
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFreeze the dynamicity: charge transfer complexation assisted control over the reaction pathway†
Nilotpal
Singha‡
,
Basab Kanti
Das‡
,
Bapan
Pramanik§
 ,
Saurav
Das§
and
Debapratim
Das
*
,
Saurav
Das§
and
Debapratim
Das
*
Department of Chemistry, Indian Institute of Technology Guwahati, Assam 781039, India. E-mail: ddas@iitg.ac.in
First published on 9th September 2019
Abstract
Charge transfer (CT) complexes between electron donor and acceptor molecules provide unique alternate D–A arrangements. However, these arrangements cannot be fully translated into chemo-selective organic transformations as the dynamicity of CT complexes in solution results in the co-existence of D–A assemblies and free monomers during the reaction time-scale. A conceptually new strategy to exploit CT complexes toward chemo-selective products by means of seizing the dynamicity of CT complexes is reported here. Aqueous CT complexes of donor and acceptor molecules bearing reactive thiol groups were frozen instantly and cryo-desiccated to get the alternate D–A assemblies intact in the solid state. Oxidation of reactive thiols in an oxygen rich solvent in the solid state resulted in the formation of the hetero-dimer exclusively. CT complexation and appropriate molecular arrangements are the key factors behind successful execution of this novel methodology. The strategy also paves the way to prepare unsymmetrical disulfide molecules from two dissimilar thiols.
Introduction
Molecular self-assemblies have the capacity to direct specific reaction pathways that are otherwise difficult to achieve.1 Weak charge-transfer (CT) interactions via molecular orbital overlap between electron donor (D) and acceptor (A) aromatic molecules have been extensively utilized for the design of various supramolecular assemblies in solution, like rotaxanes,2 catenanes,3 supra-amphiphiles,4–6 foldamers,7etc. CT assemblies have also been comprehensively used aqueous dynamic combinatorial libraries to create molecular loops, knots and catenanes.3,8–12 In solution, the association constants for D–A assemblies range between 101–103 M−1.13,14 The molecules thus remain in a dynamic motion between the aggregated (CT state) and monomeric state.15,16 The dynamic nature of CT complexes does not allow the alternate D–A arrangements to get fully translated to covalently linked D–A when appropriate organic transformation is carried out in the solution phase. Any such chemical transformation in the solution phase thus results in low yields of the desired products as the dynamicity of constituent molecules also leads to orthogonal assemblies between the same molecules (Scheme 1A) resulting in undesired products.9 In order to obtain a highly selective product, the dynamicity of the system needs to be regulated. Control over these reactions is often obtained by addition of salts or a template which can be removed once the product is formed.10,17–19 Though regulating the reaction pathway could be achieved to a certain extent, these approaches cannot stop the formation of undesired orthogonal products significantly.10,17,18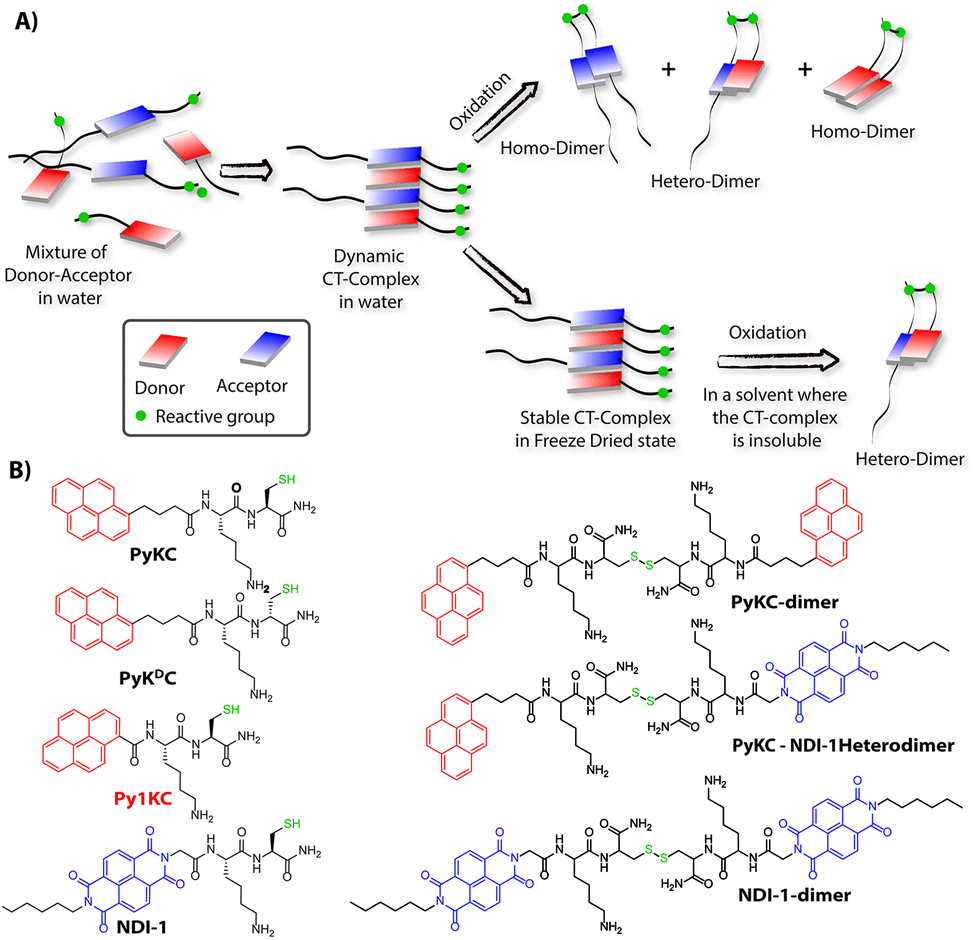 | ||
