
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective synthesis of formamides, 1,2-bis(N-heterocyclic)ethanes and methylamines from cyclic amines and CO2/H2 catalyzed by an ionic liquid–Pd/C system†
Ruipeng
Li
ab,
Yanfei
Zhao
*a,
Huan
Wang
ab,
Junfeng
Xiang
a,
Yunyan
Wu
ab,
Bo
Yu
a,
Buxing
Han
 abc and
Zhimin
Liu
*abc
abc and
Zhimin
Liu
*abc
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid, Interface and Thermodynamics, CAS Research, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: liuzm@iccas.ac.cn; lianyi302@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cPhysical Science Laboratory, Huairou National Comprehensive Science Center, China
First published on 3rd September 2019
Abstract
The reduction of CO2 with amines and H2 generally produces N-formylated or N-methylated compounds over different catalysts. Herein, we report the selective synthesis of formamides, 1,2-bis(N-heterocyclic)ethanes, and methylamines, which is achieved over an ionic liquid (IL, e.g., 1-butyl-3-methylimidazolium tetrafluoroborate, [BMIm][BF4])–Pd/C catalytic system. By simply varying the reaction temperature, formamides and methylamines can be selectively produced, respectively, in high yields. Interestingly, 1,2-bis(N-heterocyclic)ethanes can also be obtained via the McMurry reaction of the formed formamide coupled with subsequent hydrogenation. It was found that [BMIm][BF4] can react with formamide to form a [BMIm]+–formamide adduct; thus combined with Pd/C it can catalyze McMurry coupling of formamide in the presence of H2 to afford 1,2-bis(N-heterocyclic)ethane. Moreover, Pd/C–[BMIm][BF4] can further catalyze the hydrogenolysis of 1,2-bis(N-heterocyclic)ethane to access methylamine. [BMIm][BF4]–Pd/C was tolerant to a wide substrate scope, giving the corresponding formamides, 1,2-bis(N-heterocyclic)ethanes or methylamines in moderate to high yields. This work develops a new route to produce N-methylamine and opens the way to produce 1,2-bis(N-heterocyclic)ethane from cyclic amine as well.
Introduction
Carbon dioxide (CO2) is an abundant, readily available, nontoxic and renewable C1 building block, and its transformation into value-added chemicals and fuels is of great significance for green and sustainable development.1–6 The reactions of amines with CO2 in the presence of reductants such as hydrosilanes and H2 have been widely investigated, and they generally produce formamides or methylamines.7–11 For the reaction of amines with CO2/H2, the production of methylamines is more difficult than the formation of formamides,12–17 and it requires harsh reaction conditions and catalysts with very high activity.18–21 1,2-Bis(N-heterocyclic)ethanes (e.g., 1,2-bis(piperidine)ethane) are a kind of high value chemical and are generally synthesized via the reaction of cyclic amines with ethyl halides, suffering from production of a large amount of acid waste and complicated post-treatment.22,23 The synthesis of 1,2-bis(N-heterocyclic)ethanes from cyclic amines and CO2/H2 is a green and promising route, but this has not been realized yet.Compared to molecular solvents (e.g., water and organic solvents), ionic liquids (ILs) that are completely composed of ions have unique properties, such as a wide liquid window, very low vapor pressure, specific H-bonding between anions and cations, and so on, which make them promising media for chemical processes.24–28 In particular, they can be designed with specific functions via selection of suitable cations and/or anions, and have been widely applied in catalysis, showing great potential. For example, as both the solvent and catalyst, 1-ethyl-3-methylimidazolium acetate worked well for the transformation of alcohols to esters using O2 as an oxidant under metal-free conditions.29 CO2-philic ILs that can capture CO2via forming carbonates or carbamates have been reported to be excellent media and/or catalysts for CO2 transformation into value-added chemicals.30,31 1-Butyl-3-methylimidazolium ([BMIm]+) chloride combined with Rh nanoparticles and ZnCl2, could realize the efficient reduction of heteroarenes.32 Carbanion-functionalized IL along with Pd(OAc)2 was capable of catalyzing the alkoxycarbonylation reactions of CO to benzoate products under ambient conditions.33 In these reactions, ILs generate synergistic effects with other active species, promoting or catalyzing the reactions.
Herein, we report the reduction of CO2 with amine and H2 over an IL–Pd/C catalytic system, which accomplished the selective synthesis of formamides, 1,2-bis(N-heterocyclic)ethanes and methylamines. Interestingly, the combination of Pd/C with [BMIm][BF4] could realize the selective production of formamides (at 120 °C) or N-methylamines (at 160 °C) in high yields, respectively, as illustrated in Scheme 1. Moreover, 1,2-bis(N-heterocyclic)ethanes can also be obtained via the McMurry reaction of the formed formamide coupled with subsequent hydrogenation. It was found that [BMIm][BF4] could react with formamide to form a [BMIm]+–formamide adduct; thus combined with Pd/C, it can catalyse McMurry coupling of formamide in the presence of H2 to afford 1,2-bis(N-heterocyclic)ethane and further catalyse hydrogenolysis of 1,2-bis(N-heterocyclic)ethanes to access methylamine. In addition, detailed studies indicate that the IL played multiple roles in the reactions including modifying the electronic properties of the metallic Pd particles to enhance their catalytic activity and activating the amine and the intermediate via strong hydrogen bonding. [BMIm][BF4]–Pd/C was tolerant to a wide substrate scope, giving the corresponding formamides, 1,2-bis(N-heterocyclic)ethanes or methylamines in moderate to high yields. This work develops a new route to N-methylamine and opens the way to produce 1,2-bis(N-heterocyclic)ethane from cyclic amine and CO2/H2.
