
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature-controlled electrospray ionization mass spectrometry as a tool to study collagen homo- and heterotrimers†‡
Martin
Köhler
 ,
Adrien
Marchand
,
Nina B.
Hentzen
,
Jasmine
Egli
,
Alina I.
Begley
,
Helma
Wennemers
* and
Renato
Zenobi
*
,
Adrien
Marchand
,
Nina B.
Hentzen
,
Jasmine
Egli
,
Alina I.
Begley
,
Helma
Wennemers
* and
Renato
Zenobi
*
Department of Chemistry and Applied Biosciences, ETH Zurich, 8093, Zurich, Switzerland. E-mail: zenobi@org.chem.ethz.ch; wennemers@org.chem.ethz.ch
First published on 3rd September 2019
Abstract
Collagen model peptides are useful for understanding the assembly and structure of collagen triple helices. The design of self-assembling heterotrimeric helices is particularly challenging and often affords mixtures of non-covalent assemblies that are difficult to characterize by conventional NMR and CD spectroscopic techniques. This can render a detailed understanding of the factors that control heterotrimer formation difficult and restrict rational design. Here, we present a novel method based on electrospray ionization mass spectrometry to investigate homo- and heterotrimeric collagen model peptides. Under native conditions, the high resolving power of mass spectrometry was used to access the stoichiometric composition of different triple helices in complex mixtures. A temperature-controlled electrospray ionization source was built to perform thermal denaturation experiments and provided melting temperatures of triple helices. These were found to be in good agreement with values obtained from CD spectroscopic measurements. Importantly, for mixtures of coexisting homo- and heterotrimers, which are difficult to analyze by conventional methods, our technique allowed for the identification and monitoring of the unfolding of each individual species. Their respective melting temperatures could easily be accessed in a single experiment, using small amounts of sample.
Introduction
Collagen is an abundant fibrous protein in the extracellular matrix of connective tissues.1 The collagen family comprises 28 known types, which differ in their amino acid composition, supramolecular organization, and biological function. A common characteristic of all collagen types is the formation of a right-handed triple helix (Fig. 1a).1 Depending on the type of collagen, the triple helix can be composed either of three identical polypeptide chains (A3 homotrimers), or of two or three different chains (A2B or ABC heterotrimers).2 In multicellular organisms, heterotrimeric collagen sequences often mediate binding to transmembrane collagen receptors that are involved in important cell–matrix interactions.3 Synthetic heterotrimeric collagen model peptides (CMPs) are therefore valuable for understanding the structure and stability of these large assemblies, as well as for broadening the spectrum of biomedical applications.4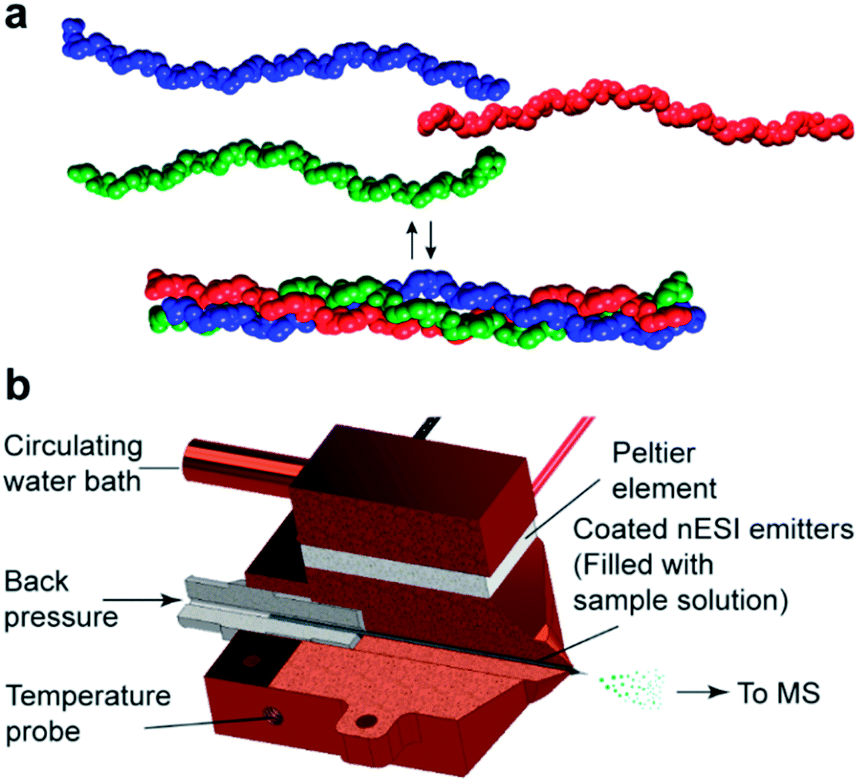 | ||
| Fig. 1 (a) Three collagen peptide chains self-assemble into a triple helix. (b) The custom-made nano-electrospray ionization (nESI) source allows for precise control of the temperature of the sample solution in the nESI emitter. The source grants the possibility to monitor thermal denaturation profiles. | ||
Collagen triple helices are comprised of three single strands containing repeating Xaa–Yaa–glycine (Gly, G) triplets, with the Xaa and Yaa positions typically occupied by proline (Pro, P) and (4R)-hydroxyproline (Hyp, O), respectively.1,5 The strands coil around each other with a single-residue offset, which defines the leading, middle, and lagging strands.2a,2b The number of possible compositions and registers of the triple helices complicate the design of specific, self-assembling heterotrimers.2a,2b For example, two different monomer chains A and B can form 4 assemblies with different compositions (AAA, BBB, AAB and ABB) and different registers of the heterotrimers. Similarly, three chains A, B, and C can result in 10 triple helices of unique composition; each of them potentially populating several registers.