| Scheme 1 (A) Schematic presentation of the CT-complexation driven protocol to control the chemo-selectivity. (B) Chemical structures of different thiol substrates used for the study and their homo and heterodimers. | ||
We hypothesized that seizing the dynamic nature of the nano-structures formed by CT interactions may allow one to regulate the reaction pathway. Slow-drying of the solutions certainly affects the aggregation pattern and does not reflect the actual nano-structures formed in the solution.20 The seizing of dynamicity thus can be achieved by bringing these nano-structures out of the solvent by means of instant freezing followed by drying as in the case of lyophilization. Owing to instant freezing, alternate D–A arrangements will remain intact in these nano-structures where the reactive groups will be in close proximity to each other. Allowing them to react in the solid state will effectively lead to the formation of a specific product free of other undesired ones (Scheme 1A). However, we also anticipated that the choice of appropriate CT partners as well as their spatial arrangement in the CT complex will also be equally important to bring the reactive groups at close enough proximity to ensure the reaction in the solid state.
To this end, unsymmetrical disulfides are of tremendous pharmaceutical importance and at the same time are synthetically extremely challenging.21–24 It is worth mentioning that disulfide bond formation is a spontaneous process and can be obtained by simple change in pH of the medium.25 However, mixing two different thiols results in a mixture of all three combinations. Moreover, there is always a possibility of dynamic disulfide exchange to occur and that has been exploited widely in dynamic covalent chemistry.25,26 Thus, methodologies to prepare asymmetric disulfides generally involve functional groups other than thiols and special reagents/reaction conditions.21,22,27–29 For the present work, we chose thiol (–SH) as the reactive group based on these facts and also to develop a simple methodology to create asymmetric disulfides exclusively from two different thiols.
Herein, we report the successful execution of the novel concept of freezing the dynamicity of CT complexes to get control over the reaction pathway in the solid state. In this process, we observed that proper execution of the methodology not only requires the CT complexation of donor and acceptor molecules but also their special orientation in the CT complex plays a crucial role.