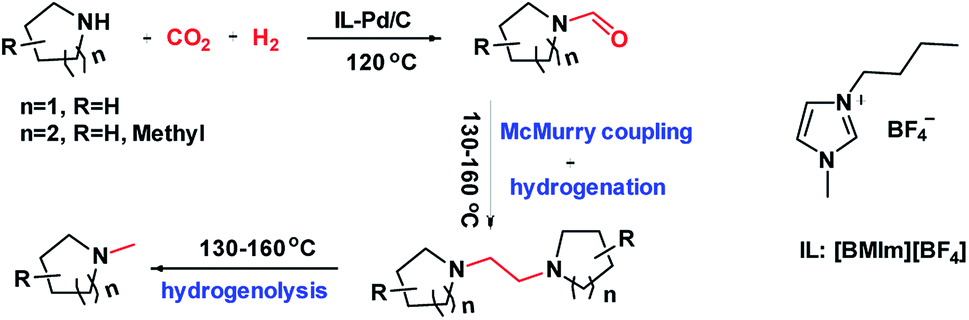 | ||
| Scheme 1 Selective reduction of CO2 with amines and H2. | ||
Results and discussion
Preparation and characterization of the Pd/C catalyst
To prepare the Pd/C catalyst with a loading of 5 wt%, a porous carbon support was first fabricated according to our previous report,34 on which Pd nanoparticles were immobilized via the equal volume impregnation method followed by hydrogen reduction. In the TEM images of the Pd/C catalyst (Fig. 1a and b), the dark dots were identified as Pd nanoparticles, which were uniformly distributed on the support with an average particle size around 1.7 nm. The N2 adsorption/desorption isotherms (Fig. 1c) indicated that the carbon support exhibited microporous and mesoporous structures with a specific surface area of 950.6 m2 g−1. The pore radius distribution curve (Fig. 1d) showed that the pore radii were centered at 15 nm according to the BJH model, a typical feature of mesoporous materials accompanied by some micropores.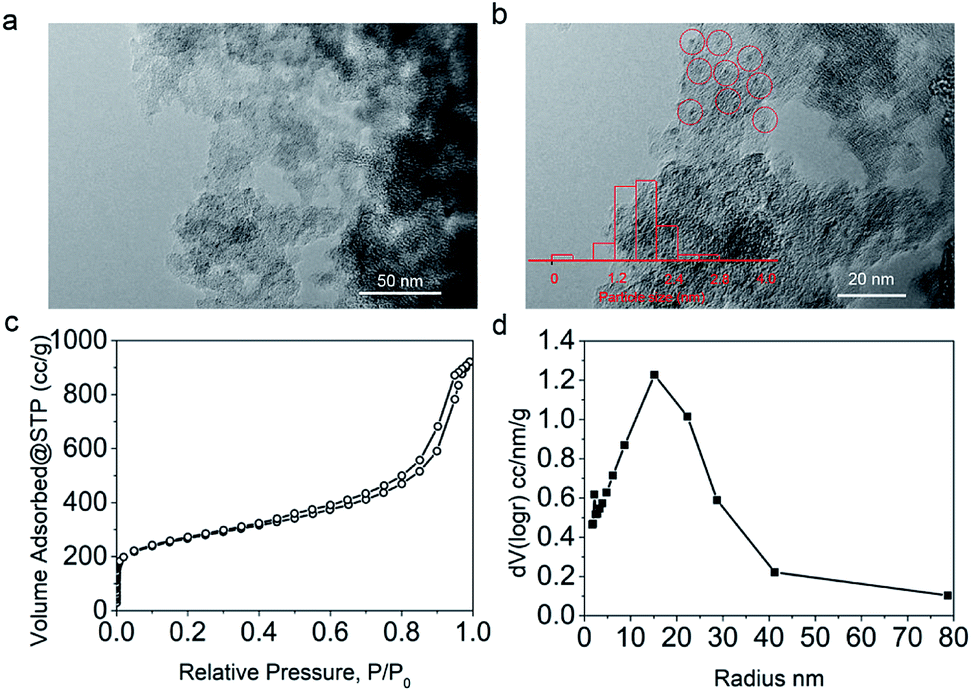 | ||
| Fig. 1 TEM (a) and HRTEM (b) images, N2 sorption isotherms (c) and pore size distribution (d) of the Pd/C catalyst used in this work. | ||
Exploration of the catalytic system for the reaction of piperidine with CO2/H2
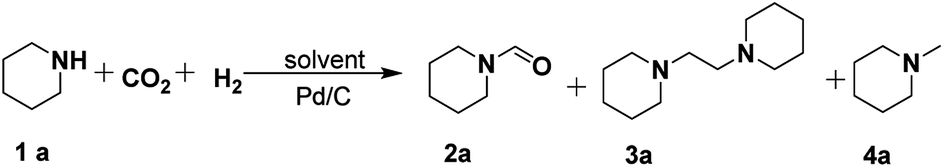
In our initial experiments, the reaction of piperidine (1a) with CO2/H2 was performed using Pd/C as the catalyst to screen the catalytic system and reaction conditions for selective formation of formylpiperidine (2a), 1,2-bis(piperidine)ethane (3a) or methylpiperidine (4a). In the absence of CO2 and H2, no product was detected. As shown in Table 1, the solvents significantly affected the product selectivity. Molecular solvents including ethanol, tetrahydrofuran (THF) and octane exclusively afforded 2a (Table 1, entries 1–3), while imidazolium-based ILs including [BMIm][BF4], [BMIm][PF6], [BMIm][NTf2] and [BMIm][Cl] could offer 2a, 3a and 4a in different yields under the experimental conditions (Table 1, entries 4–9). Among these ILs, [BMIm][BF4] showed the best performance for the production of 4a, achieving a yield of 90% within 9 h (Table 1, entry 5). However, only 2a was obtained in the ILs [P4444][BF4] and [N4444][BF4] (Table 1, entries 10 and 11). These results indicated that for the imidazolium-based ILs the cations played a key role in the formation of 4a, and the anions influenced the activity of the cations. Given that 3a is an important chemical and it has not been accessed from 1a and CO2/H2, we optimized the reaction conditions to obtain 3a as the main product (Table S1†). Combined with Pd/C the mixtures of [BMIm][BF4] with THF or with [BMIm][Cl] could afford 3a in yields higher than 50% at 160 °C (Table 1, entries 12 and 13). Considering that using THF as the reaction medium only 2a was obtained, it can be deduced that [BMIm][BF4] and Pd/C combine to catalyze the formation of 3a and 4a.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Yielde (%) | ||
| 2a | 3a | 4a | ||
| a Conditions: 1a (0.5 mmol), Pd/C (20 mg), solvent (1 mL) or IL (5 mmol), H2 (6 MPa), total pressure of 10 MPa, 160 °C, 6 h. b 9 h. c H2 (5 MPa), total pressure of 8 MPa, [BMIm][BF4] (1.7 mmol), [BMIm][Cl] (3.3 mol), 9 h. d H2 (5 MPa), total pressure of 8 MPa, [BMIm][BF4] (2.5 mmol), THF (2.5 mL), 9 h. e Yield was determined by GC using trimethoxybenzene as the internal standard. | ||||
| 1 | Ethanol | 99 | 0 | 0 |
| 2 | THF | 99 | 0 | 0 |
| 3 | Octane | 99 | 0 | 0 |
| 4 | [BMIm][BF4] | 6 | 11 | 82 |
| 5b | [BMIm][BF4] | 3 | 6 | 90 |
| 6 | [HMIm][BF4] | 17 | 22 | 49 |
| 7 | [BMIm][Cl] | 81 | 14 | 1 |
| 8 | [BMIm][PF6] | 46 | Trace | 53 |
| 9 | [BMIm][NTf2] | 67 | 13 | 18 |
| 10 | [P4444][BF4] | 99 | 0 | 0 |
| 11 | [N4444][BF4] | 99 | 0 | 0 |
| 12c | [BMIm][BF4] + [BMIm][Cl] | 16 | 57 | 26 |
| 13d | [BMIm][BF4] + THF | 1 | 77 | 22 |
Since [BMIm][BF4]–Pd/C showed the best performance for methylation of 1a with CO2/H2 to 4a, it was applied to explore the effects of temperature and reaction time on the reaction. As illustrated in Fig. 2a, the reaction temperature significantly influenced the product selectivity. At 120 °C, 2a was the sole product in an almost quantitative yield within 9 h, while both 3a and 4a were detected in the temperature range of 130–160 °C with the 4a yield increasing with temperature up to 90% at 160 °C. Notably, the yield of 3a was maintained around 20% in this temperature range, while at 160 °C it decreased significantly and 4a became the main product. From the dependence of the 2a and 3a yields on the reaction time at 160 °C (Fig. 2b), it was deduced that 2a and 3a were finally converted into 4a under the experimental conditions.
