
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New sulfuryl fluoride-derived alkylating reagents for the 1,1-dihydrofluoroalkylation of thiols†
Paul J.
Foth
,
Frances
Gu
,
Trevor G.
Bolduc
,
Sahil S.
Kanani
and
Glenn M.
Sammis
 *
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: gsammis@chem.ubc.ca
First published on 20th September 2019
Abstract
Herein, we report a new method for the one-pot synthesis of 1,1-dihydrofluoroalkyl sulfides by bubbling sulfuryl fluoride (SO2F2) through a solution of the corresponding alcohol and thiol. The reaction proceeds through a new class of bis(1,1-dihydrofluoroalkyl) sulfate reagents, to afford the desired 1,1-dihydrofluoroalkyl sulfides in 55–90% isolated yields. The bis(1,1-dihydrofluoroalkyl) sulfates are highly chemoselective for thiol alkylation, and are unreactive with competing, unprotected nucleophiles, including amines, alcohols, and carboxylic acids.
Sulfuryl fluoride (SO2F2) has been utilized since the 1960s as an industrial fumigant,1 but it has only recently attracted significant attention as a reagent for organic synthesis.1b,2 Studies have demonstrated that oxygen nucleophiles, such as alcohols (1),3,4 phenol derivatives (2),1b,5 oximes (3),6 and carboxylic acids (4),7 react with sulfuryl fluoride to form fluorosulfate derivatives (Scheme 1).2 The addition of a second equivalent of the oxygen nucleophile is kinetically slow, which allows fluorosulfate 5 to undergo subsequent transformations.8 Fluorosulfates (5) have been utilized as key reactants in a diverse range of reactions, including metal-catalyzed cross couplings,5c,9 click reactions,1b,5d deoxyfluorinations,5b alkylations,3a,4 nitrile syntheses,6 and the formation of amide bonds.7a
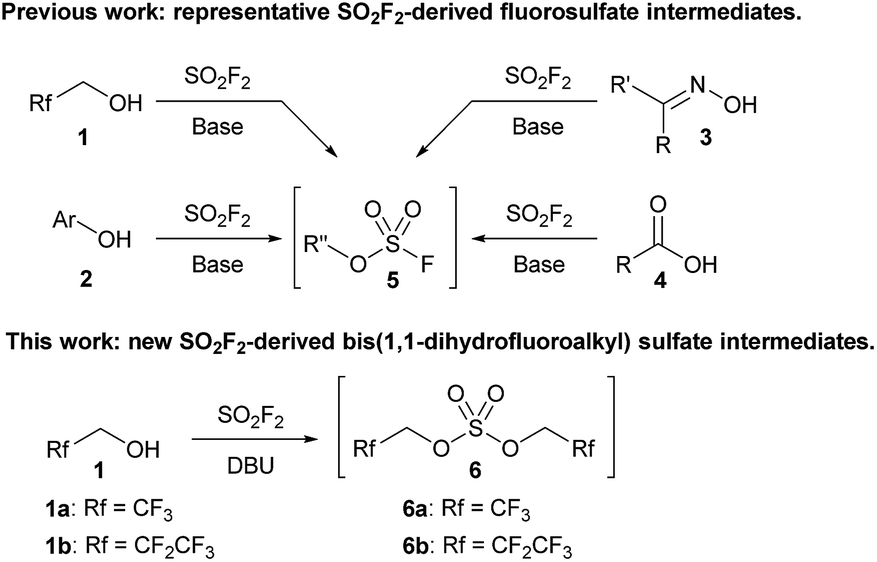 | ||
| Scheme 1 Representative examples of sulfuryl fluoride-mediated processes that utilize fluorosulfate reactive intermediates (5) and a new bis(trifluoroalkyl) sulfate (6). | ||
All current synthetic methods rely on a very similar protocol for the formation of the fluorosulfate intermediate. Sulfuryl fluoride is bubbled through a solution of the requisite oxygen nucleophile and a base, which is usually N,N-diisopropylethylamine (DIPEA), triethylamine, or a carbonate salt.1b,2–7 Despite the expansion of the use of sulfuryl fluoride, the only reactive intermediates that have been identified are fluorosulfate derivatives (5), and no other sulfuryl fluoride-derived reactive intermediates have been explored.10
We previously reported that bubbling sulfuryl fluoride through a solution of 2,2,2-trifluoroethanol (1a) and an amine base, such as DIPEA or triethylamine, afforded trifluoroethyl fluorosulfate (5a, R′′ = CH2CF3) in >90% yield.3a,11 Following up on the synthesis and reactivity of fluorosulfate 5a in new transformations, we serendipitously discovered that even moderately more basic reagents,12 such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) with a pKaH of 12,13 afforded bis(trifluoroethyl) sulfate (6a) as the major product, and only trace amounts of fluorosulfate 5a were detected by 19F NMR spectroscopy. Bis(trifluoroethyl) sulfate (6a) is an intriguing species as there are only two previous methods for its synthesis,14–16 and there are no studies investigating its reactivity.17