The assembly of synthetic CMPs into heterotrimers is therefore challenging and only a few strategies were developed to form heterotrimers.2a,2b,6 For example, salt bridges between precisely positioned lysine (Lys, K) and aspartic acid (Asp, D) residues were used to direct the self-assembly of synthetic CMPs.6,7
The analysis of mixtures consisting of different collagen homo- and heterotrimers is similarly challenging. NMR spectroscopy is currently the method of choice to characterize such mixtures. This analysis requires isotopic labeling at multiple sites of the CMPs and the resulting spectra can be rather complex.8 The characterization of every species formed is therefore time-consuming, in particular for systems whose assembly behavior is difficult to predict. The stability of collagen triple helices is typically assessed by thermal denaturation experiments using circular dichroism (CD) spectroscopy as monitoring tool.1 This method is straightforward for the analysis of CMPs that form a unique collagen triple helix. However, since the CD spectrum reflects an average of the entire molecular population, it does not allow to monitor the unfolding of different coexisting triple helices in the same solution. Hence, there is a need for an analytical tool able to simultaneously characterize and quantify coexisting collagen triple helices in complex mixtures and to estimate their relative stabilities.
Native nano-electrospray ionization (nESI) mass spectrometry (MS) identifies molecular assemblies by their mass to charge (m/z) ratios and is able to distinguish non-covalent complexes of different compositions. Native MS has proven useful in a number of applications, from the study of the interconversion of peptide structures to the elucidation of ligand binding sites on RNA sequences.9 In recent studies, it was also shown that collagen triple helices can be brought intact from the solution to the gas phase.10 In addition, in combination with a temperature-controlled ionization source, native MS has been applied to identify protein unfolding intermediates,11 and to investigate the thermodynamic properties of ligands binding to DNA structures.12
Results and discussion
Analysis of a collagen homotrimer
First, we evaluated temperature-controlled nESI MS as a tool to examine the triple helix formation and the thermal unfolding of homotrimeric collagen. Such triple helices form from a single CMP and have been extensively studied by conventional techniques.1,13 They are therefore ideal model systems to establish the nESI MS setup for the analysis of collagen triple helices. We used CMP A, consisting of seven repeating units of POG, as a representative compound (Fig. 2a). This peptide is known to form triple helices in solution, and has been studied by NMR13a and CD spectroscopy.13c,13e The CD spectrum of CMP A (50 μM in 10 mM aq. NH4Ac at pH 7) shows a minimum at 196 nm and a maximum at 225 nm that are indicative of collagen triple helices (Fig. S1‡). We subjected the same solution of CMP A to native MS using a quadrupole-time-of-flight mass spectrometer. The resulting mass spectrum shows a distribution of peaks corresponding to the m/z signals for monomeric (M) and trimeric (T) CMP A (Fig. 2a, red and blue peaks, respectively). The most abundant signals for each CMP species correspond to acetate (Ac) adduct ions, due to the low ionization efficiency of the amino acids of this peptide. At 25 °C, the predominant form of CMP A is, as expected from CD spectroscopy, the trimer, and only a low fraction of CMP A is present as monomers. Higher aggregates could not be detected at 25 °C, which supports that trimers formed by CMP A do not aggregate further. These data confirm that the collagen trimer is preserved in the gas phase and reveal that both monomers and trimers can be simultaneously observed by nESI MS.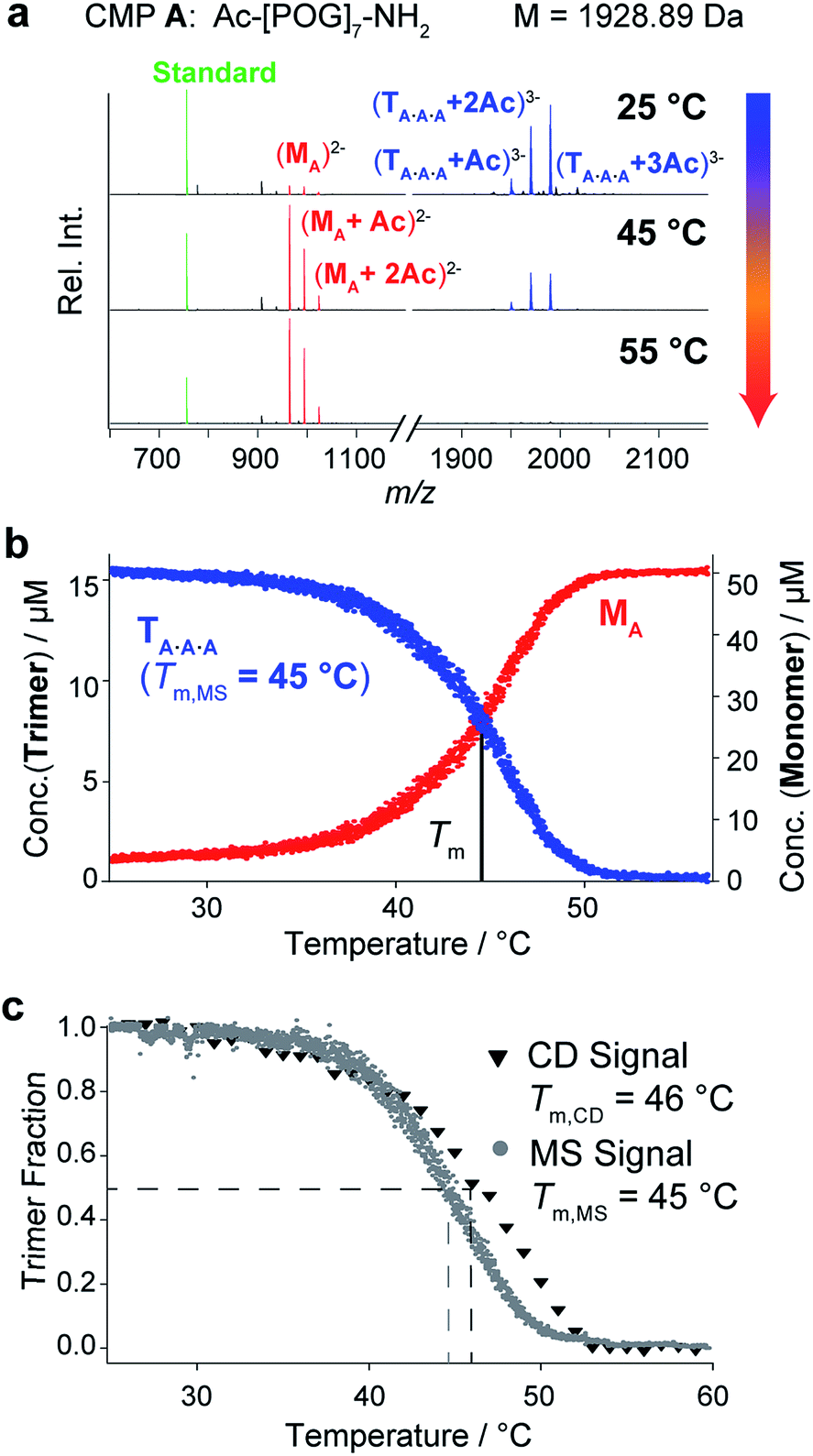 | ||