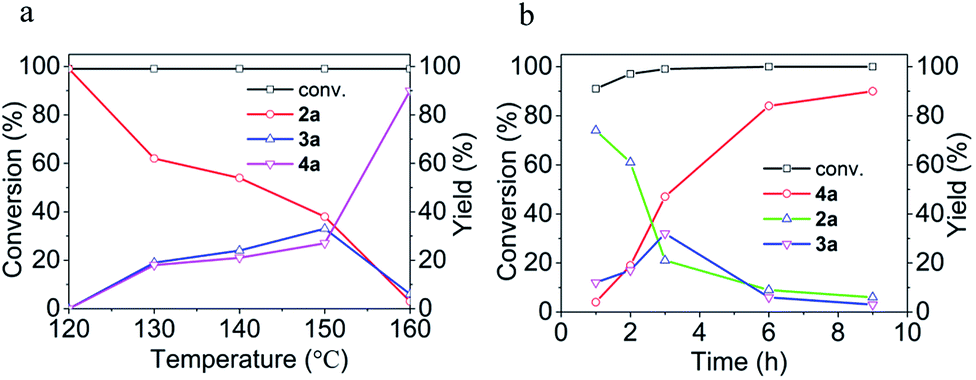 | ||
| Fig. 2 Effects of temperature (a) and reaction time (b) on the conversion of 1a and the yields of 2a, 3a and 4a. (a) 9 h. (b) 160 °C. | ||
Generality of the [BMIm][BF4]–Pd/C catalytic system
To explore the generality of the [BMIm][BF4]–Pd/C catalytic system, it was applied in the reactions of various cyclic amines with CO2/H2. As shown in Scheme 2, this catalytic system was effective for catalyzing the reactions of the tested substrates with CO2/H2, and formylated and methylated products were selectively obtained at 120 and 160 °C, respectively. Pyrrolidine, piperidine, hexamethyleneimine, and morpholine exhibited good reactivity, affording corresponding formylated and methylated products in 90–99% yields under the optimized conditions. By using substituted piperidines like 3-methylpiperidine, 4-methylpiperidine, 2-methylpiperidine, 3,5-dimethylpiperidine, 1,2,3,4-tetrahydroisoquinoline and 1,2,3,4-tetrahydroquinoline as substrates, the reaction also proceeded smoothly and selectively furnished the desired products in moderate to high yields. However, 2-methylpiperidine was converted into the corresponding formamide (2d) only in 10% yield at 120 °C and 1,2,3,4-tetrahydroquinoline led to the corresponding methylamine in low yield.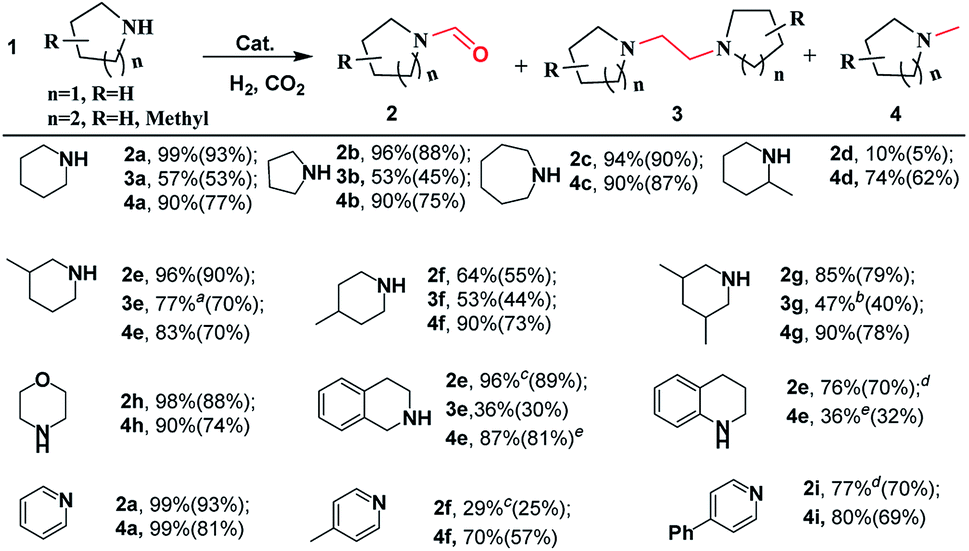 | ||
| Scheme 2 Substrate scope for selective reduction of CO2 with amine and H2 over the Pd/C catalyst. For N-formylation: substrate (0.5 mmol), Pd/C (20 mg), [BMIm][BF4] (5 mmol), H2 (3 MPa), total pressure of 5 MPa, 120 °C, 6 h; isolated yield in brackets. For methylation: H2 (6 MPa), total pressure of 10 MPa, 160 °C, 6 h; the others are the same as those for N-formylation. For accessing 1,2-bis(N-heterocyclic)ethanes: substrate (0.5 mmol), Pd/C (20 mg), [BMIm][BF4] (1.7 mmol), [BMIm][Cl] (3.3 mmol), H2 (5 MPa), total pressure of 8 MPa, 6 h. a12 h. b10 h. c140 °C, 9 h. d140 °C, 12 h, e160 °C, 24 h. | ||
After detailed screening of the reaction conditions, 1,2-bis(N-heterocyclic)ethanes could be achieved in moderate yields in most cases. The highest yield of 1,2-bis(N-heterocyclic)ethane (e.g., 3e) reached up to 77%. Interestingly, using pyridine, 4-methylpyridine, and 4-phenylpyridine as the substrates, they were first hydrogenated, and the corresponding formamides and methylamines were obtained in high yields. However, in these cases corresponding 1,2-bis(N-heterocyclic)ethanes were detected in small amounts, and the reason is unclear. In addition, [BMIm][BF4]–Pd/C was also effective in the reaction of chain secondary amines (e.g., 1,2-diaminopropane) with CO2/H2, selectively producing formamide or methylamine in excellent yields, but it was difficult to access the corresponding 1,2-bis(amino)ethane (Scheme S1†).