We elected to study the reactivity of this new bis(trifluoroethyl) sulfate intermediate (6a) for the 1,1-dihydrofluoroalkylation of thiols for two reasons: (1) the resulting fluoroalkyl sulfides are important fluorinated motifs in pharmaceuticals and agrochemicals,18–20 and (2) the more common sulfuryl fluoride-derived reagent, trifluoroethyl fluorosulfate 5a, is not an effective intermediate for thiol alkylation. Previous studies by Shreeve and coworkers indicated that the reaction between methane thiol (7), triethylamine, and 5a afforded the corresponding fluoroalkyl sulfide (8) in only 31% yield and a 2.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 preference for reactivity at carbon compared to sulfur (Scheme 2A).14
:1 preference for reactivity at carbon compared to sulfur (Scheme 2A).14
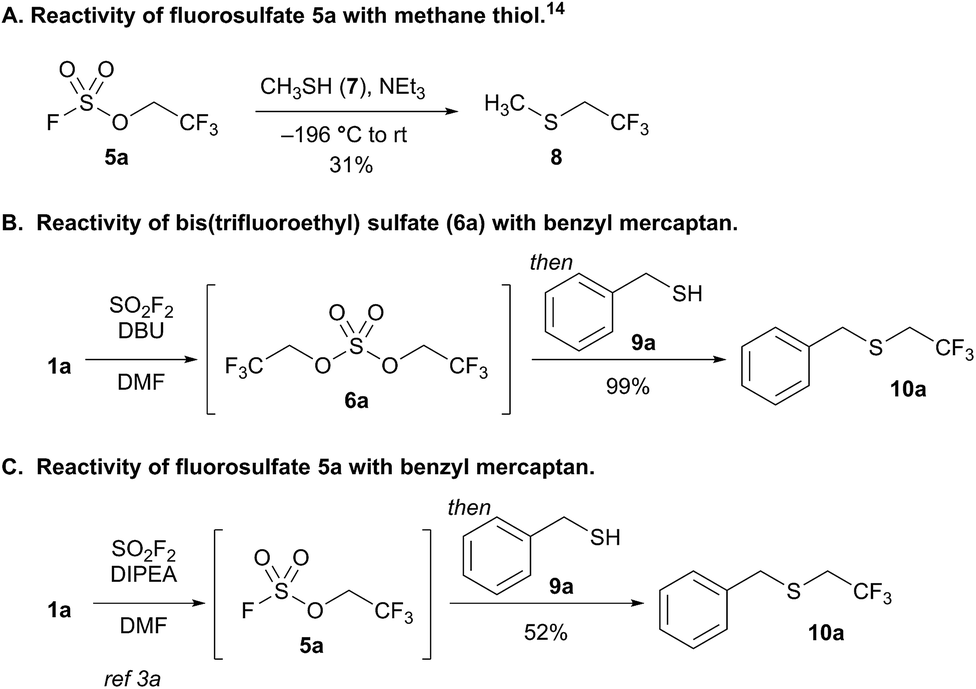 | ||
| Scheme 2 Investigations into thiol alkylation using fluorosulfate (5a) and bis(trifluoroethyl) sulfate (6a). The yields in (B) and (C) were determined by 19F NMR spectroscopy using trifluorotoluene as an internal standard. | ||
To examine the viability of bis(trifluoroethyl) sulfate (6a) as a thiol alkylating reagent, we treated a solution of 6a and DBU with benzyl mercaptan (9a), which led to a 99% 19F NMR yield of 1,1-dihydrofluoroalkylated product 10a (Scheme 2B). The analogous reaction with a solution of DIPEA and fluorosulfate 5a with 9a afforded only 52% yield of 10a (Scheme 2C), which is comparable to the result reported by Shreeve and coworkers.14 The addition of DBU and benzyl mercaptan to a solution of DIPEA and 5a improved the yield; however, the reaction led to an increase in the amount of free trifluoroethanol in solution, presumably resulting from nucleophilic attack at sulfur.21
This initial result is noteworthy as it represents the first example of the direct conversion of an unactivated 1,1-dihydrofluoroalcohol to the corresponding fluoroalkyl sulfide in a one-pot process. Thiol 1,1-dihydrofluoroalkylation can be achieved through nucleophilic displacement of activated trifluoroalkyl moieties,14,22 copper-catalyzed reactions with trifluoroalkyl iodide23 or trifluorodiazoalkanes,24 or reductive trifluoroalkylthiolations.25 All previous work relies on activated trifluoroalkyl moieties. This is particularly problematic for select activated trifluoroethyl derivatives and longer chain 1,1-dihydrofluoroalkyl groups that are only available from the corresponding alcohols, and thus require additional synthetic steps to activate.