| Fig. 2 CD spectroscopic and native MS analyses of CMP A (50 μM in 10 mM aq. NH4Ac at pH 7). (a) MS spectra of CMP A at different temperatures. The observed m/z signals correspond to the triply charged trimers (blue), the doubly charged monomers (red), and the internal standard (10 μM bradykinin 1–7, green). (b) Thermal denaturation profile of CMP A obtained by monitoring the monomer and trimer m/z signals as a function of temperature (heating rate: 1 °C min−1). (c) Overlay of the normalized thermal denaturation curves of CMP A obtained by using CD spectroscopy (triangles) and MS (dots) as monitoring tools (heating rate: 1 °C min−1). | ||
Next, we evaluated whether the thermal stability of collagen triple helices can be determined by temperature-controlled nESI MS. Whereas CD spectroscopic analyses provide spectra that are an average over all coexisting species in solution, native MS enables the distinction between monomers and trimers, and would detect additional complex stoichiometries including unfolding intermediates (dimers) or aggregation products (large multimers). Using a custom-made nESI source in which the nESI emitter, and hence the whole sample solution, is embedded inside a temperature-controlled copper block, we monitored the decrease of the integrated m/z signal of the CMP A trimer as a function of temperature from 25 °C to 55 °C. As examples, we show mass spectra of solutions of CMP A at 25 °C, 45 °C, and 55 °C (Fig. 2a). An internal standard (bradykinin peptide 1–7) was added to determine response factors and ensure reliable quantification of the monomer/trimer (M/T) ratio following a previously described procedure.14 The calculated concentrations are plotted as a function of temperature (Fig. 2b). The trimer signals are predominant at 25 °C and decrease with increasing temperature, accompanied by an increase of the monomer signals. At 55 °C, only monomers are present. No m/z signal corresponding to a dimeric unfolding intermediate was observed, which is in agreement with the two-state unfolding model of the collagen triple helix.15 Within the resolution limits of our instrumentation, we could, however, not detect any change in the quaternary structure of the trimers via ion mobility separation (change in drift time) during unfolding. The resulting thermal denaturation curve shows the sigmoidal transition typically observed for triple helix unfolding and represents the unfolding of the trimer and concomitant release of the monomer, with an inflection point at the melting temperature (Tm) of the triple helix (Fig. 2b).
Thermal denaturation studies using CD spectroscopy were also performed as a reference. Because the Tm depends on the sample concentration, buffer, and heating rate and is thus a relative measure for triple helix stability,16 the experiments were performed with the same solution of CMP A as the one used for MS, and with the same heating rate. Monitoring the decline of the signal at 225 nm provided sigmoidal transition curves with a midpoint at 46 °C (Fig. 2c, triangles). The melting temperature from CD measurements is in good agreement with the value determined by MS monitoring (Tm,CD = 46 °C vs. Tm,MS = 45 °C), as seen from the overlay of the normalized denaturation curves. The slightly higher Tm value obtained using CD spectroscopy can be attributed to a time lag in the heating of the solution due to the higher sample volumes needed for CD (250 μL vs. 10 μL for MS).
