).20 Singlet carbenes such as NHCs and CAACs are weak carbon bases in comparison, according to pKBH+ values around 23 in THF and DMSO55 or calculated PAs.34,56 The exceptional C-basicity of the title compounds is only surpassed by our PAP phosphorus superbases (pyrr)P3P (36.7) and (dma)P4P (37.2).16
Quantumchemical calculations
First and second proton affinity (PA) and gas-phase basicity (GB) of carbodiphosphoranes 1, 2, 4 and phosphine 7 are calculated utilizing M06-2X/6-11+G(2df,p)//M06-2X/6-31+G(d) theoretical model. pKBH+ values in THF are obtained using the same functional and basis set whereas solvent is treated as dielectric continuum utilizing the SMD solvation model. pKBH+ values are calculated as relative values using an isodesmic reaction approach57 where Schwesingers (dma)P4-tBu phosphazene with pKBH+ of 33.9 (ref. 20) has served as a reference base. Calculated values for protonation at central carbon atom, and in case of 7 protonation at the PIII atom as well, are presented in Table 2. It appears that the first proton affinity as well as pKBH+ values of 1 and 2 are higher than in Schwesingers (dma)P4-tBu phosphazene which has PA of 293.3 kcal mol−1 calculated at the same level of theory. Interestingly first GB of 1 is slightly lower than the GB of (dma)P4-tBu (GB = 288.2 kcal mol−1) implying that the higher pKBH+ value of 1 relative to (dma)P4-tBu is a result of a more pronounced solvation effect in the carbodiphosphorane. This is unexpected considering that the N–H bond in a protonated phosphazene has a higher polarity than the C–H bond in protonated CDP as a result of lower electronegativity of carbon relative to nitrogen. The calculated pKBH+ (THF) 39.1 of 2 would be far higher than the pKBH+ (THF) 33.9 of (dma)P4-tBu, the strongest commercially available superbase. As described isolation of neutral base 2 is not achieved experimentally as other C–H bonds in the precursor 2·H+ seemed to have a higher kinetic and thermodynamic acidity. In order to understand the deprotonation path of 2·H+ under the action of NaNH2, the reaction profile is calculated and presented in Fig. S36 in the ESI.† It appears, that the deprotonation of peripheral NMe2 group in combination with the irreversible elimination of N-methylmethanimine is thermodynamically feasible (exergonic), however, kinetically hindered by a high barrier (ΔG‡ = 32.8 kcal mol−1). This explains, that deprotonation induced degradation is competitive to deprotonation of central carbon atom at elevated temperatures, though the central carbon atom in 2·H+ is the thermodynamically most acidic site. It appears that decomposition product – phosphine 7 – has a gas-phase basicity (30.9 kcal mol−1) much lower than CDP 2. Interestingly, GB value for protonation at central carbon and PIII phosphorus of 7 is almost the same, whereas pKBH+ in THF for protonation at PIII is by 3.3 orders of magnitude lower than pKBH+ for protonation at carbon, which again indicates a more pronounced solvation effect in C-protonated CDP.Conclusions
In this work we presented the most basic uncharged carbon bases known so far. A convenient synthesis for first- and novel second-order carbodiphosphorane superbases was presented. The CDPs (pyrr)6-CDP 4 and sym-(tmg)2(dma)4-CDP 1 were synthesized as free base as well as in their mono- and bisprotonated forms. In our attempt to synthesize the even more outstanding base sym-(dmaP1)2(dma)4-CDP 2 an unexpected, but highly selective deprotonation at peripheral PNCH3 bonds induced an irreversible elimination path towards phosphine 7. This reaction is indicating a potential basicity limit for phosphazene containing superbases. Structural as well as spectroscopic features were investigated and the basicity was quantified by theoretical and experimental means. Remarkable pKBH+ values for 4 and 1 confirm them as benchmark breakers for non-ionic carbon bases on the THF basicity scale. Compared to the top Schwesinger bases, this basicity is even more outstanding, if their molecular weight below 500 g mol−1 is considered. We expect, that such simply synthesized carbodiphosphoranes with water stable protonated forms will enter the field of organic superbase catalysis.1Experimental section
General
All Reactions with air or moisture sensitive substances were carried out under inert atmosphere using standard Schlenk techniques. Air or moisture sensitive substances were stored in a nitrogen-flushed glovebox. Solvents were purified according to common literature procedures and stored under an inert atmosphere over molsieve (3 Å or 4 Å).58 Pyrrolidine and tetramethylguanidine were distilled from CaH2, triflimidic acid was purified by sublimation under argon. Bis(dichlorophosphino)methane38 (6), bis[bis(dimethylamino)phosphino]methane38 (3), tris(dimethylamino)phosphazene4 and (pyrr)P4-tBu4 were prepared according to literature-known procedures. (dma)P4-tBu was purchased as 1 M solution in n-hexane and dried in high vacuum. All other reagents were used as provided.1H, 13C, and 31P NMR spectra were recorded on a Bruker Avance III HD 250, Avance II 300, Avance III HD 300 or Avance III HD 500 spectrometer. Chemical shift δ is denoted relatively to SiMe4 (1H, 13C) or 85% H3PO4 (31P). 