With an established protocol for a one-pot, sequential reaction, investigations next focused on a one-step process, where the alkylation proceeds by bubbling sulfuryl fluoride through a solution of trifluoroethanol (1a), benzyl mercaptan (9a), and DBU. At room temperature, the reaction proceeded efficiently to afford desired trifluoroethylated product 10a in 71% yield (Table 1, entry 1). The yield increased at 40 °C (entry 2), but there was no further improvement when the reaction was run at 60 °C (entry 3). We next examined the reaction performance in different solvents (entries 4–8).26 Overall, the reaction was robust in a range of solvents, providing good yield in both polar aprotic (entries 4 and 5) and nonpolar solvents (entries 6 and 7). DCM was not as effective for this transformation, with product 10a observed in only 38% yield (entry 8).27
|
|
|||
|---|---|---|---|
| Entry | Solvent | Temp (°C) | 19F NMR yieldb (%) |
|
a All reactions were carried out following a one-pot procedure on 0.30 mmol scale of 9a and a 1:1 v/v trifluoroethanol:solvent ratio.
b Yield after 20 minutes, as determined by 19F NMR spectroscopy using trifluorotoluene as an internal standard.
|
|||
| 1 | DMF | 25 | 71 |
| 2 | DMF | 40 | 86 |
| 3 | DMF | 60 | 85 |
| 4 | THF | 40 | 81 |
| 5 | ACN | 40 | 77 |
| 6 | Hexane | 40 | 75 |
| 7 | Benzene | 40 | 67 |
| 8 | DCM | 40 | 38 |
The one-pot reaction generally affords high yields of the desired thioalkylated product regardless of the steric bulk or the electronics of the thiol (Scheme 3). Benzyl mercaptan (9a) and furfuryl mercaptan (9b) were both efficiently trifluoroethylated to form the corresponding sulfides 10a and 10b in good yields. 1-Decanethiol (9c), 2-phenylethanethiol (9d), methyl thiolglycolate (9e), and 1,9-nonanedithiol (9f) were effective substrates for this transformation, affording mono- and dialkylated products 10c–10f in 58% to 73% isolated yields. The reaction was insensitive to steric bulk alpha to the thiol, with both cyclohexyl mercaptan (9g) and triphenylmethanethiol (9h) alkylated in comparable yields (10g and 10h, respectively). Electron rich and electron poor thiophenol derivatives were well tolerated, regardless of the position of the substituents (10i-q). Importantly, longer chain 1,1-dihydrofluoroalcohols, such as 2,2,3,3,3-pentafluoropropanol (1b), were viable starting materials; however extended reaction times were required. The isolated yields of 11a and 11c were increased to 90% and 89%, respectively, by conducting the reaction in a sequential one-pot manner,28 where sulfuryl fluoride was first bubbled through a solution of DBU and trifluoroethanol followed by the addition of the requisite thiol.