These experiments demonstrate that temperature-controlled nESI MS is suitable to monitor the concentrations of monomeric and trimeric CMPs, and to determine the melting temperature of homotrimeric collagen triple helices.
Analysis of the composition and thermal stability of an ABC heterotrimer
Next, we evaluated whether nESI MS can also be used for the characterization of heterotrimeric collagen. For an initial study, we chose an ABC-type system that is closely related to one previously reported by the Hartgerink group.6e Specifically, we prepared three different CMPs, B, C and D, that contain 8 repeating units of PKG, DOG and POG, respectively (Fig. 3a). Their assembly into ABC heterotrimers is mediated by the formation of axial salt bridges between Asp and Lys in neighboring strands (Fig. 3a).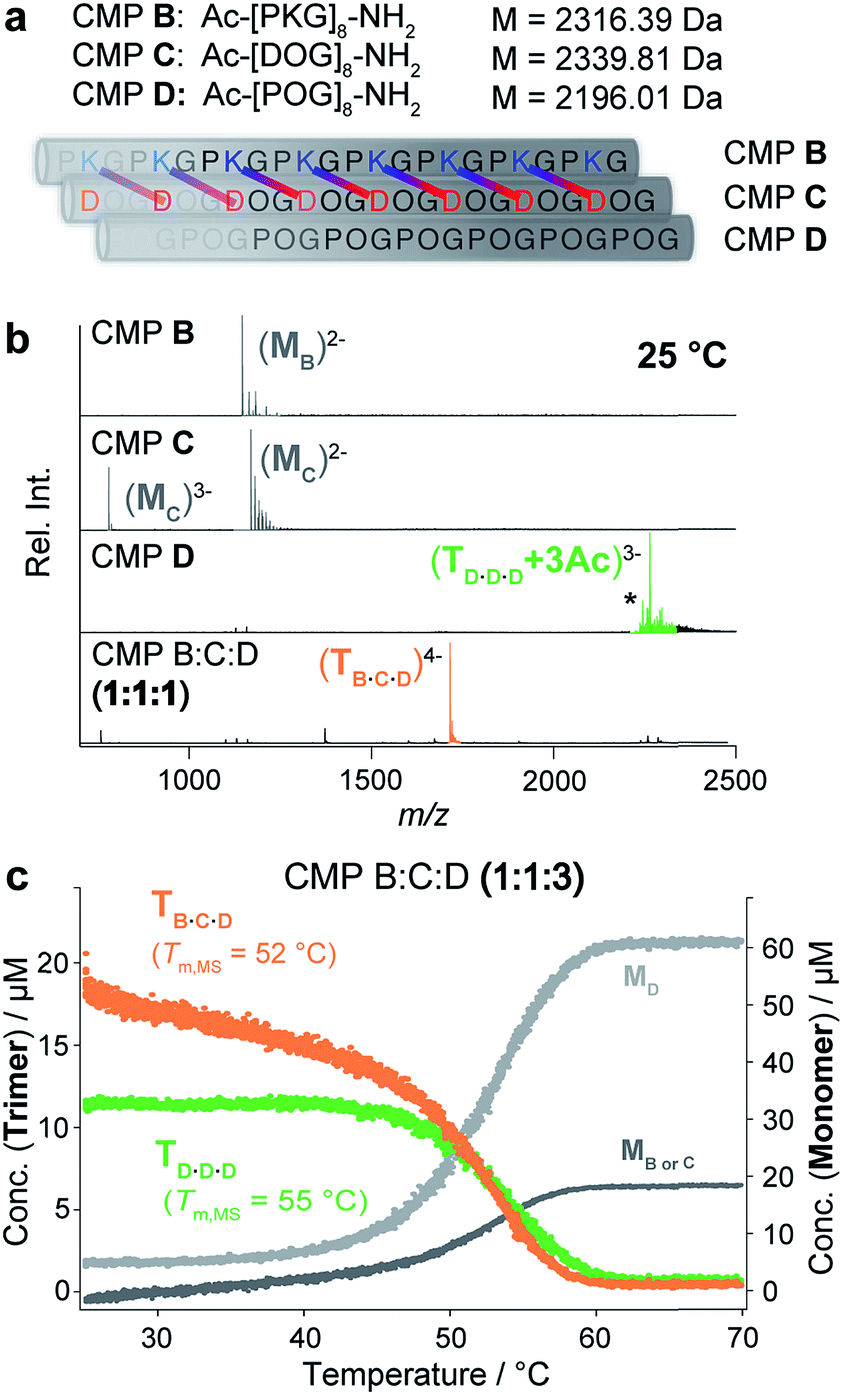 | ||
Fig. 3 (a) CMPs B, C and D and their self-assembly into a heterotrimer. The axial salt bridges formed between Asp and Lys of CMPs B and C are shown. (b) Native MS spectra of CMPs B, C and D and an annealed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of CMPs B, C and D (50 μM in 10 mM aq. NH4Ac at pH 7 and 25 °C). The most abundant peaks of CMP D correspond to acetate adducts: +2Ac (*) and +3Ac. (c) Thermal denaturation of a 1:1:3 mixture of CMPs B, C and D monitored by temperature-controlled native MS (100 μM in 10 mM aq. NH4Ac at pH 7, heating rate of 1 °C min−1) shows the unfolding of two coexisting homotrimeric D·D·D (green) and heterotrimeric B·C·D (orange) helices with Tm,MS = 52 °C (B·C·D), and Tm,MS = 55 °C (D·D·D). :1:1 mixture of CMPs B, C and D (50 μM in 10 mM aq. NH4Ac at pH 7 and 25 °C). The most abundant peaks of CMP D correspond to acetate adducts: +2Ac (*) and +3Ac. (c) Thermal denaturation of a 1:1:3 mixture of CMPs B, C and D monitored by temperature-controlled native MS (100 μM in 10 mM aq. NH4Ac at pH 7, heating rate of 1 °C min−1) shows the unfolding of two coexisting homotrimeric D·D·D (green) and heterotrimeric B·C·D (orange) helices with Tm,MS = 52 °C (B·C·D), and Tm,MS = 55 °C (D·D·D). | ||
We recorded native MS and CD spectra of each of the CMPs separately at 25 °C (50 μM in 10 mM aq. NH4Ac at pH 7). The self-assembly of the charged CMPs B and C into homotrimers is hindered by coulombic repulsion. Neither the CD nor the mass spectra of CMPs B and C indicate triple helix formation (Fig. 3b and S2a‡). In fact, the native MS spectra of CMPs B and C only show monomer signals. In contrast, the CD and MS spectra of the neutral CMP D show triple helix formation (Fig. 3b and S2a‡).
Next, we analyzed an annealed equimolar mixture of CMPs B, C and D by native MS. The resulting mass spectrum shows one abundant m/z signal corresponding to the ABC heterotrimer (Fig. 3b, bottom). This finding confirms the expected selective assembly of the CMPs into an ABC heterotrimer (B·C·D). Since CMP D self-assembles into a homotrimer, a mixture of CMPs B, C and D with a 1:1:3 ratio of B:C:D is expected to contain a mixture of both heterotrimeric (B·C·D) and homotrimeric (D·D·D) triple helices. CD spectroscopic analysis of an annealed solution of such a 1:1:3 mixture indicates triple helix formation but does not allow for distinguishing different triple helical species (Fig. S2b‡). Reassuringly, the native MS spectrum of the mixture shows two signals corresponding to m/z values of the B·C·D heterotrimer and the D·D·D homotrimer (Fig. S3‡).