The roles of the IL
To explore the roles of the IL in the reactions, the interactions of the IL with Pd/C and with 1a, 2a, and 3a were investigated. Thermogravimetric analysis on the Pd/C catalysts after adsorption of IL at 120 °C and 160 °C (Fig. S1†) indicates that the amounts of the IL adsorbed on Pd/C reached 1.28 and 0.69 wt%, respectively, and its decomposition temperature (500 °C) was much higher than that of the free IL (380 °C). These results indicate that the IL had strong interaction with Pd/C.The Pd/C adsorbing 1.28 wt% [BMIm][BF4] (denoted as IL–Pd/C) was examined by XPS. In the XPS spectrum of IL–Pd/C, the peaks at 340.8 (Pd 3d3/2) and 335.5 eV (Pd 3d5/2) attributed to Pd0 in Pd/C–IL exhibited a downshift of 0.3 eV compared with those of Pd/C (Fig. 3), indicating a possible electron transfer from the IL to the metallic Pd particles. This was also verified by the upshift of the N 1s peak of IL–Pd/C as compared to that of IL–C (Fig. S2,† IL–C refers to the IL adsorbed on a carbon support). The IR analysis (Fig. S3†) indicates that the adsorbed IL on Pd/C showed a red-shifted C–N stretching band of the IL cation from 1580 to 1575 cm−1, which is consistent with electron donation from N atoms in [BMIm]+ to the Pd particles. FT-EXAFS spectra also gave evidence that the IL was complexed with Pd (Fig. S4†). It is reasonable that the lone electron pair of N can affect the electronic state of the Pd particles attached to it because one N atom in the [BMIm]+ cation is essentially in the sp2 hybridization state.
 | ||
| Fig. 3 Pd 3d XPS spectra of (a) Pd/C and (b) IL–Pd/C. | ||
Furthermore, the interactions of [BMIm][BF4] with CO2, 1a, 2a and 3a were investigated via NMR analysis. No new signals or obvious chemical shifts were observed in the 1H, 13C, 19F and 11B NMR spectra of the mixture of [BMIm][BF4] and CO2 (Fig. S5†), indicating that this IL cannot activate CO2 noticeably. Given that 1a is easily converted to a white solid, dihydropyridine-monocarboxylic acid, once exposed to CO2, it is supposed that CO2 is activated by 1avia forming an adduct. 1H, 15N, 11B NMR (Fig. S6, S7A and B†) and FTIR (Fig. S8†) analyses on the [BMIm][BF4]–1a mixture indicate that 1a was activated by the IL via hydrogen bonding and electrostatic interaction. In the 1H NMR spectrum of the [BMIm][BF4]–2a mixture (Fig. 4a), the 1H signal of the formyl group in 2a and that of the H atoms in [BMIm]+ shifted greatly, suggesting the strong C–H⋯O interaction between the acidic H(2) of [BMIm]+ and the O atom of the formyl group in 2a. Specifically, the 13C NMR signals of the formyl group also shifted accordingly (Fig. S9†), further revealing the activation of the formyl group by the IL. In the FTIR spectrum, the absorption band at 1669 cm−1 attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of 1a shifted to 1664 cm−1 upon 1a interacting with [BMIm][BF4], and the peaks at 3162 and 3122 cm−1 belonging to the stretching vibration of C–H from the imidazole ring of [BMIm]+ shifted accordingly. The FTIR analysis gave evidence of interaction between 1a and the IL, also supporting the NMR results (Fig. 4b). As reported, reactive carbenes can be generated at the C(2) position of [BMIm]+ under mild basic conditions.35 In this work, the basic reaction environment provided by 1a or 4a may be favorable for the formation of carbenes from [BMIm]+, which may further react with 2a to form an intermediate. To confirm this hypothesis, a mixture of IL and 2a together with K2CO3 was examined. Based on 1H NMR and 1H–1H correlation spectroscopy (COSY) analysis (Fig. 4c and S10†), an intermediate from [BMIm][BF4] and 2a (denoted as [BMIm–OH–2a][BF4]) was detected (Fig. 4c), which was also confirmed by the detection of {[BMIm–OH–2a][BF4][BMIm](2a)}+ (m/z = 591.4) by electrospray-ionization mass spectrometry (ESI-MS) (Fig. S11†). The above results indicate that the IL can activate 2a to form a [BMIm]+–formamide adduct, which may be favorable for the formation of 3a and 4a. In addition, 3a can also be activated by the IL, as supported by NMR analysis with the obvious changes of chemical shifts as it mixed with the IL in the presence of 2a (Fig. S12†), which can explain why the IL promotes the hydrogenolysis of 3a.