1H and 13C NMR spectra were referenced to the solvent signals.59 Multiplicity is abbreviated as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br. (broad signal). High resolution mass spectrometry were performed on a Thermo Fisher Scientific LTQ-FT Ultra (ESI(+)) or a Jeol AccuTOF GCv (LIFDI(+) = liquid injection field desorption ionization), elemental analysis on an Elementar Vario Micro Cube. IR spectra were recorded in a glovebox on a Bruker Alpha ATR-FT-IR. CCDC 1903830 (4·2HCl + HpyrrCl), 1903833 (1·2HBF4), 1903838 (2·2HBF4), 1903840 (1), 1903841 (4·2HBF4), and 1903843 (4) contain the supplementary crystallographic data for this paper.†
General procedure for the precipitation of BF4-salts
The crude product was dissolved in a minimum amount of water and a concentrated aqueous sodium tetrafluoridoborate solution (2.0 eq.) was added. The resulting precipitate was filtered off, rinsed three times with small portions of cold water, washed with THF and dried in high vacuum.(pyrr)6-CDP·2HBF4 (4·2HBF4)
6 (3.60 g, 16.5 mmol, 1.00 eq.) was dissolved in THF (60 mL), cooled to −78 °C and pyrrolidine (17.7 mL, 216 mmol, 13.1 eq.) was added dropwise. Afterwards the cooling bath was removed and the mixture stirred for additional 6 h. Carbon tetrachloride (3.12 mL, 32.3 mmol, 1.96 eq.) was added at −78 °C and the mixture allowed to warm to room temperature overnight. The suspension was filtered under air and the filter cake extracted with THF (3 × 60 mL). The solvent was removed under reduced pressure and the residue dried in high vacuum. The crude product was converted to its tetrafluoridoborate salt as described in the general procedure and recrystallized from methanol/ethanol. 4·2HBF4 (6.38 g, 9.52 mmol, 58%) was obtained as colourless solid.[C25H50B2F8N6P2] (670.27 g mol−1) 1H NMR (500.2 MHz, CD3CN): δ (ppm) = 3.43 (t, 2JPH = 19 Hz, 2H, CH2), 3.25–3.22 (m, 24H, H1), 1.97–1.95 (m, 24H, H2, (overlapped with the solvent signal)). 13C{1H} NMR (125.8 MHz, CD3CN): δ (ppm) = 48.7 (s, C1), 26.9–26.8 (m, C2), 26.4 (t, 1JPC = 110 Hz, CH2). 31P{1H} NMR (121.5 MHz, CD3CN): δ (ppm) = 32.7. ESI(+) MS (MeOH): m/z (%) = 495.6 (100) [M – H – 2BF4]+, 583.2 (5) [M – BF4]+. ESI(+) HRMS: m/z [M – H – 2BF4]+ calcd 495.3488, found 495.3505; [M − BF4]+ calcd 583.3600, found 583.3611. Elemental analysis: calcd C 44.80%, H 7.52%, N 12.54%; found C 44.49%, H 7.50%, N 12.46%. IR (neat): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 2970 (w), 2879 (w), 1458 (w), 1251 (w), 1210 (m), 1134 (m), 1047 (vs.), 1021 (vs.), 918 (m), 870 (m), 824 (m), 779 (m), 699 (m), 581 (w), 549 (w), 517 (m) 484 (s). XRD: for single crystal X-ray structure determination suitable single crystals were obtained by slow evaporation of a concentrated solution in chloroform.
(cm−1) = 2970 (w), 2879 (w), 1458 (w), 1251 (w), 1210 (m), 1134 (m), 1047 (vs.), 1021 (vs.), 918 (m), 870 (m), 824 (m), 779 (m), 699 (m), 581 (w), 549 (w), 517 (m) 484 (s). XRD: for single crystal X-ray structure determination suitable single crystals were obtained by slow evaporation of a concentrated solution in chloroform.
sym-(tmg)2(dma)4-CDP·2HBF4 (1·2HBF4)
3 (831 mg, 3.29 mmol, 1.00 eq.) and tetramethylguanidine (1.14 g, 9.88 mmol, 3.00 eq.) were dissolved in THF (60 mL). Carbon tetrachloride (640 μL, 6.62 mmol, 2.01 eq.) was added at −78 °C and the mixture allowed to warm to room temperature overnight. The suspension was filtered under air and the filter cake extracted with THF (3 × 20 mL). The solvent was removed under reduced pressure and the residue dried in high vacuum. The crude product was converted to its tetrafluoridoborate salt as described in the general procedure and recrystallized from ethanol. 1·2HBF4 (1.08 g, 1.66 mmol, 50%) was isolated as colourless solid.[C10H50B2F8N10P2] (654.24 g mol−1) 1H NMR (500.2 MHz, CD3CN): δ (ppm) = 3.16 (t, 2JPH = 17 Hz, 2H, CH2), 2.91 (s, 24H, H1), 25.3 (d, 3JPH = 10 Hz, 24H, H2). 13C{1H} NMR (125.8 MHz, CD3CN): δ (ppm) = 161.6 (dd, 2× 2,4JPC = 2 Hz, CN3), 40.9 (s, C1), 37.1 (dd, 2× 2,4JPC = 2 Hz, C2), 25.2 (t, 1JPC = 112 Hz, CH2). 31P{1H} NMR (202.5 MHz, CD3CN): δ (ppm) = 20.8 (s, 1JPC = 113 Hz (satellites)). ESI(+) MS (MeOH): m/z (%) = 479.5 (100) [M − H − 2BF4]+. ESI(+) HRMS: m/z [M − H − 2BF4]+ calcd. 479.3622, found 479.3625. Elemental analysis: calcd C 34.88%, H 7.70%, N 21.41%; found C 34.98%, H 7.84%, N 21.39%. IR (neat): (cm−1) = 2911 (br. w.), 1539 (s), 1486 (m), 1429 (m), 1401 (m), 1356 (m), 1289 (m), 1235 (w), 1186 (m), 1161 (m), 1046 (vs.), 1034 (vs.), 979 (vs.), 933 (vs.), 784 (s), 771 (s), 739 (m), 716 (m), 690 (w), 672 (w), 618 (w), 572 (m), 519 (m), 459 (m), 437 (m). XRD: for single crystal X-ray structure determination suitable single crystals were obtained from ethanol at −25 °C.