The thermal denaturation profiles of the two coexisting B·C·D and D·D·D trimers were obtained by monitoring the change in their distinct m/z signal integrals as a function of temperature (from 25 °C to 70 °C). These signal integrals were converted into concentrations by calculating the response factors. The initial concentrations of the B·C·D and D·D·D trimers observed at 25 °C were 18 μM and 12 μM, respectively, which is in agreement with the predicted 1.5:1 ratio of B·C·D to D·D·D in a 1:1:3 mixture of CMP B, C and D. In a single experiment, their respective melting temperatures were determined as Tm,MS = 52 °C (B·C·D heterotrimer) and Tm,MS = 55 °C (D·D·D homotrimer, Fig. 3c). To confirm the accuracy of the obtained Tm values by CD spectroscopy, we separately monitored the thermal denaturation of solutions of both a 1:1:1 mixture containing only the B·C·D heterotrimer, and a solution of CMP D, containing only the D·D·D homotrimer. The melting temperatures obtained for the B·C·D heterotrimer (Tm,CD = 53 °C) and for the D·D·D homotrimer (Tm,CD = 57 °C) are in good agreement with the values obtained from the MS experiment (Fig. S2‡).§
Analysis of the composition and thermal stabilities of different coexisting triple helices in a complex mixture
Finally, we probed the effectiveness of native MS for monitoring mixtures of closely related coexisting collagen triple helices. Towards this goal we designed and synthesized CMPs E and F (Fig. 4a). Both CMPs contain one Lys and one Asp residue that can engage in interchain Lys–Asp – lateral or axial – salt bridges. Specifically, CMP F can self-assemble by formation of one lateral and two axial bridges,6a–d whereas in the homotrimeric form of CMP E, the oppositely charged residues are too far from each other to interact (Fig. 4a, left). The E·F·F and E·E·F heterotrimers can assemble by the formation of either two or three axial salt bridges (Fig. 4a, right). Thus, depending on the composition, at most three interstrand salt bridges can form. This number is considered not sufficient to favor the formation of only one specific heterotrimer2 and a mixture of these four trimeric species of different composition is expected. It is also worth noting that Asp was intentionally placed in the Yaa and Lys in the Xaa position as opposed to the more commonly used salt bridge between Asp in Xaa and Lys in Yaa. This “inverted” salt bridge stabilize the triple helix less than the “common” salt bridge,6b and should therefore favor the formation of multiple triple helices. Thermal denaturation experiments using CD spectroscopy as monitoring tool provided Tm,CD values of 46 °C and 38 °C for the pure F·F·F and E·E·E homotrimers, respectively (Fig. 4b). This finding corroborates that the formation of salt-bridges leads to an increased stability of the triple helices. Annealed solutions of mixtures containing CMPs E and F in ratios of 2:1, 1:1, and 1:2 provided single-transition melting curves with melting temperatures of Tm,CD = 41 °C (2:1 mixture), Tm,CD = 43 °C (1:1 mixture) and Tm,CD = 44 °C (1:2 mixture) (Fig. 4b). These curves represent the unfolding of the ensemble of the triple helices and provide neither insight into the composition of the mixture nor into the Tm values of different coexisting trimers. Thus, the CD experiments do not provide conclusive insight into the formation and the ratio of heterotrimers assembled from CMPs E and F.
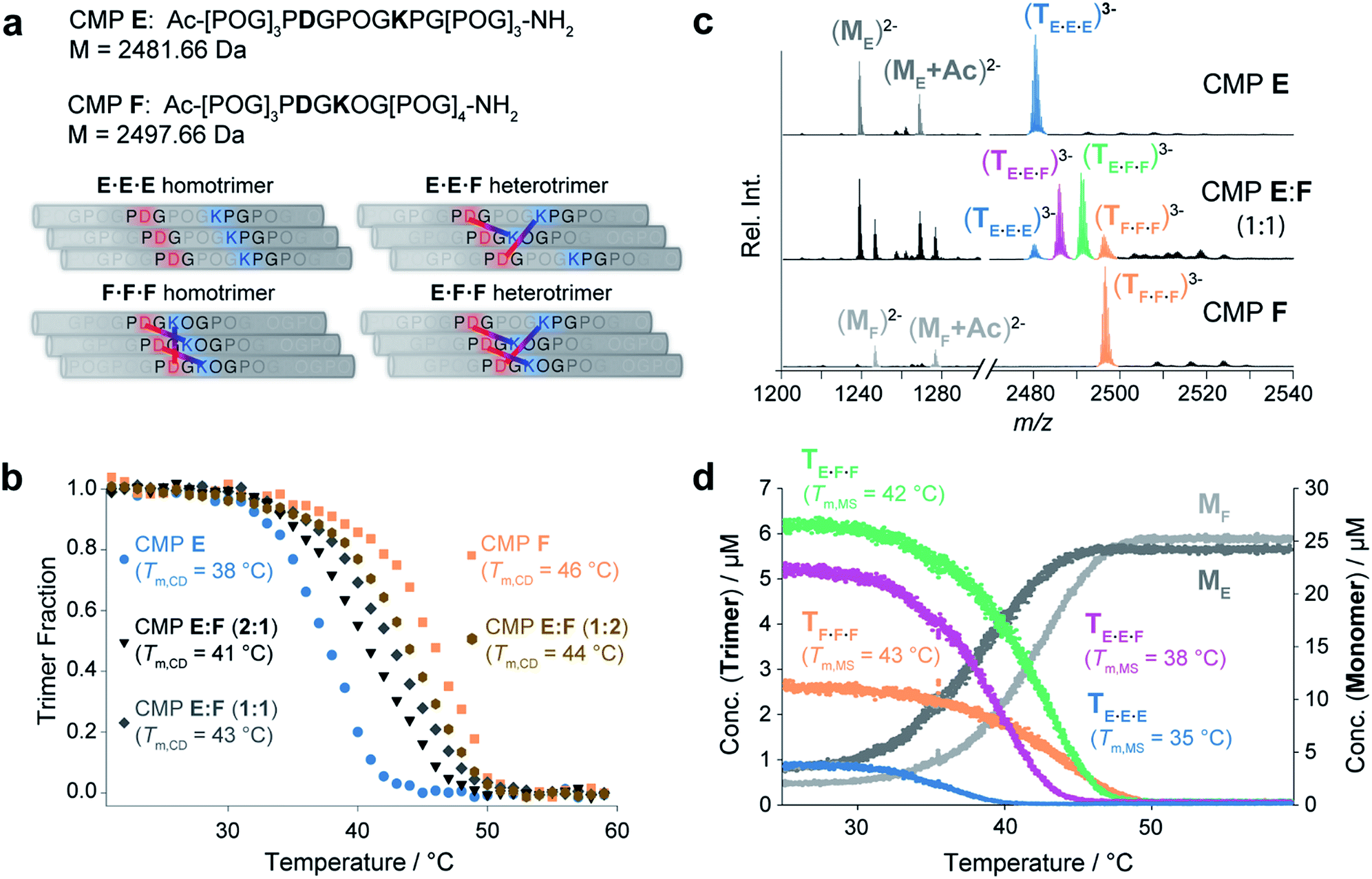 | ||
| Fig. 4 (a) Possible homotrimeric (left) and heterotrimeric (right) helices formed in a mixture of CMPs E and F. For the heterotrimers, only the most favored A2B and AB2-type heterotrimers are shown. (b) CD spectroscopic thermal denaturation studies (50 μM in 10 mM aq. NH4Ac at pH 7, heating rate of 1 °C min−1) of CMP E (blue), CMP F (orange), and annealed mixtures of CMPs E:F 2:1 (black), 1:1 (dark grey), and 1:2 (brown). (c) Native MS spectra of CMPs E and F and an annealed 1:1 mixture thereof. The detected trimers are colored as follows: E·E·E (blue), F·F·F (orange), E·F·F (green), and E·E·F (purple). (d) The thermal denaturation study of a 1:1 mixture of CMPs E and F by temperature-controlled native MS shows the unfolding of four coexisting trimeric species. Tm,MS values were determined as follows: 35 °C (E·E·E), 38 °C (E·E·F), 42 °C (E·F·F) and 43 °C (F·F·F). | ||