Results and discussion
To validate the hypothesis, we have taken pyrene (Py) and naphthalenediimide (NDI) as the donor and acceptor respectively since they showed strong CT complexation ability.30 Initially, a series (Scheme S3†) of NDI and Py containing peptide amphiphiles31 were designed and subjected to density functional theory (DFT) calculations to get energy minimized structures of these D–A pairs in the CT complex states. The molecules were designed in a way to introduce water solubility by incorporating residues like lysine (Lys) or arginine (Arg). The presence of cysteine (Cys) residues provides reactive thiol groups for disulfide formation. Incorporation of the hydrophobic tail at one end of the NDI moiety ensures that the peptide sequences remain face to face as NDI and Py units form CT complexes (Scheme 1A). Amongst all these pairs, PyKC32 and NDI-1 (Scheme 1 and S3†) were found to have the closest proximity of the –SH groups (4.07 Å, Fig. 1, S1 and Table S2†) in the energy minimized structures of the pairs as obtained from DFT calculations. We envisioned that this distance is close enough to form the S–S bond (S–S bond distance is ∼2.05 Å) during the planned solid phase oxidation. Moreover, the distance between the π-planes was found to be 3.51 Å indicating possible strong CT interactions between the molecules.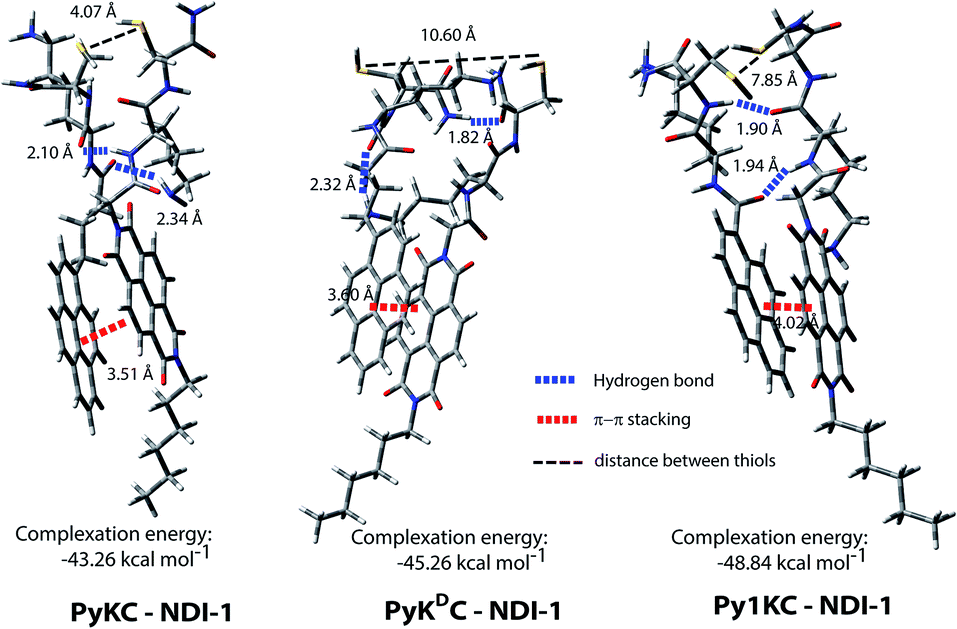 | ||
| Fig. 1 Energy minimized structures of different D–A pairs as obtained from DFT calculations showing the distances between the –SH groups and between π-rings of the donor and acceptor. | ||
Based on these results, PyKC and NDI-1 were selected and synthesized. CT complexation between these molecules was monitored using UV-visible spectroscopy. As can be seen from Fig. 2A, an equimolar mixture of the compounds resulted in the appearance of a new broad band at 550 nm which is the characteristic CT-band for Py–NDI pair.30 A concentration dependent study showed that below 0.05 mM individual concentration, no CT-band could be observed indicating 0.05 mM as the critical concentration below which these molecules probably fail to experience the CT interaction. A titration experiment monitored at the pyrene emission maximum (376) of PyKC showed a continuous quenching of the emission as the concentration of NDI-1 increased. A saturation obtained at a molar ratio of 1 suggests a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding between PyKC and NDI-1 (Fig. 2B). Isothermal titration calorimetry (ITC) data showed 1:1 complexation as well with an association constant of (1.05 ± 0.164) × 104 M−1 (Fig. 2C). Moreover, 1H NMR spectra (Fig. S2A†) of the mixture showed prominent up-field shifts along with broadening of the aromatic signals for both the components when they were mixed together in an equimolar ratio. These results confirm strong CT interactions between the Py and NDI units of PyKC and NDI-1 respectively. Additionally, some of the amide-NH and NH2 protons of both the molecules showed down-field shifts in the complex form which can only result if they are involved in hydrogen bonding. The intermolecular hydrogen bonding between PyKC and NDI-1 is also prominent from the Fourier transform infrared (FTIR) spectroscopic analyses of the pure molecules and the complex (Fig. S2B†). Both PyKC and NDI-1 showed non-hydrogen bonded N–H stretching at 3434 