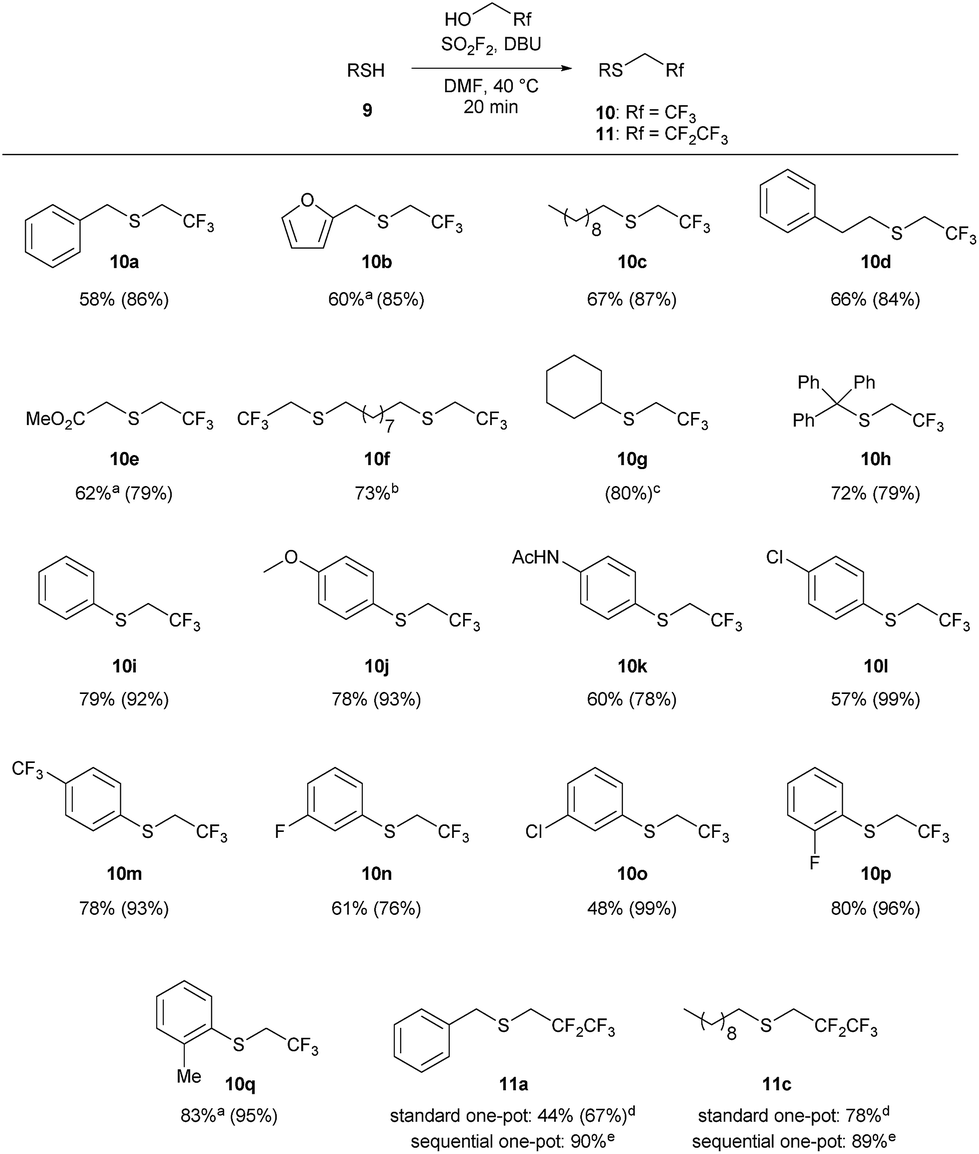 | ||
| Scheme 3 Substrate scope for the 1,1-dihydrofluoroalkylation of thiols. Reaction conditions: SO2F2 (2.9 equiv.) was bubbled through a solution of 9 (1 equiv.), DBU (5.9 equiv.) in 1:1 TFE/DMF (v/v), at 40 °C for 3 minutes, and then the reaction was stirred for an additional 17 min. All reactions were run on 1 mmol scale of thiol unless otherwise indicated. Isolated yields for the one-pot reaction are reported, with 19F NMR yields (using trifluorotoluene as the internal standard) provided in parentheses. aThe isolated yield has been corrected to account for disulfide or solvent impurities. See the ESI† for details. bThe reaction was conducted on 0.5 mmol scale. cThe product was not isolated due to volatility. dThe reaction was stirred for 2 hours. ePentafluoropropanol:DMF (1:2 v/v) was used to form the reagent, and then the thiol was added to the reaction mixture. The reaction was stirred for 30 minutes. | ||
We next examined the functional group tolerance of this new thiol 1,1-dihydrofluoroalkylation (Scheme 4). In substrates in which there is competition between alcohol and thiol alkylation, the reaction cleanly afforded good yields of the desired thiol 1,1-dihydrofluoroalkylation products (10r and 11r). Carboxylic acids were also tolerated, with good isolated yields of thiol alkylated product 10s using either the standard one-pot or the sequential one-pot protocols. The reaction was selective for the thiol over potential competing reactivity at the nitrogen atom of aniline and pyridine derivatives (10t, 11t, and 10u). As primary nitrogen derivatives were competent nucleophiles in reactions with trifluoroalkyl fluorosulfate, we next examined the reaction of L-cysteine ethyl ester. Under our sequential one-pot conditions, reactivity was only observed at sulfur to give 10v in 63% isolated yield.29
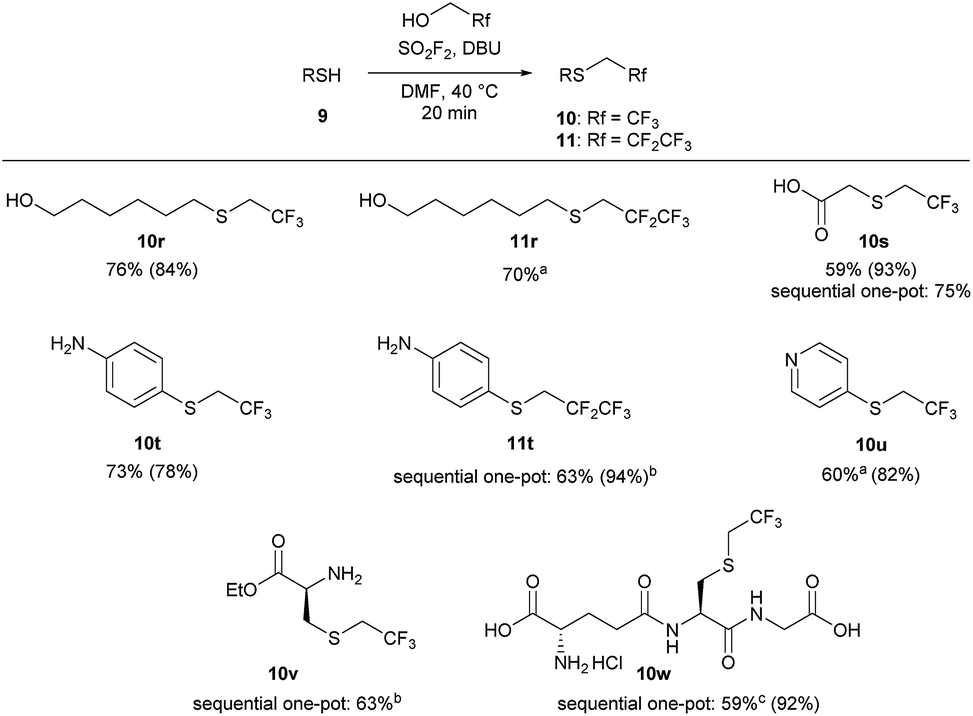 | ||