The MS spectra of pure solutions of CMPs E and F show m/z signals corresponding to the monomers and the respective homotrimers (Fig. 4c). The spectrum of a 1:1 mixture of the two CMPs shows m/z signals corresponding to the homotrimers (E·E·E and F·F·F) and confirms the formation of heterotrimers (E·F·F and E·E·F) (Fig. 4c). Thermal denaturation with the 1:1 E:F mixture provided individual unfolding curves for each trimeric species, with the following melting temperatures: Tm,MS (E·E·E) = 35 °C, Tm,MS (E·E·F) = 38 °C, Tm,MS (E·F·F) = 42 °C and Tm,MS (F·F·F) = 43 °C (Fig. 4d). The Tm,MS values of the pure homotrimers (Fig. 4d) and the sum of all trimer signals are in good agreement with the respective values obtained by CD spectroscopic monitoring (Fig. S5‡). Thus, the nESI MS measurements provided fast and reliable insight into the composition and relative ratio of coexisting collagen triple helices in a complex mixture.
Conclusions
In summary, we have established temperature-controlled native MS as a tool for studying the composition and thermal stabilities of synthetic collagen peptide assemblies. We validated our native MS setup by detailed investigations on previously reported non-covalently assembled homotrimeric and heterotrimeric CMPs, and ultimately demonstrated the value of this technique for characterizing a complex mixture of triple helices that cannot be sufficiently analyzed by CD spectroscopy. The high-resolution power of MS allowed for the identification of triple helices of different compositions. A temperature-controlled nESI source enabled the measurement of their thermal denaturation profiles in a single experiment. Theoretically, there is no limit to the number of triple helices of distinct masses that can be simultaneously monitored by native MS. The efficiency and low cost of native MS are further advantages that are unequaled by conventional tools for CMP analysis. This method opens up entirely new opportunities for the design of synthetic collagen heterotrimers and will have a far-reaching impact far beyond this field, e.g., in areas where non-covalent assemblies of closely related subunits of biomolecules form functional units.Experimental section
Peptide synthesis and purification
All peptides were synthesized using a Fmoc/tBu strategy for solid phase peptide synthesis on a Syro I peptide synthesizer (MultiSynTech, Witten, Germany) at a scale of 0.1 mmol on Rink Amide-ChemMatrix resin (loading 0.48 mmol g−1) from Biotage. Fmoc-ProHypGly-OH, Fmoc-ProLys(Boc)Gly-OH and Fmoc-Asp(OtBu)HypGly-OH (all synthesized on 2-chlorotrityl chloride resin) were used for peptide coupling reactions. Fmoc-AA-OH (3 equiv., 0.5 M in DMF) activated by treatment with HATU (3 equiv., 0.5 M in DMF) and i-Pr2NEt (6 equiv., 3 M in NMP) were added to the amino functionalized resin and coupled for 80 min at room temperature. Fmoc-deprotection was carried out by addition of piperidine (40% v/v in DMF) (4 × 1 min). The acetylation was carried out dissolving AcOH (4 equiv.), HATU (4 equiv.) and i-Pr2NEt (9 equiv.) in DMF (0.5 M). After 5 min of preactivation, the solution was added to a suspension of the amino functionalized resin in DMF. The mixture was agitated for 90 min and washed with DMF (5×). To cleave the synthesized peptide, the resin was treated for 60 min with a mixture of TFA/H2O/TIS (95:2.5:2.5) and then filtered. This procedure was repeated for 30 min and the collected filtrates were concentrated under reduced pressure. After addition of cold Et2O, a white solid precipitated, which was isolated after centrifugation and decantation of the supernatant. The solid was resuspended in Et2O, the suspension was sonicated, centrifuged, and the supernatant was decanted. The residual white solid was dried under reduced pressure, dissolved in water and lyophilized to obtain a white foam. The crude product was purified by reverse-phase HPLC on a Dionex UHPLC (Ultimate 3000, Thermo Fisher Scientific, Waltham, Massachusetts, USA) equipped with a Jupiter 4 μm Proteo 90 Å 250 × 10 mm column from Phenomenex (Torrance, California, USA). Acetonitrile (solvent A) and water containing 1% acetonitrile and 0.1% TFA (solvent B) were used as eluents at a flow rate of 5 mL min−1 and oven temperature of 65 °C. All peptides were C-terminally amidated and N-terminally acetylated.