O of 1a shifted to 1664 cm−1 upon 1a interacting with [BMIm][BF4], and the peaks at 3162 and 3122 cm−1 belonging to the stretching vibration of C–H from the imidazole ring of [BMIm]+ shifted accordingly. The FTIR analysis gave evidence of interaction between 1a and the IL, also supporting the NMR results (Fig. 4b). As reported, reactive carbenes can be generated at the C(2) position of [BMIm]+ under mild basic conditions.35 In this work, the basic reaction environment provided by 1a or 4a may be favorable for the formation of carbenes from [BMIm]+, which may further react with 2a to form an intermediate. To confirm this hypothesis, a mixture of IL and 2a together with K2CO3 was examined. Based on 1H NMR and 1H–1H correlation spectroscopy (COSY) analysis (Fig. 4c and S10†), an intermediate from [BMIm][BF4] and 2a (denoted as [BMIm–OH–2a][BF4]) was detected (Fig. 4c), which was also confirmed by the detection of {[BMIm–OH–2a][BF4][BMIm](2a)}+ (m/z = 591.4) by electrospray-ionization mass spectrometry (ESI-MS) (Fig. S11†). The above results indicate that the IL can activate 2a to form a [BMIm]+–formamide adduct, which may be favorable for the formation of 3a and 4a. In addition, 3a can also be activated by the IL, as supported by NMR analysis with the obvious changes of chemical shifts as it mixed with the IL in the presence of 2a (Fig. S12†), which can explain why the IL promotes the hydrogenolysis of 3a.
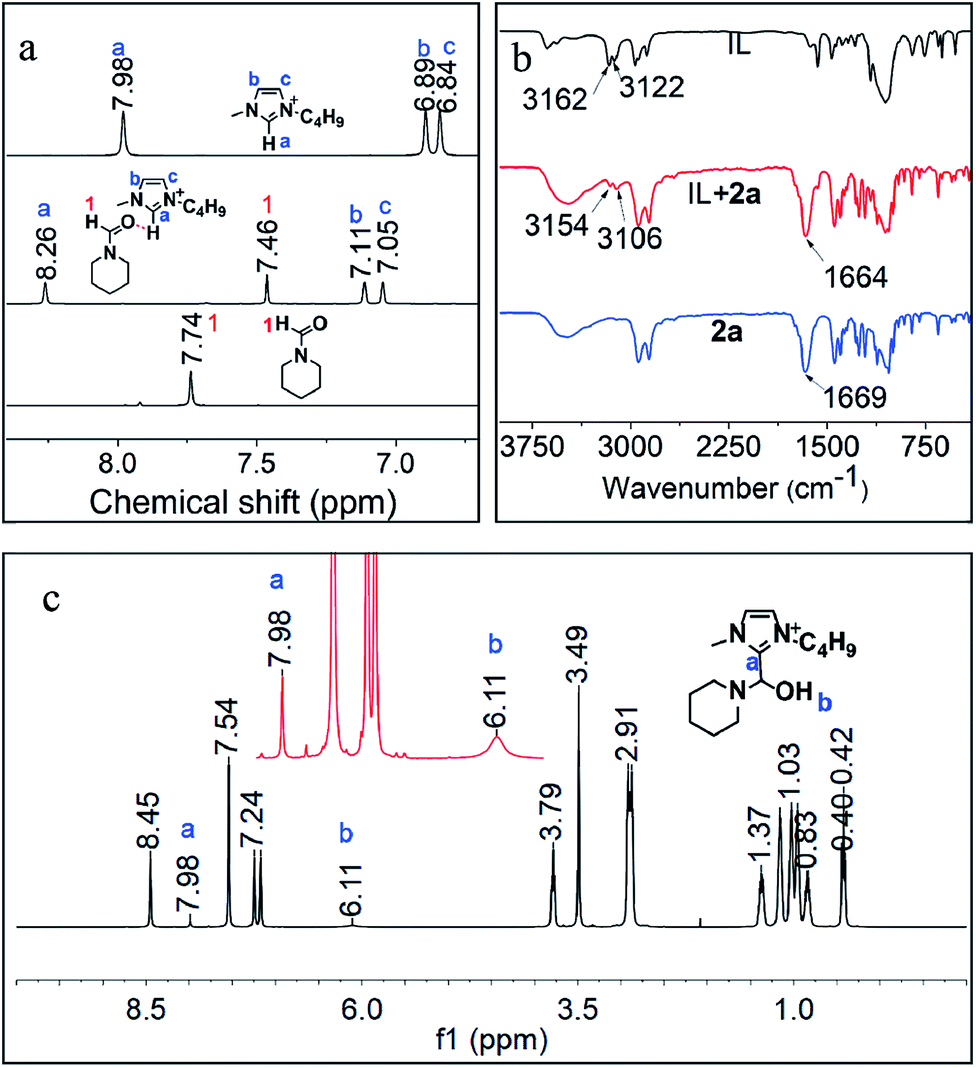 | ||
| Fig. 4 1H NMR (a) and FT-IR spectra (b) of [Bmim][BF4], 2a and their mixture, and 1H NMR (c) of the mixture of IL and 2a together with K2CO3. | ||
Possible reaction pathway
To gain deep insight into the reaction pathway of the formation of 3a and 4a, control experiments were performed. Previous reports revealed that 2a could be catalytically converted to 4avia hydrogenation under appropriate conditions.36 However, in this work taking 2a as the substrate to react with CO2/H2 or only with H2 over Pd/C in [BMIm][BF4], no 4a was detectable (Scheme S2A and B†). This indicates that Pd/C could not catalyze the direct hydrogenation of 2a to 4a in [BMIm][BF4], which excludes the pathway of 2a reduction with H2, and implies that 2a undergoes transformation to 3a to form 4a.As is known, McMurry coupling is a reductive reaction, in which two ketone or aldehyde groups are coupled to form an alkene using a titanium chloride compound (e.g., titanium(IV) chloride) and a reducing agent (e.g., zinc) in the presence of a base.37 In the process of 1a reacting with CO2/H2, the reaction system is always kept in a basic environment due to the presence of the amine feedstock or the formed methylamine, which might facilitate the occurrence of the McMurry reaction of the formed formamide. To verify this, we treated 2a in the presence of 1a or 4a over Pd/C in [BMIm][BF4] (Scheme 3a and b), and 3a was excitingly obtained in appreciable yields in both cases, different from the results shown in Scheme S2A and B.† This suggests that the McMurry reaction of 2a may occur to produce 1,2-bis(piperidine)ethylene although it was not detected in the reaction process, because it can be rapidly hydrogenated to 3a catalyzed by Pd/C (Scheme 3c). Notably, on prolonging the reaction time to 12 h, both 2a and 3a disappeared, and 4a became the sole product (Scheme 3b). This indicates that 3a was further converted into 4avia hydrogenolysis under the experimental conditions, which was confirmed by the reaction of 3a with H2 over Pd/C in [BMIm][BF4] containing t-BuOK to produce 4a (Scheme 3d). Generally, the C–N bond breaks more easily than the C–C bond. However, in this work the C–C bond rather than the C–N bond in 3a was broken. To give a reasonable explanation for this, Gaussian calculation was performed, which showed that the acidic hydrogen atom H(2) of [BMIm]+ could interact with an N atom of 3a, causing the length of the C–C bond in 3a to become longer (Fig. S13†). This may be responsible for the cleavage of the C–C bond in 3a to give 4a. [BMIm][BF4] played a crucial role in the hydrogenolysis of 3a to 4a, deduced from the fact that without the IL the hydrogenolysis of 3a did not occur even in the presence of the base t-BuOK (Scheme S2C†).