| Scheme 4 Functional group tolerance of the 1,1-dihydrofluoroalkylation reaction. Reaction conditions: SO2F2 (2.9 equiv.) was bubbled through a solution of 9 (1 equiv.), DBU (5.9 equiv.) in 1:1 TFE/DMF (v/v), at 40 °C for 3 minutes, and then the reaction was stirred for an additional 17 min. All reactions were run on a 1 mmol scale unless otherwise indicated. Isolated yields for the one-pot reaction are reported, with 19F NMR yields (using trifluorotoluene as the internal standard) provided in parentheses. aThe one-pot reaction was stirred for 2 hours. bThe sequential, one-pot reaction was stirred for 30 minutes after addition of thiol. cThe product was converted to an HCl salt, and the reported yield has been corrected for solvent impurities. | ||
Finally, we investigated whether we could achieve selective 1,1-dihydrofluoroalkylation using glutathione (9w). Glutathione is a challenging substrate as it has two carboxylic acids, an amine, and two amides that may interfere with the desired thiol fluoroalkylation. Gratifyingly, under our sequential, one-pot reaction conditions, the thiol was selectively alkylated in 92% 19F NMR yield and 59% isolated yield. Under similar reaction conditions, trifluoroethyl triflate only afforded moderate yields of 10w.
Intrigued by the chemoselectivity of the bis(trifluoroethyl) sulfate reagent (6a), we next investigated its selectivity compared to trifluoroethyl fluorosulfate (5a)30 in a competition experiment between benzyl mercaptan (9a) and piperidine (12)31 (Scheme 5).32 Addition of 9a and 12 to a preformed solution of trifluoroethyl fluorosulfate and DIPEA afforded only a slight preference for thiol alkylation (Scheme 5A). Trifluoroethanol (1a) was liberated in the course of the reaction, which is likely the result of the addition of thiol to the sulfur center of the fluorosulfate reagent.33,34 Better selectivity for thiol versus amine alkylation could be achieved by adding DBU with 9a and 12;35 however, there was also a concomitant increase in the amount of 1a (Scheme 5B). Increasing the amount of fluorosulfate reagent 5a resulted in more amine alkylated product (13), but did not lead to a significantly better yield of 10a. In contrast, formation of bis(trifluoroethyl) sulfate (6a) followed by addition of 9a and 12 led to 90% yield of thiol alkylated product 10a, and only trace amounts of 13 and trifluoroethanol (Scheme 5C). Further increasing the equivalents of the alkylating reagent led to near quantitative yield of 10a (>97%). Even when pyrrolidine (14), a more nucleophilic amine,36 was used in a competition experiment, trifluoroethyl sulfide 10a was obtained almost exclusively (Scheme 5D).37