sym-(dmaP1)2(dma)4-CDP·2HBF4 (2·2HBF4)
3 (1.55 g, 6.14 mmol, 1.00 eq.) and tris(dimethylamino)phosphazene (3.28 g, 18.4 mmol, 3.00 eq.) were dissolved in THF (60 mL). Carbon tetrachloride (1.19 mL, 12.3 mmol, 2.00 eq.) was added at −78 °C and the mixture allowed to warm to room temperature overnight. The suspension was filtered under air and the filter cake extracted with THF (3 × 20 mL). The solvent was removed under reduced pressure and the residue dried in high vacuum. The crude product was converted to its tetrafluoridoborate salt as described in the general procedure and recrystallized from ethanol/n-hexane. 2·2HBF4 (2.58 g, 3.31 mmol, 54%) was isolated as colourless solid.[C21H62B2F8N12P4] (780.31 g mol−1) 1H NMR (500.2 MHz, CD3CN): δ (ppm) = 2.87 (t, 2JPH = 19 Hz, 2H, CH2), 2.68 (d, 3JPH = 11 Hz, 24H, H2), 2.65 (d, 3JPH = 10 Hz, 36H, H1). 13C{1H} NMR (125.8 MHz, CD3CN): δ (ppm) = 37.3 (m, C1, C2), 25.6 (tt, 1JPC = 122 Hz, 3JPC = 7 Hz, CH2). 31P{1H} NMR (202.5 MHz, CD3CN): δ (ppm) = 23.2–22.7 (m, P1), 20.6–20.3 (m, P2). ESI(+) MS (MeOH): m/z (%) = 303.5 (25) [M − 2BF4]2+, 605.6 (60) [M − H − 2BF4]+, 693.5 (100) [M − BF4]+. ESI(+) HRMS: m/z [M − 2BF4]2+ calcd 303.2080, found 303.2088; [M − H − 2BF4]+ calcd 605.4087, found 605.4104; [M − BF4]+ calcd 693.4195, found 693.4215. Elemental analysis: calcd C 32.32%, H 8.01%, N 21.54%; found C 31.94%, H 7.70%, N 21.18%. IR (neat): (cm−1) = 2886 (w), 1539 (s), 1486 (m), 1429 (m), 1401 (m), 1356 (m), 1298 (m), 1234 (m), 1186 (w), 1161 (m), 1047 (vs.), 1035 (vs.), 979 (vs.), 933 (s), 784 (s), 771 (s), 739 (m), 715 (m), 690 (m), 672 (m), 572 (m), 519 (m), 459 (m), 439 (m). XRD: for single crystal X-ray structure determination suitable single crystals were obtained from ethanol/n-hexane at −25 °C.
(pyrr)6-CDP (4)
A solution of potassium bis(trimethylsilyl)amide (558 mg, 2.80 mmol, 2.09 eq.) in THF (15 mL) was added to a suspension of 4·2HBF4 (938 mg, 1.34 mmol, 1.00 eq.) in THF (40 mL) and stirred for 16 h at room temperature. All volatiles were removed in vacuo, the residue dissolved in n-hexane (20 mL) and filtered over Celite. The filter cake was extracted with n-hexane (2 × 15 mL) and the filtrate evaporated to dryness. 4 (481 mg, 973 μmol, 70%) was isolated as colourless solid. [C25H48N6P2] (494.65 g mol−1) 1H NMR (500.2 MHz, C6D6): δ (ppm) = 3.33–3.23 (m, 24H, H1), 1.75–1.64 (m, 24H, H2). 13C{1H} NMR (125.8 MHz, C6D6): δ (ppm) = 47.4 (s, C1), 28.9 (s, C2), −1.6 (t, 1JPC = 280 Hz, PCP). 31P{1H}-NMR (202.5 MHz, C6D6): δ (ppm) = 11.5. LIFDI(+) MS (n-hexane): m/z (%) = 495.4 (100) [M + H]+. LIFDI(+) HRMS: m/z [M + H]+ calcd 495.34939, found 495.35037. Elemental analysis: calcd C 60.70%, H 9.78%, N 16.99%; found C 60.39%, H 9.62%, N 17.42%. IR (neat): (cm−1) = 2952 (m), 2836 (m), 1492 (w), 1435 (s), 1338 (m), 1319 (m), 1289 (w), 1191 (m), 1134 (m), 1046 (vs.), 1000 (vs.), 980 (vs.), 909 (s), 870 (m), 742 (m), 546 (vs.), 497 (vs.). XRD: for single crystal X-ray structure determination suitable single crystals were obtained from n-hexane at −25 °C.