and 3433 cm−1 respectively, whereas, the CT complex of the compounds showed a combination of non-hydrogen bonded and hydrogen bonded stretching at 3433 and 3270 cm−1 respectively. Evidence of hydrogen bonding is also present in the amide-I and amide-II stretching region. The prominent amide signals of the parent compounds become a broad peak at ∼1663 cm−1 which suggests possible intermolecular hydrogen bonding. In this regard, analyses of the CT structure obtained from DFT calculation also suggest possible hydrogen bonding (2.10 Å) between the C
:1 binding between PyKC and NDI-1 (Fig. 2B). Isothermal titration calorimetry (ITC) data showed 1:1 complexation as well with an association constant of (1.05 ± 0.164) × 104 M−1 (Fig. 2C). Moreover, 1H NMR spectra (Fig. S2A†) of the mixture showed prominent up-field shifts along with broadening of the aromatic signals for both the components when they were mixed together in an equimolar ratio. These results confirm strong CT interactions between the Py and NDI units of PyKC and NDI-1 respectively. Additionally, some of the amide-NH and NH2 protons of both the molecules showed down-field shifts in the complex form which can only result if they are involved in hydrogen bonding. The intermolecular hydrogen bonding between PyKC and NDI-1 is also prominent from the Fourier transform infrared (FTIR) spectroscopic analyses of the pure molecules and the complex (Fig. S2B†). Both PyKC and NDI-1 showed non-hydrogen bonded N–H stretching at 3434 and 3433 cm−1 respectively, whereas, the CT complex of the compounds showed a combination of non-hydrogen bonded and hydrogen bonded stretching at 3433 and 3270 cm−1 respectively. Evidence of hydrogen bonding is also present in the amide-I and amide-II stretching region. The prominent amide signals of the parent compounds become a broad peak at ∼1663 cm−1 which suggests possible intermolecular hydrogen bonding. In this regard, analyses of the CT structure obtained from DFT calculation also suggest possible hydrogen bonding (2.10 Å) between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the Lys residue of PyKC and NH of the Lys residue of NDI-1 (Fig. 1). Another possible hydrogen bond is also found between the side chain NH2 of Lys (NDI-1) and the carbonyl group of the butyric acid residue of PyKC (2.34 Å). All these experimental and theoretical calculations strongly suggest multiple intermolecular hydrogen bonding interactions between PyKC and NDI-1. It is to be noted that the calculated complexation energy value is relatively higher than that of a typical CT complex. The presence of these additional stabilizing interactions also explains this higher value.
O of the Lys residue of PyKC and NH of the Lys residue of NDI-1 (Fig. 1). Another possible hydrogen bond is also found between the side chain NH2 of Lys (NDI-1) and the carbonyl group of the butyric acid residue of PyKC (2.34 Å). All these experimental and theoretical calculations strongly suggest multiple intermolecular hydrogen bonding interactions between PyKC and NDI-1. It is to be noted that the calculated complexation energy value is relatively higher than that of a typical CT complex. The presence of these additional stabilizing interactions also explains this higher value.
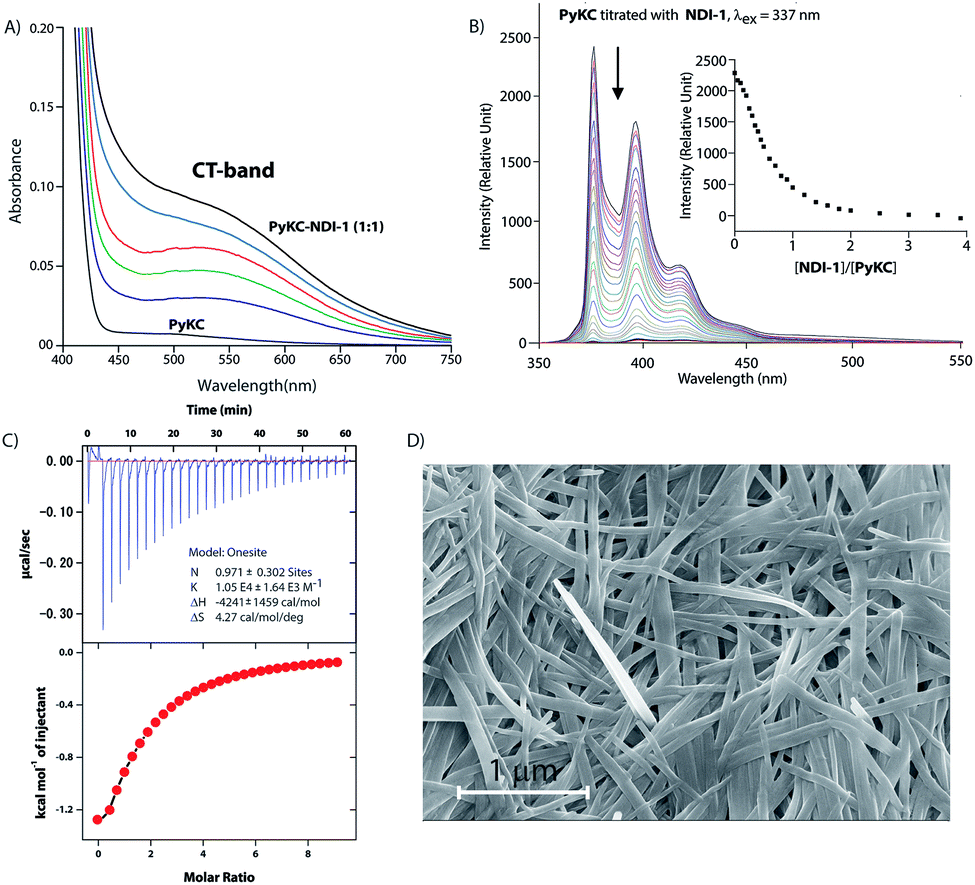 | ||