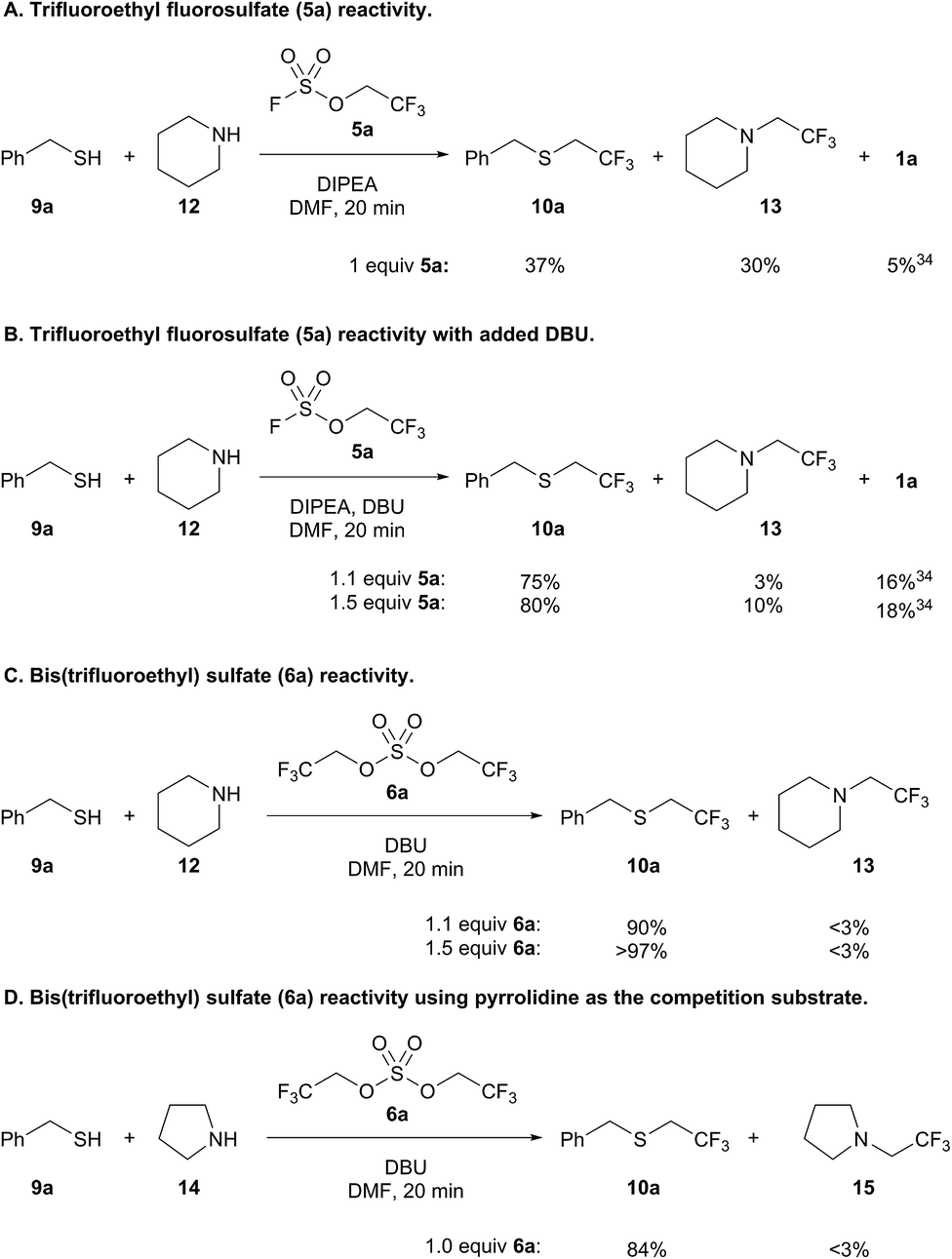 | ||
| Scheme 5 Competition experiments with sulfur and nitrogen nucleophiles. | ||
Overall, we have developed a new method for the 1,1-dihydrofluoroalkylation of thiols using a previously unexplored, sulfuryl fluoride derived bis(trifluoroethyl) sulfate reagent (6a). This protocol enables the one-pot activation and thiolation of 1,1-dihydrofluoroalcohols to afford industrially relevant moieties in high yields, regardless of the sterics or electronics of the starting thiol. In situ generated bis(trifluoroethyl) sulfate (6a) is highly selective for thiols, even in the presence of unprotected alcohols, carboxylic acids, or amines, allowing for possible late-stage functionalization. Compared to trifluoro-ethyl fluorosulfate, the new bis(trifluoroethyl) sulfate reagent displays superior thiol alkylation chemoselectivity over both competing amine alkylation and reactivity at the sulfate center. Efforts to further explore this new class of bis(1,1-dihydrofluoroalkyl) reagents in the context of other reactions are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of British Columbia (UBC), the Natural Sciences and Engineering Research Council of Canada (NSERC), the NSERC CREATE Sustainable Synthesis Program for support for PJF, an NSERC CGSM scholarship for TGB, and a UBC Work Learn International Undergraduate Research Award to SSK.Notes and references
- (a) E. E. Kenaga, J. Econ. Entomol., 1957, 50(1), 1–6 CrossRef CAS; (b) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53(36), 9430–9448 CrossRef CAS PubMed; (c) Dow AgroSciences Technical Bulletin, “Sulfuryl Fluoride Gas Fumigant”, April 2002.
- For a representative review, see: L. Revathi, L. Ravindar, J. Leng, K. P. Rakesh and H.-L. Qin, Asian J. Org. Chem., 2018, 7(4), 662–682 CrossRef CAS.
- For select examples of 1,1-dihydrofluoroalcohol activation, see: (a) M. Epifanov, P. J. Foth, F. Gu, C. Barrillon, S. S. Kanani, C. S. Higman, J. E. Hein and G. M. Sammis, J. Am. Chem. Soc., 2018, 140(48), 16464–16468 CrossRef CAS PubMed; (b) M. R. Johnson, WO Pat. 124 456, 2014.
- For a representative example, see: A. Ishii, T. Yamazaki and M. Yasumoto, US Pat. 8 426 645B2, Central Glass Company, Ltd., 2013.