sym-(tmg)2(dma)4-CDP (1)
A mixture of 1·2HBF4 (190 mg, 290 μmol, 1.00 eq.) and sodium amide (113 mg, 2.90 mmol, 10.0 eq.) was stirred in THF (15 mL) for 16 h at room temperature. The suspension was filtered over Celite and the filter cake extracted with THF (3 × 5 mL). All volatiles were removed in vacuo, n-hexane (10 mL) added to the residue, filtered again over Celite and extracted with n-hexane (3 × 4 mL). Evaporation of the solvent and drying in high vacuum yielded 1 (86 mg, 0.17 mmol, 60%) as colourless solid. [C19H48N10P2] (478.61 g mol−1) 1H NMR (500.2 MHz, C6D6): δ (ppm) = 2.88 (dd, 2× 3,5JPH = 5 Hz, 24H, H2), 2.73 (s, 24H, H1). 13C{1H} NMR (125.8 MHz, C6D6): δ (ppm) = 156.0 (s, CN3), 40.1 (s, C1), 38.3 (s, C2), 9.5 (t, 1JPC = 209 Hz, PCP). 31P{1H} NMR (121.5 MHz, C6D6): δ (ppm) = 18.2. LIFDI(+) MS (n-hexane): m/z (%) = 479.4 (100) [M + H]+. LIFDI(+) HRMS: m/z [M + H]+ calcd 479.36169, found 479.36229. Elemental analysis: calcd C 47.68%, H 10.11%, N 29.27%; found C 47.54%, H 9.96%, N 29.47%. IR (neat): (cm−1) = 3006 (w), 2847 (m), 2810 (m), 2778 (m), 1566 (vs.), 1496 (s), 1472 (m), 1453 (m), 1440 (m), 1421 (m), 1358 (vs.), 1281 (m), 1251 (m), 1235 (m), 1211 (m), 1173 (m), 1128 (s), 1052 (m), 971 (s), 949 (vs.), 917 (m), 860 (vs.), 796 (m), 748 (m), 685 (s), 652 (s), 629 (vs.), 568 (m), 527 (s), 452 (s). XRD: for single crystal X-ray structure determination suitable single crystals were obtained from n-hexane at −25 °C.
Attempted synthesis of sym-(dmaP1)2(dma)4-CDP (2)
A mixture of 2·2HBF4 (136 mg, 174 μmol, 1.0 eq.) and freshly ground sodium amide (75 mg, 1.9 mmol, 11 eq.) was suspended in THF (15 mL) and stirred for 72 h at 60 °C. The solid was removed by filtration over Celite and the filtrate evaporated to dryness. The residue was dissolved in n-pentane (20 mL), cleared via syringe filtration, the solvent removed and the residue dried in high vacuum to give 7 as pale yellow high viscous oil.[C19H55N10P4] (561.62 g mol−1) 1H NMR (300.3 MHz, C6D6): δ (ppm) = 2.99 (d, 3JPH = 9 Hz, 12H, H4), 2.88 (d, 3JPH = 10 Hz, 12H, H3), 2.83 (d, 3JPH = 11 Hz, 12H, H2), 2.32 (d, 3JPH = 10 Hz, 18H, H1), 0.42 (dddd, 2× 2JPH = 3 Hz, 2× 4JPH = 2 Hz, 1H, CH). 13C{1H} NMR (75.5 MHz, C6D6): δ (ppm) = 38.5 (dd, 2JPC = 4 Hz, 4JPC = 3 Hz, C3), 38.4 (d, 2JPC = 16 Hz, C4), 38.1 (dd, 2JPC = 4 Hz, 4JPC = 1 Hz, C2) 37.1 (d, 2JPC = 4 Hz, C1), 13.0 (ddd, 1JPC = 187 Hz, 1JPC = 186 Hz, 3JPC = 2 Hz, CH). 31P{1H} NMR (121.5 MHz, C6D6): δ (ppm) = 109.9 (d, 2JPP = 100 Hz, P4), 39.9 (dd, 2JPP = 50 Hz, 2JPP = 41 Hz, P2), 37.0 (dd, 2JPP = 100 Hz, 2JPP = 41 Hz, P3), 15.1 (d, 2JPP = 50 Hz, P1). LIFDI(+) MS (n-hexane): m/z (%) = 561.4 (100) [M]+. LIFDI(+) HRMS: m/z [M]+ calcd 561.35924, found 561.35562.
(pyrr)6-CDP·HTFSI (4·HTFSI)
4 (8.954 mg, 18.10 μmol, 1.04 eq.) and triflimidic acid (4.911 mg, 17.46 μmol, 1.00 eq.) were mixed in THF-d8 (0.5 mL) and used for analytics.[C27H49F6N7O4P2S2] (775.79 g mol−1) 1H NMR (500.2 MHz, THF-d8): δ (ppm) = 3.20–3.17 (m, 24H, H1), 1.88–1.85 (m, 24H, H2), 0.93 (t, 2JPH = 7 Hz, 1H, CH). 13C{1H} NMR (125.8 MHz, THF-d8): δ (ppm) = 121.1 (q, 1JFC = 323 Hz, CF3), 47.8 (s, C1), 26.9 (dd, 2× JPC = 4 Hz, C2), 10.3 (t, 1JPC = 192 Hz, CH). 31P{1H} NMR (121.5 MHz, THF-d8): δ (ppm) = 40.1. LIFDI(+) MS (THF): m/z (%) = 495.4 (100) [M − TFSI]+. LIFDI(+) HRMS: m/z [M − TFSI]+ calcd 495.34939, found 495.35146.