| Fig. 2 (A) UV-visible spectra of PyKC (0.1 mM) in the presence of increasing amounts of NDI-1 showing the appearance of the CT-band. (B) Emission spectra of PyKC when titrated with NDI-1 showing the quenching of the emission. Inset: the changes in emission intensity at 376 nm against the molar ratio of PyKC and NDI-1. (C) Thermogram (top) and binding isotherm (bottom) of titration of PyKC with NDI-1 at 298 K. (D) FESEM image of a freeze dried sample of an aqueous mixture of PyKC with NDI-1 (1:1) containing 1% TFA. | ||
As the CT-complexation between PyKC and NDI-1 is established, the solution phase disulfide formation of the CT complex was tested. The pH of an equimolar (1 mM) aqueous solution of these two molecules was then adjusted to neutral to allow the disulfide bonding. After 24 h of incubation under ambient conditions the solution was monitored by analytical HPLC using calibration plots of all three possible dimers (Scheme 1B). Although the formation of the heterodimer (PyKC–NDI-1 heterodimer) was found to be predominant, other two homo-dimers (PyKC-dimer and NDI-1-dimer) formed in a substantially good proportion and followed the statistical ratio (2:1:1) of a dynamic system (Scheme 1 and Table S3†).
In order to verify our hypothesis mentioned earlier, the aqueous solution of a 1:1 mixture (in the presence of 1% trifluoroacetic acid (TFA)), to prevent disulfide formation (see control experiments in the ESI†)33) of PyKC and NDI-1 (1 mM individual concentrations) was instantly frozen using liquid nitrogen and lyophilized to get the solid. HPLC and ESI-MS analyses of the solid showed the absence of any dimer. FESEM images of the dried sample revealed the formation of high aspect ratio nano-tapes as can be seen in Fig. 2D. The nanostructure obtained is a result of the CT-interaction between NDI and Py which are stacked in an alternate fashion and grown along the long axis. At this point, it was important to find a suitable solvent for the planned oxidation reaction using the lyophilized powder. The essential criteria were that the solvent must have sufficiently high oxygen solubility and none of the components and the lyophilized powder must be soluble in the solvent. A thorough literature survey showed that cyclohexane or hexane could be appropriate choices for this purpose as their oxygen solubilities were found to be significantly high (at 298.2 K and 101.3 kPa, 1.28 × 10−3 and 1.25 × 10−3 mol dm−3 for cyclohexane and hexane respectively).34 As expected, no noticeable solubility was observed for both PyKC and NDI-1 as well as lyophilized powder in these two solvents. Based on these observations, the solid nano-tapes were then suspended in cyclohexane. After shaking the mixture at 100 rpm at room temperature for 24 h, the suspension was centrifuged and dried. The material was re-dissolved in water containing 1% TFA to avoid any further oxidation and immediately analyzed by analytical HPLC. Interestingly, a single peak corresponding to the PyKC–NDI-1 heterodimer with 98.2% yield (Fig. 3A) was obtained. ESI-MS of fractions from this peak confirmed the heterodimer formation (Fig. S3†). Negligible amounts of the homo-dimers (>0.25%) were obtained supporting the successful display of the hypothesis. Further, a time dependent study was performed keeping the experimental conditions similar. As can be seen from Fig. 3A and B, the reaction completes within 20 h.