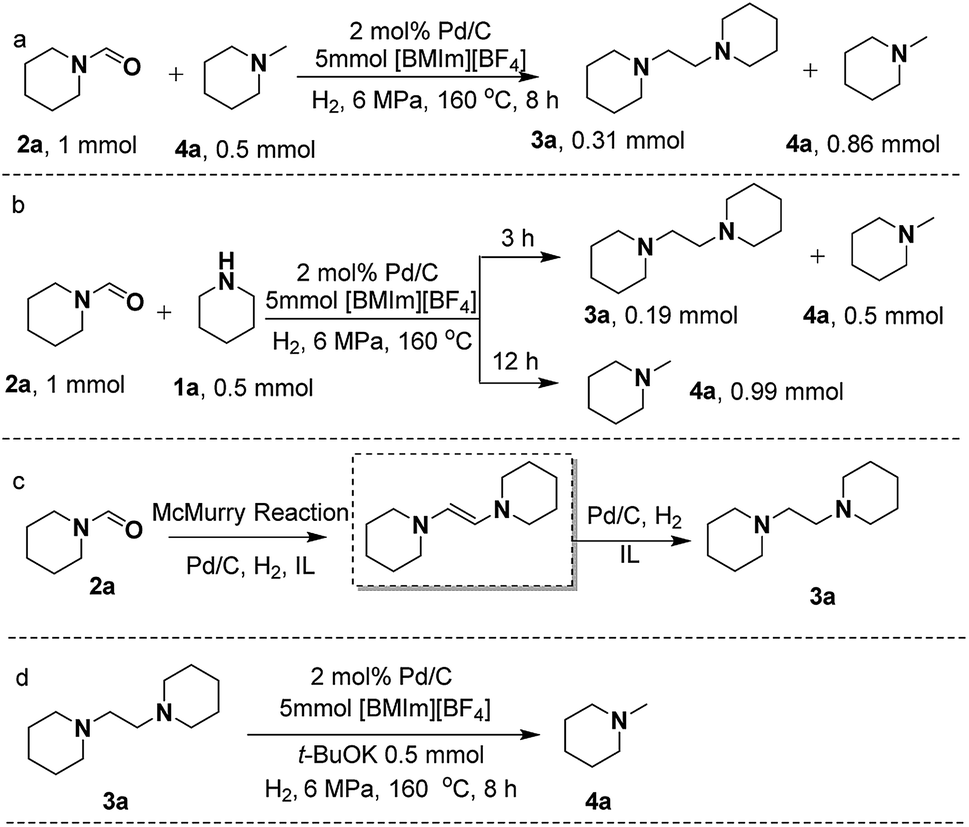 | ||
| Scheme 3 Control experiments. Conversion of 2a to 3a in the presence of 4a (a). Conversion of 2a to 3a and/or 4a in the presence of 1a for different reaction times (b). The possible reaction pathway from 2a to 3a (c). Conversion of 3a to 4a in the presence of t-BuOK (d). | ||
To identify possible intermediates in the reaction process, the reaction solution was analyzed by means of electrospray ionization mass spectrometry (ESI-MS.) As shown in Fig. S14,† {H[BMIm][BF4](1a)}+ (m/z = 312.2), {H[BMIm][BF4](3a)}+ (m/z = 423.3), {[BMIm][N-(N-piperidinoglycolyl)piperidine](4a)}+ (m/z = 464.4), {[BMIm-[N-(N-piperidinoglycolyl)piperidine](2a)}+ (m/z = 476.4), {[BMIm–OH–2a][BF4][BMIm](2a)}+ (m/z = 591.4) and {([BMIm][BF4])2[BF4][N-(N-piperidinoglycolyl)piperidine]}− (m/z = 765.4) were detected, suggesting that under the experimental conditions, [BMIm–OH–2a][BF4] and N-(N-piperidinoglycolyl)piperidine were the key intermediates and [BMIm][BF4] played a vital role in activating and stabilizing them.
Based on the experimental results, a possible pathway is proposed, as shown in Scheme 4. Initially, 1a is activated by the IL via hydrogen bonding and electrostatic interaction and undergoes formylation with CO2/H2 to yield 2a over Pd/C. Subsequently, an intermediate [BMIm–OH–2a][BF4] is formed from 2a and [BMIm]+ in the presence of 1a, and the McMurry reaction proceeds through coupling of [BMIm–OH–2a][BF4] with 2a to produce N-(N-piperidinoglycolyl)piperidine, followed by hydrogenation to form 3a. Finally, hydrogenolysis of 3a results in the formation of 4a. Notably, this is the first time that the McMurry reaction of formamide over Pd/C using H2 as a reductant has been realized, very different from the traditional routes.