sym-(tmg)2(dma)4-CDP·HTFSI (1·HTFSI)
1 (9.273 mg, 19.38 μmol, 1.00 eq.) and triflimidic acid (5.517 mg, 19.62 μmol, 1.01 eq.) were mixed in THF-d8 (0.5 mL) and used for analytics.[C21H49F6N11O4P2S2] (759.75 g mol−1) 1H NMR (300.3 MHz, THF-d8): δ (ppm) = 2.90 (s, 24H, H1), 2.67–2.64 (m, 24H, H2), 0.55 (t, 2JPH = 4 Hz, 1H, CH). 13C{1H} NMR (75.5 MHz, THF-d8): δ (ppm) = 161.1 (s, CN3), 121.1 (q, 1JFC = 322 Hz, CF3), 40.3 (s, C1), 37.7 (dd, 2× 2,4JPC = 2 Hz, C2), 9.3 (t, 1JPC = 185 Hz, CH). 31P{1H} NMR (121.5 MHz, C6D6): δ (ppm) = 37.1. LIFDI(+) MS (THF): m/z (%) = 479.4 (100) [M − TFSI]+. LIFDI(+) HRMS: m/z [M − TFSI]+ calcd 479.36169, found 479.36232.
sym-(dmaP1)2(dma)4-CDP·HBF4 (2·HBF4)
A mixture of 2·2HBF4 (600 mg, 769 μmol, 1.00 eq.) and finely ground sodium amide (321 mg, 8.23 mmol, 10.7 eq.) was suspended in THF (20 mL), cooled to −78 °C and ammonia (ca. 40 mL) was condensed in. The mixture was allowed to warm to room temperature overnight, the solid removed by centrifugation and the supernatant evaporated to dryness. The residue was dissolved in dichloromethane (40 mL) and filtered over Celite. All volatiles were removed in vacuo, the residue washed with diethyl ether (2 × 40 mL) and dried in high vacuum. 2·HBF4 (365 mg, 527 μmol, 69%) was isolated as colorless solid. [C21H61BF4N10P4] (692.50 g mol−1) 1H NMR (500.2 MHz, CD3CN): δ (ppm) = 2.64 (d, 3JPH = 10 Hz, 36H, H1), 2.60–2.57 (m, 24H, H2), 0.25 (tt, 2JPH = 6 Hz, 4JPH = 3 Hz, 1H, CH). 13C{1H} NMR (125.8 MHz, CD3CN): δ (ppm) = 37.9 (d, 2JPC = 2 Hz, C2), 37.4 (d, 2JPC = 5 Hz, C1), 12.6 (tt, 1JPC = 194 Hz, 3JPC = 4 Hz, CH). 31P{1H} NMR (121.5 MHz, CD3CN): δ (ppm) = 34.3–33.6 (m, P2), 16.5–15.8 (m, P1). LIFDI(+) MS (THF): m/z (%) = 605.4 (100) [M − BF4]+. LIFDI(+) HRMS: m/z [M − BF4]+ calcd 605.40926, found 605.41147. Elemental analysis: calcd C 36.42%, H 8.88%, N 24.27%; found C 36.25%, H 8.59%, N 24.21%. IR (neat): (cm−1) = 3000 (w), 2883 (m), 2846 (m), 2804 (m), 1458 (m), 1288 (s), 1243 (m), 1183 (m), 1167 (m), 1092 (m), 1048 (s), 976 (vs.), 955 (vs.), 845 (m), 823 (m), 770 (m), 740 (s), 715 (s), 660 (s), 598 (m), 551 (w), 527 (m), 498 (s), 454 (m), 420 (w).
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by Philipps-Universität Marburg.Notes and references
- T. Ishikawa, Superbases for organic synthesis. Guanidines, amidines and phosphazenes and related organocatalysts, Wiley, Chichester, West Sussex, U.K, 2009 Search PubMed.
- E. D. Raczyńska, J.-F. Gal and P.-C. Maria, Chem. Rev., 2016, 116, 13454 CrossRef PubMed.
- R. Schwesinger and H. Schlemper, Angew. Chem., Int. Ed. Engl., 1987, 26, 1167 ( Angew. Chem. , 1987 , 99 , 1212 ) CrossRef.
- R. Schwesinger, H. Schlemper, C. Hasenfratz, J. Willaredt, T. Dambacher, T. Breuer, C. Ottaway, M. Fletschinger, J. Boele, H. Fritz, D. Putzas, H. W. Rotter, F. G. Bordwell, A. V. Satish, G.-Z. Ji, E.-M. Peters, K. Peters, H. G. von Schnering and L. Walz, Liebigs Ann./Recl., 1996, 1996, 1055 CrossRef.
- B. Kovačević and Z. B. Maksić, Org. Lett., 2001, 3, 1523 CrossRef PubMed.
- A. A. Kolomeitsev, I. A. Koppel, T. Rodima, J. Barten, E. Lork, G.-V. Röschenthaler, I. Kaljurand, A. Kütt, I. Koppel, V. Mäemets and I. Leito, J. Am. Chem. Soc., 2005, 127, 17656 CrossRef CAS PubMed.
- (a) R. A. Kunetskiy, S. M. Polyakova, J. Vavřík, I. Císařová, J. Saame, E. R. Nerut, I. Koppel, I. A. Koppel, A. Kütt, I. Leito and I. M. Lyapkalo, Chem.–Eur. J., 2012, 18, 3621 CrossRef CAS; (b) K. Vazdar, R. Kunetskiy, J. Saame, K. Kaupmees, I. Leito and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 1435 ( Angew. Chem. , 2014 , 126 , 1459 ) CrossRef CAS.
- E. D. Nacsa and T. H. Lambert, J. Am. Chem. Soc., 2015, 137, 10246 CrossRef CAS PubMed.