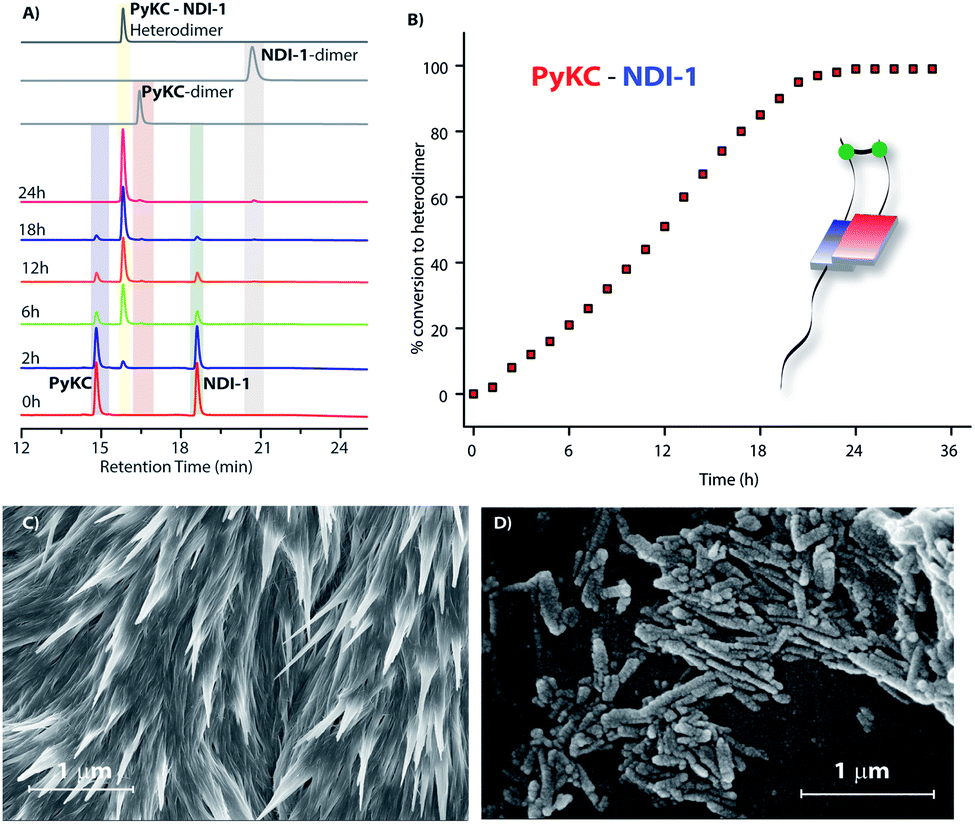 | ||
| Fig. 3 (A) Time dependent chromatograms showing the exclusive formation of the PyKC–NDI-1 heterodimer using the present protocol. (B) % conversion to the heterodimer with time as obtained from (A). (C) and (D) FESEM images of the freeze dried samples obtained from the combinations of NDI-1 with (C) PyKDC and (D) Py1KC. | ||
The solid retained its tape like morphology after the reaction (Fig. S4†). Interestingly, the concentration at which the starting materials were mixed to form CT complexes plays a significant role in the success of the presented method. At 0.1 mM starting concentration, both conversion and the chemo-selectivity were similar to those for the 1 mM starting concentration. As we reduced the starting concentration to 0.01 mM, no selectivity toward the heterodimer was observed and the overall conversion also dropped to ∼20% (20 h, Table S3†). At this concentration, neither any CT band appeared in the absorption spectra nor could any particular morphology be seen in the lyophilised solid.
The dithiol formation in cyclohexane is definitely assisted by the dissolved oxygen. It is to be noted that the parent compounds as well as their lyophilized CT complex are insoluble in cyclohexane mainly due to the presence of polar Lys residues. However, the hydrophobic segments of the CT complex plausibly make the complex partially dissolved which further strengthens the CT complex. This partial solubilisation does not impart any change in the alternate D–A arrangement and consequently to the morphology but allows the dissolved oxygen molecules to oxidize the thiol groups which are at close proximity.
To further optimize the reaction protocol, other organic solvents (hexane, dichloromethane, and benzene) were used in place of cyclohexane.34 In term of the reaction completion time, cyclohexane and hexane showed a similar result while in other solvents the reaction was found to be little slower as the oxygen solubility in these solvents is lower (Table S3†). To compare with the aerial oxidation, the solid was kept in open air for 24 h and analyzed to observe that though heterodimer was the sole product, only ∼5% material was converted within this time frame.