- For representative examples, see: (a) W. C. J. Firth, J. Polym. Sci., Part B: Polym. Phys., 1972, 10, 637–641 CrossRef CAS; (b) S. D. Schimler, M. A. Cismesia, P. S. Hanley, R. D. J. Froese, M. J. Jansma, D. C. Bland and M. S. Sanford, J. Am. Chem. Soc., 2017, 139(4), 1452–1455 CrossRef CAS PubMed; (c) P. S. Hanley, M. S. Ober, A. L. Krasovskiy, G. T. Whiteker and W. J. Kruper, ACS Catal., 2015, 5(9), 5041–5046 CrossRef CAS; (d) J. Dong, K. B. Sharpless, L. Kwisnek, J. S. Oakdale and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53(36), 9466–9470 CrossRef CAS PubMed.
- (a) J. Gurgar, J. Baker and V. V. Fokin, Chem.–Eur. J., 2019, 25(8), 1906–1909 CrossRef PubMed; (b) W.-Y. Fang and H.-L. Qin, J. Org. Chem., 2019, 84(9), 5803–5812 CrossRef CAS PubMed.
- For representative examples, see: (a) S.-M. Wang, C. Zhao, X. Zhang and H.-L. Qin, Org. Biomol. Chem., 2019, 17, 4087–4101 RSC; (b) A. Ishii, M. Yasumoto and U. Koji, JP Pat 155 248, 2009.
- For an early report on the stability of aryl fluorosulfates, see ref. 5a.
- A. Ishii, T. Ishimaru and T. Yamazaki, JP Pat. 001 653 A, 2013.
- While other minor sulfuryl fluoride-derived products have been observed (see ref. 3a), they have only been detected in <10% yield.
- When non-fluorinated alcohols are used, the corresponding alkyl fluorosulfate is highly reactive and leads to numerous degradation products. The 1,1-dihydrofluoroalkyl group makes the corresponding fluorosulfate more stable and less prone to degradation. For a study on the effect of halides alpha to the electrophilic center of SN2 reactions, see: J. Hine and W. H. Brader Jr, J. Am. Chem. Soc., 1953, 75(16), 3964–3966 CrossRef CAS.
- Stronger bases, such as KO-tBu and KHMDS, provide comparable results as DBU. Presumably, this is because more alkoxide is formed in the reaction, thus accelerating the conversion of 5a to 6a. For full base optimization, see the ESI.†.
- D. Granitza, M. Beyermann, H. Wenschuh, H. Haber, L. A. Carpino, G. A. Truran and M. Bienert, J. Chem. Soc., Chem. Commun., 1995, 21, 2223–2224 RSC.
- Shreeve et al., synthesized fluorosulfate 5a in two separate steps using SOCl2 followed by ClF. Fluorosulfate 5a could be converted to the corresponding bis(trifluoroethyl) sulfate (6a) by running the reaction under solvent-free conditions using triethylamine and warming the reaction from −196 °C to room temperature. No subsequent reactivity studies were performed on 6a. S. A. Kinkead, R. C. Kumar and J. M. Shreeve, J. Am. Chem. Soc., 1984, 106(24), 7496–7500 CrossRef CAS.
- For a synthesis of bis(trifluoroethyl) sulfate (6a) from trifluoroethanol and sulfuryl chloride, see: W. V. Cohen, J. Org. Chem., 1961, 26(10), 4021–4026 CrossRef CAS.
- We have previously detected the formation of bis(trifluoroethyl) sulfate (6a) as a minor byproduct in the formation of trifluoroethyl fluorosulfate (5a). See ref. 3a.
- While a SciFinder® search indicates that bis(trifluoroethyl) sulfate (6a) was reported (WO Pat. 066 559, 2000), the reagent is not mentioned in this patent..
- For selected reviews on fluoroalkyl sulfides see: (a) S. Swallow, Prog. Med. Chem., 2015, 54, 65–133 Search PubMed; (b) F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105(3), 827–856 CrossRef CAS PubMed.
- For selected patents containing trifluoroethyl sulfur compounds see: (a) A. Adrien Ép. Köhler, B. Alig, A. Becker, A. Voerste, U. Göergens, R. Fischer, W. A. Moradi, S. Cerezo-Galvez, J. Neumann, K. Ilg, H.-G. Schwarz, T. Gomibuchi, M. Ito, D. Yamazaki, K. Shibuya and E. Shimojo, WO Pat. 092 350, 2013; (b) F. Setsu, E.-J. Umemura, K. Sasaki, K. Tadauchi, T. Okutomi, K. Ohtsuka and S. Takahata, WO Pat. 042 188, 2003; (c) F. Kaiser, S. Gross, J. Langewald and A. Narine, WO Pat. 030 262, 2013; (d) B. Alig, S. Cerezo-Galvez, R. Fischer, A. Köhler, J. J. Hahn, K. Ilg, P. Lösel, O. Malsam and D. Portz, WO Pat. 004 028, 2015.