Sample preparation
Lyophilized CMPs were dissolved in ammonium acetate solution (10 mM, pH 7.0) at a final concentration of 50–100 μM. The solutions were annealed at 80 °C for 15 min and kept at 4 °C for at least 5 days prior to CD spectroscopic and mass spectrometric analysis. 10 μM peptide standard (bradikinin 1–7) was added as an internal standard.Circular dichroism spectroscopy
CD measurements were performed with a Chirascan CD spectrometer (Applied Photophysics, U.K.) equipped with a Peltier temperature control system, using quartz cuvettes with a path length of 0.1 cm (Hellma Analytics, Müllheim, Germany). Full wavelength scans were recorded for each sample at 7 °C from 180 to 260 nm using a spectral bandwidth of 1 nm, time constant of 5 s and a step resolution of 1 nm. The ellipticity at the wavelength of maximum absorption (225 nm) was then monitored as a function of temperature recording a background scan at the same wavelength. Heating rates of 1 °C min−1 were used to generate thermal unfolding curves to determine transition temperatures (Tm).Mass spectrometric analysis
All measurements were performed on a Synapt G2-S quadrupole-time-of-flight mass spectrometer (Waters, Manchester, UK). A special element of our set-up, which is not commercially available, is a custom-made, copper encased Peltier controlled thermal nESI source (Fig. 1b), which allows the solution in the spray needle to be temperature controlled between ≈15 and 85 °C. The heating was provided with a Peltier element, which was cooled against a circulating water bath. Temperature ramps were programmed on LabView (version 17.0, National Instruments) using a proportional–integral–derivative (PID). Borosilicate emitters (ES388, Thermo Fisher Scientific, Waltham, USA) were filled with 8–10 μL of peptide solution and mounted in the temperature-controlled source. The measurements were performed in negative ion mode using a capillary voltage of 0.9 to 1.6 kV and a nitrogen backpressure of 0.2 to 0.4 bar. For a gentle desolvation and transmission of the ions, the source block temperature was set to 25 °C, the trap and transfer collision energies were set to 4 and 2 V, respectively. The mass spectrometer is equipped with a travelling-wave ion mobility separation (IMS). The wave velocity in the IMS was set to 1000 m s−1 at a wave height of 40 V and a nitrogen gas flow of 50 mL min−1. Data were acquired over a range of 100–5000 m/z. The instrument was calibrated externally using a cesium iodide solution of 25 mg mL−1 in acetonitrile and doubly distilled water (1:1). Melting curves were obtained using a 1 °C min−1 temperature ramp.
Processing of mass spectrometric data
Overlapping signal adducts of monomeric and/or folded species combining different peptide strands that ionize with higher charge states can be separated using ion mobility separation (IMS). Due to signal overlap of the species investigated (for example: singly charged CMP monomer and triply charged CMP trimer), IMS data (example shown in Fig. S6‡) were used to extract the signal intensities of the different species via DriftScope (version 2.8, Waters). The extracted signals were converted from a function of time to a function of the temperature measured by the controller. Determination of the electrospray response factors for quantification was performed using the internal standard and mass balance equations according to the procedure described by Gabelica et al.14 The corresponding matrix algebra was solved using MathCad (version 14.0, PTC). Tm values were calculated by determining the temperature at the inflection point of the melting curves using a sigmoidal fit.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Swiss National Science Foundation for financial support of this research (SNF grants 200020_178765 (R. Z.) and 200020_178805 (H. W.)). N. H. is grateful for an AFR PhD Fellowship from the Fonds National de la Recherche Luxembourg (FNR). We thank Heinz Benz for building the PID controller and for his help in LabView coding, as well as Jonas Metternich for his help with MatLab scripting. We also thank Christian Marro for the construction of the source. M. K. is deeply grateful for all the help and support of Wolfgang and Ute Strumpf.Notes and references
- (a) M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed; (b) S. Ricard-Blum, Cold Spring Harbor Perspect. Biol., 2011, 3, a004978 Search PubMed; (c) J. Bella, Biochem. J., 2016, 473, 1001–1025 CrossRef CAS PubMed.
- (a) J. A. Fallas, L. E. O'Leary and J. D. Hartgerink, Chem. Soc. Rev., 2010, 39, 3510–3527 RSC; (b) A. A. Jalan and J. D. Hartgerink, Curr. Opin. Chem. Biol., 2013, 17, 960–967 CrossRef CAS PubMed; (c) U. Sharma, L. Carrique, S. Vadon-Le Goff, N. Mariano, R. N. Georges, F. Delolme, P. Koivunen, J. Myllyharju, C. Moali, N. Aghajari and D. J. Hulmes, Nat. Commun., 2017, 8, 14671 CrossRef PubMed; (d) A. S. DiChiara, R. C. Li, P. H. Suen, A. S. Hosseini, R. J. Taylor, A. F. Weickhardt, D. Malhotra, D. R. McCaslin and M. D. Shoulders, Nat. Commun., 2018, 9, 4206 CrossRef PubMed.
- (a) B. Leitinger, Annu. Rev. Cell Dev. Biol., 2011, 27, 265–290 CrossRef CAS PubMed; (b) B. Sacca and L. Moroder, J. Pept. Sci., 2002, 8, 192–204 CrossRef CAS PubMed; (c) J. Emsley, C. G. Knight, R. W. Farndale and M. J. Barnes, J. Mol. Biol., 2004, 335, 1019–1028 CrossRef CAS PubMed; (d) J. Emsley, C. G. Knight, R. W. Farndale, M. J. Barnes and R. C. Liddington, Cell, 2000, 101, 47–56 CrossRef CAS PubMed.
- (a) B. An, Y. S. Lin and B. Brodsky, Adv. Drug Delivery Rev., 2016, 97, 69–84 CrossRef CAS PubMed; (b) G. B. Fields, Org. Biomol. Chem., 2010, 8, 1237–1258 RSC.
- J. A. Ramshaw, N. K. Shah and B. Brodsky, J. Struct. Biol., 1998, 122, 86–91 CrossRef CAS PubMed.
- (a) A. V. Persikov, J. A. M. Ramshaw, A. Kirkpatrick and B. Brodsky, J. Mol. Biol., 2002, 316, 385–394 CrossRef CAS PubMed; (b) F. Wei, J. A. Fallas and J. D. Hartgerink, Macromol. Rapid Commun., 2012, 33, 1445–1452 CrossRef CAS PubMed; (c) J. A. Fallas, J. H. Dong, Y. Z. J. Tao and J. D. Hartgerink, J. Biol. Chem., 2012, 287, 8039–8047 CrossRef CAS PubMed; (d) H. Zheng, C. Lu, J. Lan, S. Fan, V. Nanda and F. Xu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6207–6212 CrossRef CAS PubMed; (e) V. Gauba and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 15034–15041 CrossRef CAS PubMed.