As discussed in the introduction, we assumed that the spatial arrangement of the –SH group within the CT complex might play a crucial role in the success of the methodology. To test that, other analogues were prepared (Scheme 1B). The stereochemistry of the Cys residue of PyKC was reversed to make PyKDC and tested by combining it with NDI-1. Though the CT-band appeared in the UV-Visible spectra and 1:1 binding was observed from the fluorescence experiment (Fig. S5 and S6†), the microscopic images of the freeze dried samples showed needle like aggregates (Fig. 3C). These observations indicate a possible different aggregation pattern compared to the PyKC–NDI-1 mixture. Further, the spacer between the pyrene and peptide sequence is reduced in the case of Py1KC. Similar to the previous case, 1:1 CT complexation was confirmed from UV-Visible and emission experiments (Fig. S5 and S7†) but small rod like aggregates were observed in the morphology (Fig. 3D) for the Py1KC–NDI-1 pair. Importantly, when the present protocol was applied for these two combinations, mixtures of all three possible products were observed with relatively low conversion rates (3–5% in 24 h). Next, powder XRD analyses were performed for all three combinations (Fig. S8†). The lyophilized powders of both, PyKC–NDI-1 and PyKDC–NDI-1 showed strong π–π interactions with similar stacking distances (3.58 and 3.60 Å respectively). However, in the case of Py1KC–NDI-1 pair, the distance was significantly higher (3.93 Å). Due to the absence of any spacer between Py and the peptide unit, for Py1KC, the stacking is presumably not as strong as in the other two cases.
To get an explanation for the failures in the cases of PyKDC–NDI-1 and Py1KC–NDI-1 pairs, additionally, the DFT calculations for energy minimized structures of these pairs were performed. The distance between the Py and NDI planes calculated for PyKDC–NDI-1 was found to be 3.60 Å which is very similar to that obtained from PXRD analyses and close to what found in the case of PyKC–NDI-1 (3.58 Å). However, the orientation of the –SH groups is entirely in the opposite direction to that of the PyKC–NDI-1 pair and the distance between the –SH groups was found to be considerably high (10.60 Å). In the case of Py1KC, the π-planes were found to be 4.02 Å apart which is similar to the PXRD data of 3.93 Å. Due to the absence of any spacer, the –SH groups stay far apart with an inter-atomic (S–S) distance of 7.85 Å.
In order to react in the solid phase, the functional groups must be in close proximity. In the case of PyKDC–NDI-1, due to the change in stereochemistry of Cys, –SH groups are oriented in opposite directions and far from each other which prevent them from forming disulfide bonds. On the other hand, owing to the shorter spacer in Py1KC, an unfavorable packing for Py1KC–NDI-1 keeps the –SH groups far apart. All these data explain the lower yield and the lack of selectivity observed for these two pairs using the present prototype. It is clear that our initial assumption based on the DFT calculation of the PyKC–NDI-1 pair was valid. The close proximity of the –SH groups for PyKC–NDI-1 allows solid phase oxidation whereas that is certainly not the case for the other two pairs. Thus, though CT interactions are present, the other two combinations failed to result in the formation of heterodimers selectively. These results evidently demonstrate the critical role of appropriate orientation of the functional groups in order to successfully implement the methodology.
Conclusions
To summarize, a novel approach is depicted here to control the reaction pathway utilizing CT interactions. Freezing the dynamicity of CT-aggregates is the key to achieve the control. However, appropriate orientation and packing of the reacting molecules is essential for the successful execution of the described methodology. Importantly, the new method opens up the possibility to create new molecules which are otherwise difficult to achieve like the asymmetric disulfides depicted here. As the presented protocol is fundamentally a new concept, it is worth mentioning that the method is limited to only those systems where the thiol groups are at very close proximity. Thus, for effective execution of this method, the D–A complex must have an appropriate molecular orientation. We are in the process of utilizing the methodology with other functional groups and the results will be communicated in due course of time.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DD acknowledges financial support from SERB (EMR/2016/000857), India, UKIERI (DST/INT/UK/P-119/2016), the Alexander von Humboldt Foundation, Germany, and the DST-FIST program.Notes and references
- C.-H. Tung, L.-Z. Wu, L.-P. Zhang and B. Chen, Acc. Chem. Res., 2003, 36, 39–47 CrossRef CAS PubMed.
- M. Wolf, A. Ogawa, M. Bechtold, M. Vonesch, J. A. Wytko, K. Oohora, S. Campidelli, T. Hayashi, D. M. Guldi and J. Weiss, Chem. Sci., 2019, 10, 3846–3853 RSC.
- J. M. Spruell, A. Coskun, D. C. Friedman, R. S. Forgan, A. A. Sarjeant, A. Trabolsi, A. C. Fahrenbach, G. Barin, W. F. Paxton, S. K. Dey, M. A. Olson, D. Benítez, E. Tkatchouk, M. T. Colvin, R. Carmielli, S. T. Caldwell, G. M. Rosair, S. G. Hewage, F. Duclairoir, J. L. Seymour, A. M. Z. Slawin, W. A. Goddard III, M. R. Wasielewski, G. Cooke and J. F. Stoddart, Nat. Chem., 2010, 2, 870 CrossRef CAS PubMed.