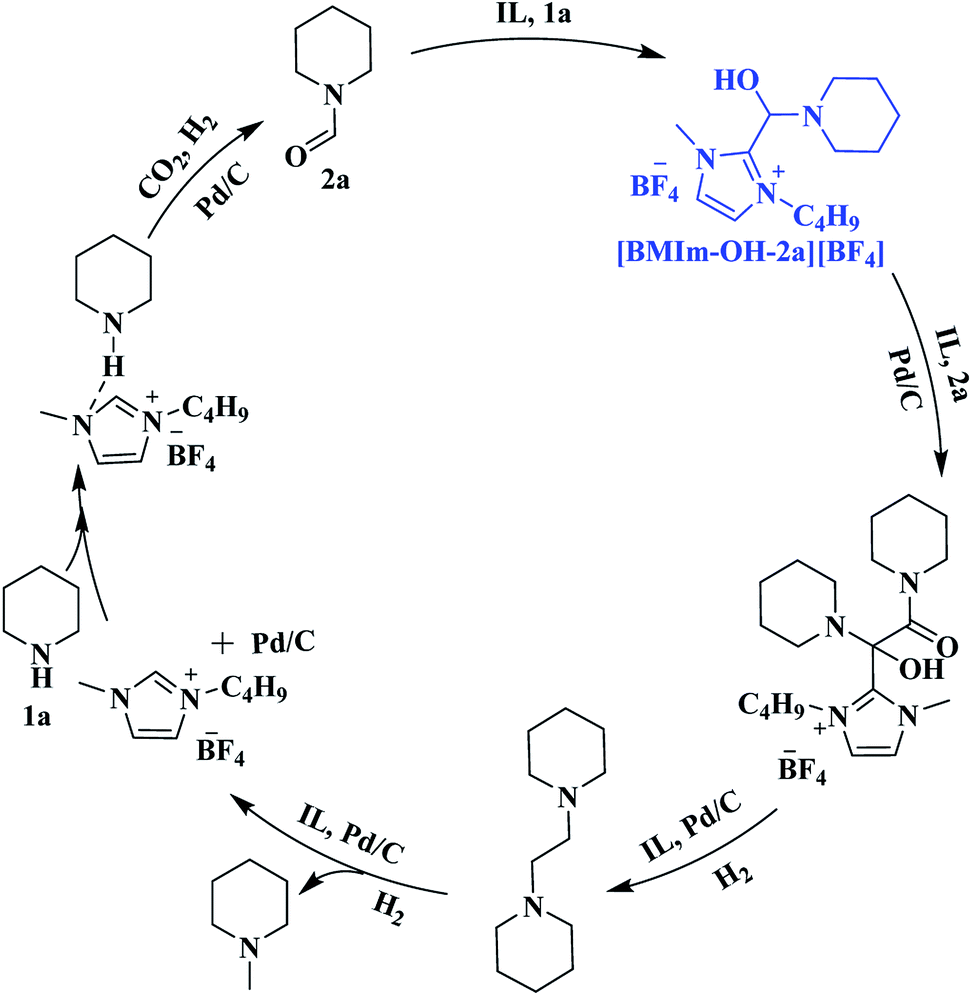 | ||
| Scheme 4 Plausible pathways for the formation of 2a, 3a and 4a. | ||
Conclusions
In summary, selective reduction of CO2 with cyclic amines and H2 was realized over the [BMIm][BF4]–Pd/C catalytic system, and 1,2-bis(N-heterocyclic)ethanes were obtained via the McMurry reaction of formamide coupled with subsequent hydrogenation. [BMIm][BF4] displayed multiple functions including improving the catalytic activity of Pd particles and activating the amine substrate and the formed formamide intermediate. [BMIm][BF4]–Pd/C was tolerant to a wide substrate scope, giving the corresponding formamides, 1,2-bis(N-heterocyclic)ethanes or methylamines in moderate to high yields. We believe that these findings provide insights into achieving cooperativity between the IL and metal catalysts.Experimental
General procedures for the reaction of N-containing compounds with CO2/H2
All reactions were performed in a stainless steel autoclave equipped with a Teflon tube (16 mL inner volume) and a magnetic stirrer. In a typical experiment, piperidine (0.5 mmol), [BMIm][BF4] (5 mmol) and Pd/C (20 mg) were successively loaded into the autoclave under an N2 atmosphere, and then the autoclave was sealed. H2 (6 MPa) and CO2 were charged successively into the reactor until the total pressure reached 10 MPa at room temperature. The autoclave was moved to a heating furnace at 433 K. After the desired reaction time, the reactor was cooled down in ice water and the gas inside was vented slowly. Trimethoxybenzene (internal standard) and diethyl ether were added to the reaction solution, stirred vigorously and centrifuged. The upper liquid was extracted for analysis.Product analysis
Liquid samples were analysed using a gas chromatograph (Agilent 4890D) equipped with an ultra-inert capillary column (19091S-433UI HP-5 ms). NMR spectra were collected using a coaxial insert NMR tube with d6-DMSO in the internal tube as a reference, recorded using Bruker 400 HD (1H NMR, 11B NMR and 13C NMR spectra) and Bruker 600 instruments (15N NMR and 19F NMR spectra and the 1H–1H correlation spectrum (1H–1H COSY). 11B NMR and 19F NMR spectra were obtained using the same equipment in succession. 1H NMR data recorded in d6-DMSO were listed with the residual DMSO at 2.50 ppm, while 13C NMR data were listed with the residual DMSO at 39.51 ppm; 1H NMR data in d-CDCl3 were listed with the residual CHCl3 at 7.26 ppm, while 13C NMR data were listed with the residual CHCl3 at 77.17 ppm. FT-IR spectra were recorded using a Bruker Tensor-27 instrument, scanning from 400 to 4000 cm−1. Transmission electron microscopy (TEM) images were obtained using a JEOL-2100F. X-ray photoelectron spectroscopy (XPS) was performed on an ESCALab220i-XL electron spectrometer from VG Scientific at a pressure of 3 × 10−9 mbar using 300 W Al-Kα radiation. The binding energies (BEs) were referenced to the C 1s line at 284.8 eV from adventitious carbon. X-ray absorption data at the Pd K-edge of the samples were recorded at room temperature in fluorescence mode with a silicon drift fluorescence detector at beam line BL14W1 of the Shanghai Synchrotron Radiation Facility (SSRF), China. The electron storage ring was operated at 3.5 GeV. Data processing was performed using the program ATHENA. The electrospray ionization mass spectrometry (ESI-MS) data (in both negative and positive modes) were collected using a Bruker 9.4T Solarix instrument.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2017YFA0403102 and 2017YFA0403003), Natural Science Foundation of China (21890761, 21533011, and 21773266), Chinese Academy of Sciences (QYZDY-SSW-SLH013-2), and Beijing Municipal Science & Technology Commission (Z181100004218004).Notes and references
- M. Y. He, Y. H. Sun and B. X. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS PubMed.
- D. X. Yang, Q. G. Zhu, C. J. Chen, H. Z. Liu, Z. M. Liu, Z. J. Zhao, X. Y. Zhang, S. J. Liu and B. X. Han, Nat. Commun., 2019, 10, 677 CrossRef PubMed.
- Z. Lian, D. U. Nielsen, A. T. Lindhardt, K. Daasbjerg and T. Skrydstrup, Nat. Commun., 2016, 7, 13782 CrossRef PubMed.
- Y. M. Ni, Z. Y. Chen, Y. Fu, Y. Liu, W. L. Zhu and Z. M. Liu, Nat. Commun., 2018, 9, 3457 CrossRef PubMed.
- C. X. Qian, W. Sun, D. L. H. Hung, C. Y. Qiu, M. Makaremi, S. G. Hari Kumar, L. Wan, M. Ghoussoub, T. E. Wood, M. K. Xia, A. A. Tountas, Y. F. Li, L. Wang, Y. C. Dong, I. Gourevich, C. V. Singh and G. A. Ozin, Nat. Catal., 2019, 2, 46 CrossRef CAS.
- C. S. Yang, R. T. Mu, G. S. Wang, J. M. Song, H. Tian, Z. J. Zhao and J. L. Gong, Chem. Sci., 2019, 10, 3161 RSC.
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC.
- Y. H. Li, X. J. Cui, K. W. Dong, K. Junge and M. Beller, ACS Catal., 2017, 7, 1077 CrossRef CAS.
- R. H. Lam, C. M. A. McQueen, I. Pernik, R. T. McBurney, A. F. Hill and B. A. Messerle, Green Chem., 2019, 21, 538 RSC.
- X.-D. Li, S.-M. Xia, K.-H. Chen, X.-F. Liu, H.-R. Li and L.-N. He, Green Chem., 2018, 20, 4853 RSC.
- R. L. Nicholls, J. A. McManus, C. M. Rayner, J. A. Morales-Serna, A. J. P. White and B. N. Nguyen, ACS Catal., 2018, 8, 3678 CrossRef CAS.
- O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934 CrossRef CAS PubMed.
- H. Lv, Q. Xing, C. T. Yue, Z. Q. Lei and F. W. Li, Chem. Commun., 2016, 52, 6545 RSC.
- M. A. Affan and P. G. Jessop, Inorg. Chem., 2017, 56, 7301–7305 CrossRef CAS PubMed.
- Y. J. Zhang, H. L. Wang, H. K. Yuan and F. Shi, ACS Sustainable Chem. Eng., 2017, 5, 5758 CrossRef CAS.
- U. Jayarathne, N. Hazari and W. H. Bernskoetter, ACS Catal., 2018, 8, 1338 CrossRef CAS.
- X. J. Cui, X. C. Dai, Y. Zhang, Y. Q. Deng and F. Shi, Chem. Sci., 2014, 5, 649 RSC.
- K. Beydoun, G. Ghattas, K. Thenert, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2014, 53, 11010 CrossRef CAS PubMed.
- K. Kon, S. M. A. H. Siddiki, W. Onodera and K.-i. Shimizu, Chem.–Eur. J., 2014, 20, 6264 CrossRef CAS PubMed.
- I. Sorribes, J. R. Cabrero-Antonino, C. Vicent, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 13580 CrossRef CAS PubMed.
- C. Fang, C. L. Lu, M. H. Liu, Y. L. Zhu, Y. Fu and B. L. Lin, ACS Catal., 2016, 6, 7876 CrossRef CAS.
- H. Yang and F. C. Thyrion, Int. J. Chem. Kinet., 1994, 26, 309 CrossRef CAS.
- S. A. Gamzaeva, P. S. Mamedova, K. M. Allakhverdieva, G. K. Velieva, M. A. Akhundova and M. A. Allakhverdiev, Russ. J. Appl. Chem., 2009, 82, 1577 CrossRef CAS.
- K. H. Chen, G. L. Shi, W. D. Zhang, H. R. Li and C. M. Wang, J. Am. Chem. Soc., 2016, 138, 14198 CrossRef CAS PubMed.
- Z. R. Zhang, J. L. Song and B. X. Han, Chem. Rev., 2017, 117, 6834 CrossRef CAS PubMed.
- A. Weilhard, M. I. Qadir, V. Sans and J. Dupont, ACS Catal., 2018, 8, 1628 CrossRef CAS.
- Y. F. Zhao, Z. Z. Yang, B. Yu, H. Y. Zhang, H. J. Xu, L. D. Hao, B. X. Han and Z. M. Liu, Chem. Sci., 2015, 6, 2297 RSC.
- X. X. Sang, J. L. Zhang, J. F. Xiang, J. Cui, L. R. Zheng, J. Zhang, Z. H. Wu, Z. H. Li, G. Mo, Y. Xu, J. L. Song, C. C. Liu, X. N. Tan, T. Luo, B. X. Zhang and B. X. Han, Nat. Commun., 2017, 8, 175 CrossRef PubMed.
- M. Y. Liu, Z. R. Zhang, H. Z. Liu, Z. B. Xie, Q. Q. Mei and B. X. Han, Sci. Adv., 2018, 4, eaas9319 CrossRef CAS PubMed.
- Y. F. Zhao, B. Yu, Z. Z. Yang, H. Y. Zhang, L. D. Hao, X. Gao and Z. M. Liu, Angew. Chem., Int. Ed., 2014, 53, 5922 CrossRef CAS PubMed.
- Y. F. Zhao, Y. Y. Wu, G. F. Yuan, L. D. Hao, X. Gao, Z. Z. Yang, B. Yu, H. Y. Zhang and Z. M. Liu, Chem.–Eur. J., 2016, 11, 2735 CrossRef CAS.
- A. Karakulina, A. Gopakumar, İ. Akçok, B. L. Roulier, T. LaGrange, S. A. Katsyuba, S. Das and P. J. Dyson, Angew. Chem., Int. Ed., 2015, 55, 292 CrossRef PubMed.
- D. J. Tao, F. F. Chen, Z. Q. Tian, K. Huang, S. M. Mahurin, D. E. Jiang and S. Dai, Angew. Chem., Int. Ed., 2017, 56, 6843 CrossRef CAS PubMed.
- Z. S. Ma, H. Y. Zhang, Z. Z. Yang, Y. F. Zhao, B. Yu and Z. M. Liu, J. Mater. Chem. A, 2014, 2, 19324 RSC.
- V. K. Aggarwal, I. Emme and A. Mereu, Chem. Commun., 2002, 1612 RSC.
- X. L. Du, G. Tang, H. L. Bao, Z. Jiang, X. H. Zhong, D. S. Su and J. Q. Wang, ChemSusChem, 2015, 8, 3489 CrossRef CAS PubMed.
- X. F. Duan, J. Zeng, J. W. Lü and Z. B. Zhang, J. Org. Chem., 2006, 71, 9873 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and tables. See DOI: 10.1039/c9sc03242h |
| This journal is © The Royal Society of Chemistry 2019 |