- B. Kovačević, Z. B. Maksić and R. Vianello, J. Chem. Soc., Perkin Trans. 2, 2001, 886 RSC.
- J. F. Kögel, B. Oelkers, B. Kovačević and J. Sundermeyer, J. Am. Chem. Soc., 2013, 135, 17768 CrossRef PubMed.
- R. W. Alder, P. S. Bowman, W. R. S. Steele and D. R. Winterman, Chem. Commun., 1968, 723 RSC.
- (a) V. Raab, J. Kipke, R. M. Gschwind and J. Sundermeyer, Chem.–Eur. J., 2002, 8, 1682 CrossRef CAS PubMed; (b) V. Raab, E. Gauchenova, A. Merkoulov, K. Harms, J. Sundermeyer, B. Kovačević and Z. B. Maksić, J. Am. Chem. Soc., 2005, 127, 15738 CrossRef CAS PubMed; (c) L. Belding and T. Dudding, Chem.–Eur. J., 2014, 20, 1032 CrossRef CAS PubMed; (d) L. Belding, P. Stoyanov and T. Dudding, J. Org. Chem., 2016, 81, 6 CrossRef CAS PubMed.
- (a) H. A. Staab, T. Saupe and C. Krieger, Angew. Chem., Int. Ed. Engl., 1983, 22, 731 ( Angew. Chem. , 1983 , 95 , 748 ) CrossRef; (b) T. Saupe, C. Krieger and H. A. Staab, Angew. Chem., Int. Ed. Engl., 1986, 25, 451 ( Angew. Chem. , 1986 , 98 , 460 ) CrossRef; (c) M. A. Zirnstein and H. A. Staab, Angew. Chem., Int. Ed. Engl., 1987, 26, 460 ( Angew. Chem. , 1987 , 99 , 460 ) CrossRef; (d) R. Schwesinger, M. Mißfeldt, K. Peters and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1987, 26, 1165 ( Angew. Chem. , 1987 , 99 , 1210 ) CrossRef; (e) A. F. Pozharskii, O. V. Ryabtsova, V. A. Ozeryanskii, A. V. Degtyarev, O. N. Kazheva, G. G. Alexandrov and O. A. Dyachenko, J. Org. Chem., 2003, 68, 10109 CrossRef CAS PubMed; (f) A. V. Degtyarev, O. V. Ryabtsova, A. F. Pozharskii, V. A. Ozeryanskii, Z. A. Starikova, L. Sobczyk and A. Filarowski, Tetrahedron, 2008, 64, 6209 CrossRef CAS; (g) J. F. Kögel, N.-J. Kneusels and J. Sundermeyer, Chem. Commun., 2014, 50, 4319 RSC; (h) A. F. Pozharskii, V. A. Ozeryanskii, V. Y. Mikshiev, A. S. Antonov, A. V. Chernyshev, A. V. Metelitsa, G. S. Borodkin, N. S. Fedik and O. V. Dyablo, J. Org. Chem., 2016, 81, 5574 CrossRef CAS PubMed; (i) R. J. Schwamm, R. Vianello, A. Marsavelski, M. A. Garcia, R. M. Claramunt, I. Alkorta, J. Saame, I. Leito, C. M. Fitchett, A. J. Edwards and M. P. Coles, J. Org. Chem., 2016, 81, 7612 CrossRef CAS PubMed; (j) J. F. Kögel, B. Kovačević, S. Ullrich, X. Xie and J. Sundermeyer, Chem.–Eur. J., 2017, 23, 2591 CrossRef PubMed.
- (a) C. Lensink, S. K. Xi, L. M. Daniels and J. G. Verkade, J. Am. Chem. Soc., 1989, 111, 3478 CrossRef CAS; (b) J. Münchenberg, H. Thönnessen, P. G. Jones and R. Schmutzler, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 123, 57 CrossRef; (c) P. Mehlmann, C. Mück-Lichtenfeld, T. T. Y. Tan and F. Dielmann, Chem.–Eur. J., 2017, 23, 5929 CrossRef CAS PubMed.
- A. P. Marchenko, G. N. Koidan, A. M. Pinchuk and A. V. Kirsanov, J. Gen. Chem. USSR, 1984, 54, 1581 Search PubMed.
- S. Ullrich, B. Kovačević, X. Xie and J. Sundermeyer, Angew. Chem., Int. Ed., 2019, 58, 10335 ( Angew. Chem. , 2019 , 131 , 10443 ) CrossRef CAS PubMed.
- R. Schwesinger, C. Hasenfratz, H. Schlemper, L. Walz, E.-M. Peters, K. Peters and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1993, 32, 1361 ( Angew. Chem. , 1993 , 105 , 1420 ) CrossRef.
- I. Leito, I. A. Koppel, I. Koppel, K. Kaupmees, S. Tshepelevitsh and J. Saame, Angew. Chem., Int. Ed., 2015, 54, 9262 ( Angew. Chem. , 2015 , 127 , 9394 ) CrossRef CAS.
- (a) S. Goumri-Magnet, O. Guerret, H. Gornitzka, J. B. Cazaux, D. Bigg, F. Palacios and G. Bertrand, J. Org. Chem., 1999, 64, 3741 CrossRef CAS; (b) I. A. Koppel, R. Schwesinger, T. Breuer, P. Burk, K. Herodes, I. Koppel, I. Leito and M. Mishima, J. Phys. Chem. A, 2001, 105, 9575 CrossRef CAS.