- J. H. Mondal, S. Ahmed, T. Ghosh and D. Das, Soft Matter, 2015, 11, 4912–4920 RSC.
- D. Jiao, J. Geng, X. J. Loh, D. Das, T.-C. Lee and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 9633–9637 CrossRef CAS PubMed.
- K. V. Rao, K. Jayaramulu, T. K. Maji and S. J. George, Angew. Chem., Int. Ed., 2010, 49, 4218–4222 CrossRef CAS PubMed.
- K. Jalani, S. Dhiman, A. Jain and S. J. George, Chem. Sci., 2017, 8, 6030–6036 RSC.
- H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2010, 49, 5331–5334 CrossRef CAS PubMed.
- F. B. L. Cougnon, N. Ponnuswamy, N. A. Jenkins, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2012, 134, 19129–19135 CrossRef CAS PubMed.
- F. B. L. Cougnon, N. A. Jenkins, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2012, 51, 1443–1447 CrossRef CAS PubMed.
- F. B. L. Cougnon and J. K. M. Sanders, Acc. Chem. Res., 2012, 45, 2211–2221 CrossRef CAS PubMed.
- A. Coskun, J. M. Spruell, G. Barin, A. C. Fahrenbach, R. S. Forgan, M. T. Colvin, R. Carmieli, D. Benítez, E. Tkatchouk, D. C. Friedman, A. A. Sarjeant, M. R. Wasielewski, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 4538–4547 CrossRef CAS PubMed.
- M. R. Molla and S. Ghosh, Chem.–Eur. J., 2012, 18, 9860–9869 CrossRef CAS PubMed.
- R. Zaini, A. C. Orcutt and B. R. Arnold, Photochem. Photobiol., 1999, 69, 443–447 CrossRef CAS.
- J.-Y. Wang, J. Yan, L. Ding, Y. Ma and J. Pei, Adv. Funct. Mater., 2009, 19, 1746–1752 CrossRef CAS.
- C. Wang, S. Yin, S. Chen, H. Xu, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2008, 47, 9049–9052 CrossRef CAS PubMed.
- P. T. Corbett, J. K. M. Sanders and S. Otto, J. Am. Chem. Soc., 2005, 127, 9390–9392 CrossRef CAS PubMed.
- H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10466–10470 CrossRef CAS PubMed.
- H. Y. Au-Yeung, G. Dan Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2009, 131, 16030–16032 CrossRef CAS PubMed.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS PubMed.
- P. Huang, P. Wang, S. Tang, Z. Fu and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 8115–8119 CrossRef CAS PubMed.
- R. Hunter, M. Caira and N. Stellenboom, J. Org. Chem., 2006, 71, 8268–8271 CrossRef CAS PubMed.
- D. H. Dethe, A. Srivastava, B. D. Dherange and B. V. Kumar, Adv. Synth. Catal., 2018, 360, 3020–3025 CrossRef CAS.
- X. Qiu, X. Yang, Y. Zhang, S. Song and N. Jiao, Org. Chem. Front., 2019, 6, 2220–2225 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- Y. Dou, X. Huang, H. Wang, L. Yang, H. Li, B. Yuan and G. Yang, Green Chem., 2017, 19, 2491–2495 RSC.
- X. Xiao, J. Xue and X. Jiang, Nat. Commun., 2018, 9, 2191 CrossRef PubMed.
- O. Schäfer and M. Barz, Chem.–Eur. J., 2018, 24, 12131–12142 CrossRef PubMed.
- B. Pramanik, S. Ahmed, N. Singha, B. K. Das, P. Dowari and D. Das, Langmuir, 2019, 35, 478–488 CrossRef CAS PubMed.
- A. Dasgupta and D. Das, Langmuir, 2019, 35, 10704–10724 CrossRef CAS PubMed.
- N. Singha, A. Srivastava, B. Pramanik, S. Ahmed, P. Dowari, S. Chowdhuri, B. K. Das, A. Debnath and D. Das, Chem. Sci., 2019, 10, 5920–5928 RSC.
- H. A. Scheraga, W. J. Wedemeyer and E. Welker, in Methods in Enzymology, ed. A. W. Nicholson, Academic Press, 2001, vol. 341, pp. 189–221 Search PubMed.
- T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Ind. Eng. Chem. Res., 2014, 53, 19331–19337 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03417j |
| ‡ These authors contributed equally. |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |