
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical C–H bond activation via cationic iridium hydride pincer complexes†
Brian M.
Lindley‡
 a,
Andrew G.
Walden‡
ab,
Ann Marie
Brasacchio
ac,
Andrea
Casuras
d,
Nicholas
Lease
d,
Chun-Hsing
Chen
a,
Alan S.
Goldman
d and
Alexander J. M.
Miller
*a
a,
Andrew G.
Walden‡
ab,
Ann Marie
Brasacchio
ac,
Andrea
Casuras
d,
Nicholas
Lease
d,
Chun-Hsing
Chen
a,
Alan S.
Goldman
d and
Alexander J. M.
Miller
*a
aDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, USA. E-mail: ajmm@email.unc.edu
bOglethorpe University, Atlanta, Georgia 30319, USA
cHigh Point University, High Point, NC 27262, USA
dRutgers, The State University of New Jersey, New Brunswick, New Jersey 08903, USA
First published on 20th August 2019
Abstract
A C–H bond activation strategy based on electrochemical activation of a metal hydride is introduced. Electrochemical oxidation of (tBu4PCP)IrH4 (tBu4PCP is [1,3-(tBu2PCH2)-C6H3]−) in the presence of pyridine derivatives generates cationic Ir hydride complexes of the type [(tBu4PCP)IrH(L)]+ (where L = pyridine, 2,6-lutidine, or 2-phenylpyridine). Facile deprotonation of [(tBu4PCP)IrH(2,6-lutidine)]+ with the phosphazene base tert-butylimino-tris(pyrrolidino)phosphorane, tBuP1(pyrr), results in selective C–H activation of 1,2-difluorobenzene (1,2-DFB) solvent to generate (tBu4PCP)Ir(H)(2,3-C6F2H3). The overall electrochemical C–H activation reaction proceeds at room temperature without need for chemical activation by a sacrificial alkene hydrogen acceptor. This rare example of undirected electrochemical C–H activation holds promise for the development of future catalytic processes.
Introduction
The activation of C–H bonds has become one of the most powerful tools in chemical synthesis.1,2 Among the diverse mechanistic pathways available to transition metal complexes for C–H bond activation, C–H oxidative addition at a low-valent metal center is perhaps the most prominent. This reaction underpins thermal C–H functionalization by iridium pincer complexes that have proven to be especially prolific catalysts.3 The archetypal dihydride complex (tBu4PCP)Ir(H)2 and its H2 adduct (tBu4PCP)IrH4 (1, Scheme 1)4 catalyze an array of reactions based on alkane dehydrogenation, as well as C–O and C–F activation reactions that are initiated by sp3 C–H bond activation.3 Efficient dehydrogenation catalysis requires a sacrificial alkene (e.g. tert-butylethylene, TBE) to generate a three-coordinate iridium(I) intermediate (tBu4PCP)Ir that rapidly activates hydrocarbon C–H bonds. Catalyst activation by the sacrificial alkene is the rate-determining step in some (tBu4PCP)Ir-catalyzed C–H functionalization systems.3,5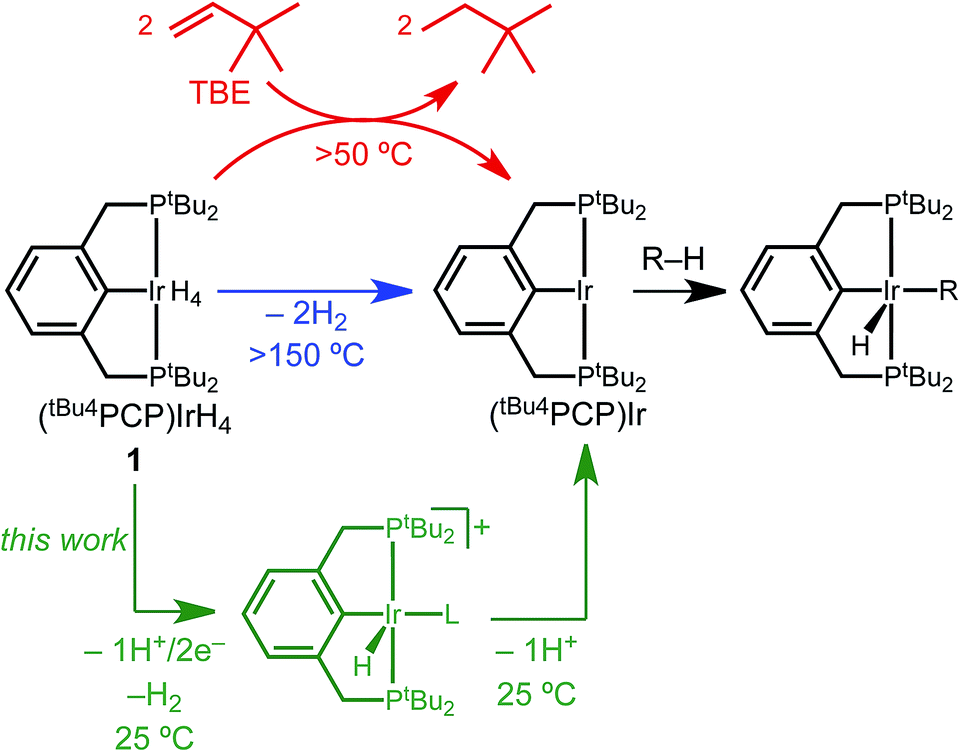 | ||
| Scheme 1 Comparison of thermal and electrochemical C–H activation by pincer iridium complexes. | ||
Electrochemical oxidation of pincer iridium hydride complexes (Scheme 1, green) represents a possible alternative to the use of sacrificial alkene hydrogen acceptor reagents. Sequential removal of H+ and e− equivalents would result in a (perhaps counterintuitive) change in formal oxidation state from Ir(III) to Ir(I) upon oxidation, generating the key three-coordinate intermediate (tBu4PCP)Ir.