- J. Saame, T. Rodima, S. Tshepelevitsh, A. Kütt, I. Kaljurand, T. Haljasorg, I. A. Koppel and I. Leito, J. Org. Chem., 2016, 81, 7349 CrossRef CAS.
- J. F. Kögel, D. Margetić, X. Xie, L. H. Finger and J. Sundermeyer, Angew. Chem., Int. Ed., 2017, 56, 3090 ( Angew. Chem. , 2017 , 129 , 3136 ) CrossRef.
- (a) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS; (b) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 ( Angew. Chem. , 2002 , 114 , 1342 ) CrossRef CAS; (c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (d) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678 CrossRef CAS PubMed; (e) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994 CrossRef CAS.
- (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 ( Angew. Chem. , 2005 , 117 , 5851 ) CrossRef CAS; (b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS; (c) Z. R. Turner, Chem.–Eur. J., 2016, 22, 11461 CrossRef CAS; (d) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 ( Angew. Chem. , 2017 , 129 , 10180 ) CrossRef CAS PubMed; (e) C. M. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255 CrossRef CAS PubMed.
- (a) C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206 ( Angew. Chem. , 2008 , 120 , 3250 ) CrossRef CAS PubMed; (b) A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 3210 ( Angew. Chem. , 2008 , 120 , 3254 ) CrossRef PubMed; (c) O. Kaufhold and F. E. Hahn, Angew. Chem., Int. Ed., 2008, 47, 4057 ( Angew. Chem. , 2008 , 120 , 4122 ) CrossRef CAS PubMed; (d) C. Pranckevicius, L. Liu, G. Bertrand and D. W. Stephan, Angew. Chem., Int. Ed., 2016, 55, 5536 ( Angew. Chem. , 2016 , 128 , 5626 ) CrossRef CAS; (e) W.-C. Chen, W.-C. Shih, T. Jurca, L. Zhao, D. M. Andrada, C.-J. Peng, C.-C. Chang, S.-K. Liu, Y.-P. Wang, Y.-S. Wen, G. P. A. Yap, C.-P. Hsu, G. Frenking and T.-G. Ong, J. Am. Chem. Soc., 2017, 139, 12830 CrossRef CAS PubMed; (f) S.-C. Chan, P. Gupta, X. Engelmann, Z. Z. Ang, R. Ganguly, E. Bill, K. Ray, S. Ye and J. England, Angew. Chem., Int. Ed., 2018, 57, 15717 ( Angew. Chem. , 2018 , 130 , 15943 ) CrossRef CAS PubMed.
- (a) W. C. Kaska, D. K. Mitchell and R. F. Reichelderfer, J. Organomet. Chem., 1973, 47, 391 CrossRef CAS; (b) W. Petz and G. Frenking, in Transition Metal Complexes of Neutral eta1-Carbon Ligands, ed. R. Chauvin and Y. Canac, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, vol. 30, pp. 49–92 Search PubMed; (c) W. Petz, Coord. Chem. Rev., 2015, 291, 1 CrossRef CAS; (d) J. E. Münzer, N.-J. H. Kneusels, B. Weinert, B. Neumüller and I. Kuzu, Dalton Trans., 2019, 48, 11076 RSC.
- (a) R. Tonner and G. Frenking, Chem.–Eur. J., 2008, 14, 3260 CrossRef CAS PubMed; (b) R. Tonner and G. Frenking, Chem.–Eur. J., 2008, 14, 3273 CrossRef CAS; (c) M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Nat. Chem., 2009, 1, 295 CrossRef CAS PubMed.
- F. Ramirez, N. B. Desai, B. Hansen and N. McKelvie, J. Am. Chem. Soc., 1961, 83, 3539 CrossRef CAS.
- O. Gasser and H. Schmidbaur, J. Am. Chem. Soc., 1975, 97, 6281 CrossRef CAS.
- R. Appel, U. Baumeister and F. Knoch, Chem. Ber., 1983, 116, 2275 CrossRef CAS.
- H. Schmidbaur, O. Gasser and M. S. Hussain, Chem. Ber., 1977, 110, 3501 CrossRef CAS.
- R. Appel and K. Waid, Z. Naturforsch. B Chem. Sci., 1981, 36, 131 Search PubMed.
- A. P. Marchenko, G. N. Koidan, V. A. Oleinik, I. S. Zal'tsman and A. M. Pinchuk, J. Gen. Chem. USSR, 1988, 58, 1485 Search PubMed.
- R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038 ( Angew. Chem. , 2006 , 118 , 8206 ) CrossRef CAS PubMed.
- R. Tonner, G. Heydenrych and G. Frenking, ChemPhysChem, 2008, 9, 1474 CrossRef CAS PubMed.
- (a) R. Appel, F. Knoll, W. Miche, W. Morbach, H.-D. Wihler and H. Veltmann, Chem. Ber., 1976, 109, 58 CrossRef CAS; (b) R. Appel, F. Knoll, H. Schöler and H.-D. Wihler, Angew. Chem., Int. Ed. Engl., 1976, 15, 702 ( Angew. Chem. , 1976 , 88 , 769 ) CrossRef; (c) R. Appel and H. Veltmann, Tetrahedron Lett., 1977, 18, 399 CrossRef; (d) A. P. Marchenko, G. N. Koidan, V. A. Oleinik, A. A. Kudryavtsev and A. M. Pinchuk, J. Gen. Chem. USSR, 1987, 57, 1916 Search PubMed; (e) V. A. Oleinik, A. P. Marchenko, G. N. Koidan and A. M. Pinchuk, J. Gen. Chem. USSR, 1988, 58, 625 Search PubMed; (f) G. N. Koidan, A. P. Marchenko, V. A. Oleinik and A. M. Pinchuk, J. Gen. Chem. USSR, 1988, 58, 1304 Search PubMed.