- (a) V. Gauba and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 8921 CrossRef CAS; (b) L. E. Russell, J. A. Fallas and J. D. Hartgerink, J. Am. Chem. Soc., 2010, 132, 3242 CrossRef CAS PubMed; (c) L. E. R. O'Leary, J. A. Fallas and J. D. Hartgerink, J. Am. Chem. Soc., 2011, 133, 5432–5443 CrossRef PubMed; (d) I. C. Tanrikulu, A. Forticaux, S. Jin and R. T. Raines, Nat. Chem., 2016, 8, 1008–1014 CrossRef CAS PubMed; (e) B. Sarkar, L. E. O'Leary and J. D. Hartgerink, J. Am. Chem. Soc., 2014, 136, 14417–14424 CrossRef CAS PubMed.
- (a) B. Madhan, J. X. Xiao, G. Thiagarajan, J. Baum and B. Brodsky, J. Am. Chem. Soc., 2008, 130, 13520–13521 CrossRef CAS PubMed; (b) A. A. Jalan and J. D. Hartgerink, Biomacromolecules, 2013, 14, 179–185 CrossRef CAS PubMed.
- (a) A. C. Leney and A. J. R. Heck, J. Am. Soc. Mass Spectrom., 2017, 28, 5–13 CrossRef CAS PubMed; (b) L. Q. Shi, A. E. Holliday, H. L. Shi, F. F. Zhu, M. A. Ewing, D. H. Russell and D. E. Clemmer, J. Am. Chem. Soc., 2014, 136, 12702–12711 CrossRef CAS PubMed; (c) E. M. Schneeberger and K. Breuker, Angew. Chem., Int. Ed., 2017, 56, 1254–1258 CrossRef CAS PubMed.
- (a) M. Lalande, C. Comby-Zerbino, M. Bouakil, P. Dugourd, F. Chirot and J. C. Poully, Chem.–Eur. J., 2018, 24, 13728–13733 CrossRef CAS PubMed; (b) J. Egli, C. Siebler, M. Köhler, R. Zenobi and H. Wennemers, J. Am. Chem. Soc., 2019, 141, 5607–5611 CrossRef CAS PubMed.
- T. J. El-Baba, D. W. Woodall, S. A. Raab, D. R. Fuller, A. Laganowsky, D. H. Russell and D. E. Clemmer, J. Am. Chem. Soc., 2017, 139, 6306–6309 CrossRef CAS PubMed.
- A. Marchand, F. Rosu, R. Zenobi and V. Gabelica, J. Am. Chem. Soc., 2018, 140, 12553–12565 CrossRef CAS PubMed.
- (a) M. H. Li, P. Fan, B. Brodsky and J. Baum, Biochemistry, 1993, 32, 7377–7387 CrossRef CAS PubMed; (b) R. S. Erdmann and H. Wennemers, J. Am. Chem. Soc., 2010, 132, 13957–13959 CrossRef CAS PubMed; (c) S. K. Holmgren, L. E. Bretscher, K. M. Taylor and R. T. Raines, Chem. Biol., 1999, 6, 63–70 CrossRef CAS PubMed; (d) S. K. Holmgren, K. M. Taylor, L. E. Bretscher and R. T. Raines, Nature, 1998, 392, 666–667 CrossRef CAS PubMed; (e) M. D. Shoulders, K. A. Satyshur, K. T. Forest and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 559–564 CrossRef CAS PubMed; (f) A. V. Persikov, J. A. Ramshaw, A. Kirkpatrick and B. Brodsky, Biochemistry, 2000, 39, 14960–14967 CrossRef CAS PubMed; (g) C. Siebler, R. S. Erdmann and H. Wennemers, Angew. Chem., Int. Ed., 2014, 53, 10340–10344 CrossRef CAS PubMed; (h) Y. Zhang, R. M. Malamakal and D. M. Chenoweth, J. Am. Chem. Soc., 2015, 137, 12422–12425 CrossRef CAS PubMed.
- V. Gabelica, F. Rosu and E. De Pauw, Anal. Chem., 2009, 81, 6708–6715 CrossRef CAS PubMed.
- (a) J. Engel, H. T. Chen, D. J. Prockop and H. Klump, Biopolymers, 1977, 16, 601–622 CrossRef CAS PubMed; (b) S. Frank, R. A. Kammerer, D. Mechling, T. Schulthess, R. Landwehr, J. Bann, Y. Guo, A. Lustig, H. P. Bachinger and J. Engel, J. Mol. Biol., 2001, 308, 1081–1089 CrossRef CAS PubMed; (c) A. V. Persikov, Y. Xu and B. Brodsky, Protein Sci., 2004, 13, 893–902 CrossRef CAS PubMed.
- K. Mizuno, S. P. Boudko, J. Engel and H. P. Bachinger, Biophys. J., 2010, 98, 3004–3014 CrossRef CAS PubMed.
Footnotes |
| † The data used in this publication are freely accessible in a curated data archive at ETH Zurich (https://www.research-collection.ethz.ch) under the DOI: 10.3929/ethz-b-000319305 |
| ‡ Electronic supplementary information (ESI) available: CD spectroscopy scans and thermal denaturation profiles of CMP A and mixtures of CMP B–F (S1, S2). Native MS spectra of different mixtures of CMP A–F (S3, S4, S6). Comparison of melting curves obtained via CD spectroscopy and MS of CMP E, F mixture (S5). Overview of all Tm values obtained via CD spectroscopy and MS (Table S1). See DOI: 10.1039/c9sc03248g |
| § Note that the B·C·D heterotrimer is less stable than the D·D·D homotrimer since replacement of the canonical Pro and Hyp residues by non-proline amino acids disfavors the PPII helix of the single strands and thus destabilizes the triple helix. |
| This journal is © The Royal Society of Chemistry 2019 |