Electrochemical C–H functionalization has recently enabled impressive advances in synthetic organic chemistry, either via direct electrochemical activation of organic substrates,6,7 or via electrochemical regeneration of organometallic complexes to close a catalytic cycle.8–10 The latter processes are notably limited to substrates with directing groups that pre-organize and accelerate the C–H bond activation. To our knowledge, there are no conclusive examples of electrocatalytic C–H bond activation of sp2 or sp3 C–H bonds lacking directing groups, and no examples of electrochemically driven intermolecular C–H activation with the versatile family of (PCP)Ir-based complexes.11
We present a detailed investigation of the electrochemical C–H bond activation of 1,2-difluorobenzene by tetrahydride 1. In demonstrating the elementary steps of Scheme 1, a series of stabilized cationic iridium hydride complexes was isolated and characterized and found to cleanly activate the arene upon deprotonation. The overall reaction utilizes electrochemistry to achieve C–H bond activation of a directing-group-free substrate at room temperature, without a sacrificial chemical hydrogen acceptor.
Results and discussion
Electrochemical synthesis of a cationic iridium hydride
Several factors guided the selection of conditions that would be suitable for electrochemical C–H bond activation by pincer iridium hydrides. First, we recognized that the three-coordinate intermediate (tBu4PCP)Ir reacts with the C–H bonds of most common solvents, leading us to consider conditions of large substrate excess (or substrate as solvent). A second concern was the ability of the substrate-solvent to dissolve a supporting electrolyte and present a suitable electrochemical potential window, given that many potential substrates are non-polar and thus unsuitable for preparative scale electrochemistry. With these constraints in mind, we selected 1,2-difluorobenzene (1,2-DFB) as an arene substrate12 without directing groups that is suitably polar to dissolve commonly employed electrolytes such as [nBu4N][PF6] and is stable over a wide potential window, especially in the oxidative direction.13–15 The tetrahydride 1 was the focus of investigations, based on excellent stability observed in control experiments (whereas the dihydride underwent partial decomposition in polar solvents). Based on our prior observation that oxidation of (tBu4PCP)Ir(H)n (n = 2, 4) in THF leads to decomposition in the absence of a stabilizing ligand,16 we hypothesized that key cationic intermediates might need to be stabilized. Neutral ligands that provide good σ donation and weak π back-donation were targeted as candidates to stabilize cationic Ir(III) but readily dissociate upon deprotonation to neutral Ir(I).We initiated our studies by exploring whether cationic complexes stabilized by pyridine and its analogues could be accessed electrochemically and serve as intermediates in the C–H bond activation of 1,2-DFB. It has been recognized previously that oxidation can greatly increase the acidity of metal hydrides,17 so we anticipated pyridine could act as a base and a ligand. Pyridine itself was found to react with 1 (see ESI Section 3†), however, so we turned to the bulkier congener 2,6-lutidine.
Cyclic voltammetry (CV) of 1 in 1,2-DFB containing 0.2 M [nBu4N][PF6] electrolyte and decamethylferrocene as an internal standard revealed an irreversible oxidation at approximately 0.35 V vs. Cp2Fe+/0 at 100 mV s−1. The iridium complex is significantly easier to oxidize than 2,6-lutidine, which is oxidized beyond 0.7 V vs. Cp2Fe+/0.
The CV data guided the choice of conditions for preparative electrochemical synthesis. The tetrahydride 1 and 11 equiv. 2,6-lutidine were subjected to controlled potential electrolysis (CPE) at 0.90 V vs. Ag+/0 (approx. 0.4 V vs. Cp2Fe+/0) in 1,2-DFB with [nBu4N][PF6] electrolyte. During the 1.5 h electrolysis, charge equivalent to 2.1e− per Ir was passed and a color change from pale to dark yellow was observed. Analysis by 1H NMR spectroscopy confirmed formation of a new species assigned as the target cationic 2,6-lutidine (lut) adduct, [(tBu4PCP)Ir(H)(lut)]+ in 83% 1H NMR yield based on the mesitylene internal standard (Scheme 2).
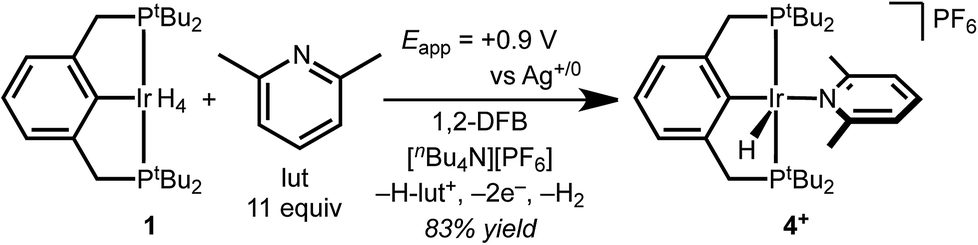 | ||
| Scheme 2 Electrosynthesis of cationic iridium hydride 4+. | ||
Independent synthesis and characterization of cationic iridium hydride complexes
An independent synthetic route to cationic hydrido pyridine complexes was designed based on halide abstraction from (tBu4PCP)Ir(H)(Cl) (2). As shown in Scheme 3, equimolar mixtures of 2 and substituted pyridines were treated with NaBArF4 (ArF = 3,5-bis(trifluoromethyl)phenyl) in CH2Cl2. This route was used to obtain pyridine complex [(tBu4PCP)Ir(H)(py)][BArF4] (3+), 2,6-lutidine complex [(tBu4PCP)Ir(H)(lut)][BArF4] (4+), and 2-phenylpyridine (2-Phpy) complex [(tBu4PCP)Ir(H)(2-Phpy)][BArF4] (5+). For pyridine, stoichiometric control is important to prevent further binding of pyridine to 3+ (see ESI Section 3†), while further binding of lut and 2-Phpy to 4+ and 5+ was not observed.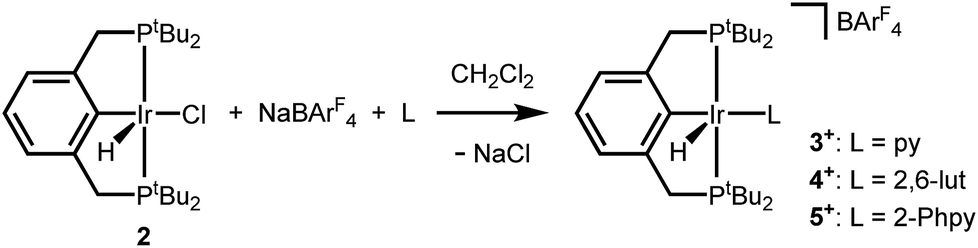 | ||
| Scheme 3 Synthesis of cationic hydride complexes. | ||
Cationic hydrides supported by the archetypal tBu4PCP ligand are surprisingly rare,18,19 so these species were fully characterized. Multinuclear NMR spectra of 3+ are consistent with a square pyramidal geometry with an apical hydride ligand, on the basis of the highly upfield-shifted 1H NMR signal for the hydride at −45.4 ppm.18 Complexes 4+ (−31.4 ppm) and 5+ (−20.1 ppm) feature hydride 1H NMR signals that are shifted downfield relative to that of 3+ (−45.4 ppm). Hydride chemical shifts in octahedral Ir(III) complexes reflect the presence and nature of ligands trans to the hydride donor,20,21 so the large span of chemical shifts indicated unexpectedly significant changes in geometry (particularly given that the hydride of 3+ is assigned to be trans to a vacant coordination site).