- For the effect of fluoroalkyl moieties on the properties in a molecule, as well as the importance of fluorine in the pharmaceutical and agrochemical industries, see: (a) W. A. Sheppard, J. Am. Chem. Soc., 1963, 85(9), 1314–1318 CrossRef CAS; (b) B. Manteau, S. Pazenok, J.-P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140–158 CrossRef CAS; (c) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115(2), 765–825 CrossRef CAS PubMed; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (e) P. Jeschke, ChemBioChem, 2004, 5(5), 570–589 CrossRef CAS PubMed; (f) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114(4), 2432–2506 CrossRef CAS PubMed; (g) B. Manteau, S. Pazenok, J.-P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140–158 CrossRef CAS; (h) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317(5846), 1881–1886 CrossRef PubMed; (i) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127(3), 303–319 CrossRef CAS.
- See the ESI† for full reaction details.
- For representative examples, see: (a) Z.-Y. Long and Q.-Y. Chen, J. Fluorine Chem., 1998, 91(1), 95–98 CrossRef CAS; (b) J. Hine and R. G. Ghirardelli, J. Org. Chem., 1958, 23(10), 1550–1552 CrossRef CAS; (c) T. Nakai, K. Tanaka, H. Setoi and N. Ishikawa, Bull. Chem. Soc. Jpn., 1977, 50(11), 3069–3070 CrossRef CAS; (d) R. F. Langler and N. A. Morrison, Can. J. Chem., 1987, 65(10), 2385–2389 CrossRef CAS; (e) A. Adin, J. J. Looker, S. Y. Farid, I. R. Gould, S. A. Godleski, J. R. Lenhard, A. A. Muenter, L. C. Vishwakarma and P. A. Zielinski, US Pat. 6 054 260, 2000; (f) T. Umemoto and Y. Gotoh, J. Fluorine Chem., 1986, 31(2), 231–236 CrossRef CAS; (g) Y. Pustovit, A. Alexenko, S. Trofymchuk, O. Lukin and A. A. Tolmachev, Synthesis, 2010, 7, 1159–1165 CrossRef.
- S. Chen, M. Zhang, X. Liao and Z. Weng, J. Org. Chem., 2016, 81(17), 7993–8000 CrossRef CAS PubMed.
- S. Hyde, J. Veliks, B. Liegault, D. Grassi, M. Taillefer and V. Gouverneur, Angew. Chem., Int. Ed., 2016, 55(11), 3785–3789 CrossRef CAS PubMed.
- (a) R. Wang, L. Jiang and Y. Wenbin, J. Org. Chem., 2018, 83(15), 7789–7798 CrossRef CAS PubMed; (b) L. Jian, W. Yi and Q. Liu, Adv. Synth. Catal., 2016, 358(23), 3700–3705 CrossRef.
- The reaction also works well using 1:2 and 1:4 TFE/DMF solvent ratios, which correlates to approximately 13 and 8 equivalents of TFE, respectively. For optimization of the amount of trifluoroethanol relative to solvent, see the ESI.†.
- The alkylation step is slower in DCM than in more polar solvents, such as DMF.
- Under the standard one-pot conditions, sulfuryl fluoride can react with the thiol, which leads to disulfides, among other products. The sequential, one-pot conditions prevent this side reaction from occurring. For a discussion of this disulfide, see the ESI.†.
- 19F NMR spectroscopic analysis of the crude reaction mixture indicated that no amine 1,1-dihydrofluoroalkylation products were observed.
- Reactivity was compared to fluoroalkyl fluorosulfates, such as 5a, because they are the only other sulfuryl fluoride-derived reagent that can be generated from any 1,1-dihydrofluoroalcohol, and used in a one-pot process.
- Piperidine was selected for the competition experiment as it was an effective substrate for amine trifluoroethylation (see ref. 3a).
- For full reactions conditions, see the ESI.†.
- As attack at the sulfur atom with nitrogen was not typically seen under these conditions (see ref. 3a), the reagent degradation is likely to be due to thiol attack at sulfur.
- The amount of trifluoroethanol is due to nucleophile-mediated degradation of the alkylating reagent. Therefore, the percentage of trifluoroethanol was calculated relative to the total amount of benzyl mercaptan.
- Similar moderate selectivity for S- versus N-trifluoroalkylation was observed with trifluoroethyl triflate. See the ESI† for details.
- T. Kanzian, T. A. Nigst, A. Maier, S. Pichl and H. Mayr, Eur. J. Org. Chem., 2009, 36, 6379–6385 CrossRef.
- The competition experiment was run using an equimolar amount of benzyl mercaptan (9a) and pyrrolidine, and 1.0 equiv. of sulfate 6a. No amine alkylation product was detected by 19F NMR spectroscopy using trifluorotoluene as an internal standard. See the ESI† for full reaction details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, methods, and optimization data; NMR, IR, and MS data including 1H, 13C, and 19F NMR spectra. See DOI: 10.1039/c9sc03570b |
| This journal is © The Royal Society of Chemistry 2019 |