- F. Ramirez and N. McKelvie, J. Am. Chem. Soc., 1957, 79, 5829 CrossRef CAS.
- (a) J. Münchenberg, O. Böge, A. K. Fischer, P. G. Jones and R. Schmutzler, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 86, 103 CrossRef; (b) J. F. Kögel, N. C. Abacılar, F. Weber, B. Oelkers, K. Harms, B. Kovačević and J. Sundermeyer, Chem.–Eur. J., 2014, 20, 5994 CrossRef PubMed.
- Z. S. Novikova, A. A. Prishchenko and I. F. Lutsenko, J. Gen. Chem. USSR, 1977, 47, 707 Search PubMed.
- T. Rodima, I. Kaljurand, A. Pihl, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2002, 67, 1873 CrossRef CAS PubMed.
- I. Kaljurand, T. Rodima, A. Pihl, V. Mäemets, I. Leito, I. A. Koppel and M. Mishima, J. Org. Chem., 2003, 68, 9988 CrossRef CAS PubMed.
- A. P. Marchenko, G. N. Koidan and A. M. Pinchuk, J. Gen. Chem. USSR, 1984, 54, 2405 Search PubMed.
- K. Issleib and M. Lischewski, J. Prakt. Chem., 1970, 312, 135 CrossRef CAS.
- (a) T. Scherpf, C. Schwarz, L. T. Scharf, J.-A. Zur, A. Helbig and V. H. Gessner, Angew. Chem., Int. Ed., 2018, 57, 12859 ( Angew. Chem. , 2018 , 130 , 13041 ) CrossRef CAS PubMed; (b) P. Weber, T. Scherpf, I. Rodstein, D. Lichte, L. T. Scharf, L. J. Gooßen and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 3203 ( Angew. Chem. , 2019 , 131 , 3235 ) CrossRef CAS PubMed.
- P. J. Quinlivan and G. Parkin, Inorg. Chem., 2017, 56, 5493 CrossRef CAS PubMed.
- H. Schmidbaur, G. Hasslberger, U. Deschler, U. Schubert, C. Kappenstein and A. Frank, Angew. Chem., Int. Ed. Engl., 1979, 18, 408 ( Angew. Chem. , 1979 , 91 , 437 ) CrossRef.
- E. A. V. Ebsworth, T. E. Fraser, D. W. H. Rankin, O. Gasser and H. Schmidbaur, Chem. Ber., 1977, 110, 3508 CrossRef CAS.
- (a) A. T. Vincent and P. J. Wheatley, J. Chem. Soc., Dalton Trans., 1972, 617 RSC; (b) G. E. Hardy, J. I. Zink, W. C. Kaska and J. C. Baldwin, J. Am. Chem. Soc., 1978, 100, 8001 CrossRef CAS.
- W. Petz, M. Fahlbusch, E. Gromm and B. Neumüller, Z. Anorg. Allg. Chem., 2008, 634, 682 CrossRef CAS.
- W. Petz, K. Dehnicke and B. Neumüller, Z. Anorg. Allg. Chem., 2011, 637, 1761 CrossRef CAS.
- R. Ganguly and V. Jevtovic, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2017, 73, 1259 CrossRef CAS PubMed.
- (a) S. Ullrich, CCDC 1903847: Experimental Crystal Structure Determination; (b) S. Ullrich, CCDC 1903849: Experimental Crystal Structure Determination.
- H. Vogt, D. Wulff-Molder, F. Ritschl, M. Mücke, U. Skrabei and M. Meisel, Z. Anorg. Allg. Chem., 1999, 625, 1025 CrossRef CAS.
- S. Ullrich, CCDC 1903848: Experimental Crystal Structure Determination.
- M. Gruber, W. Bauer, H. Maid, K. Schöll and R. R. Tykwinski, Inorg. Chim. Acta, 2017, 468, 152 CrossRef CAS.
- (a) R. W. Alder, P. R. Allen and S. J. Williams, Chem. Commun., 1995, 1267 RSC; (b) Y.-J. Kim and A. Streitwieser, J. Am. Chem. Soc., 2002, 124, 5757 CrossRef CAS PubMed; (c) N. Wang, J. Xu and J. K. Lee, Org. Biomol. Chem., 2018, 16, 8230 RSC.
- (a) R. W. Alder, M. E. Blake and J. M. Oliva, J. Phys. Chem. A, 1999, 103, 11200 CrossRef CAS; (b) R. Tonner and G. Frenking, Angew. Chem., Int. Ed., 2007, 46, 8695 ( Angew. Chem. , 2007 , 119 , 8850 ) CrossRef CAS PubMed; (c) A. A. Tukov, A. T. Normand and M. S. Nechaev, Dalton Trans., 2009, 7015 RSC.
- S. Sastre, R. Casasnovas, F. Muñoz and J. Frau, Phys. Chem. Chem. Phys., 2016, 18, 11202 RSC.
- W. L. F. Armarego and D. D. Perrin, Purification of laboratory chemicals, Butterworth Heinemann, Oxford, Boston, 4th edn, 1996 Search PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1903830, 1903833, 1903838, 1903840, 1903841 and 1903843. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03565f |
| This journal is © The Royal Society of Chemistry 2019 |