Single crystals of complexes 3+, 4+, and 5+ were obtained, enabling a comparative XRD study evaluating the molecular geometry with each pyridine ligand in the solid state (Fig. 1). Cationic hydrido pyridine complex 3+ features a square pyramidal structure, with a 179.8° angle between the aryl backbone C, central Ir, and the pyridine N, similar to the C–Ir–C(O) angle in [(tBu4PCP)Ir(H)(CO)]+.18 The same C–Ir–N angle in the 2,6-lutidine complex 4+ is 174.5°, while in the structure of 2-phenylpyridine complex 5+ the pyridyl nitrogen is canted dramatically, nearly reaching a position cis to the phenyl backbone (C–Ir–N 99°). The phenyl ring of 2-Phpy is held proximate to the site trans to the phenyl backbone in 5+. DFT geometry optimization studies (see ESI Section 7†) on the series of complexes shows a widening H–Ir–N angle from 90.6° for 3+ to 112.6° for 4+ and 175.9° for 5+. This perturbation from a cis orientation of pyridine and hydride donors to a trans orientation is fully consistent with the observed downfield hydride 1H NMR chemical shifts. The computed structure of 4+ is more dramatically bent than observed in the experimental XRD study, with evidence from DFT for a lutidine methyl C–H agostic interaction trans to the hydride (Ir–C 2.95 Å).
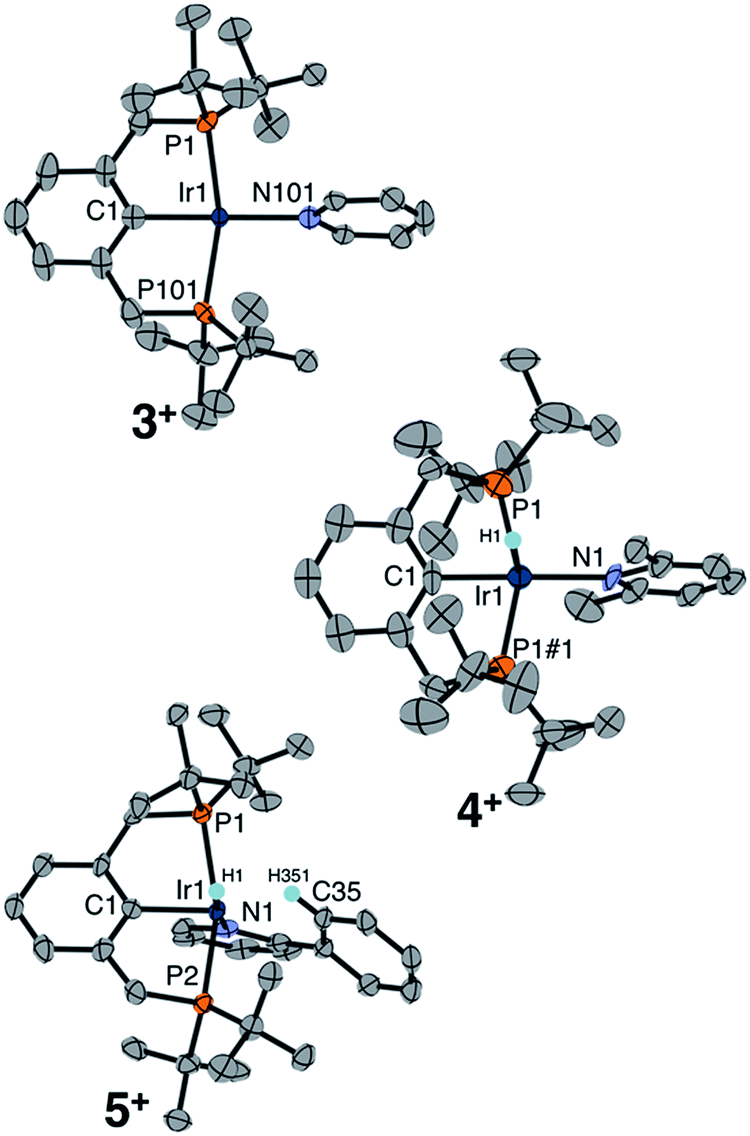 | ||
| Fig. 1 Structural representation of 3+, 4+, and 5+ from XRD with ellipsoids rendered at 50% probability. Most hydrogen atoms and all BArF4 counter ions omitted for clarity. Hydride hydrogen atom of 3+ was not located in the difference map. Selected bond distances (Å) and angles (°), 3+: Ir1–P1 2.324(2), Ir1–P101 2.329(5), Ir1–N101 2.135(7), P1–Ir1–P1#1 163.6(1), C1–Ir1–N1 179.99(4); 4+: Ir1–P1 2.345(2), Ir1–N1 2.131(9), Ir1–C1 2.008(11), P1–Ir1–P1#1 157.11(15), C1–Ir1–N1 174.5(5); 5+: Ir1–P1 2.365(2), Ir1–P2 2.345(2), Ir1–N1 2.192(5), Ir1–C1 2.038(6), P1–Ir1–P2 158.02(6), C1–Ir1–N1 98.7(2). | ||
The proximity of the 2-Phpy phenyl ring to the Ir center in 5+ (Ir–C35 2.565(6) Å) is consistent with an agostic interaction involving an aromatic proton on the phenyl substituent of 2-Phpy. While no upfield resonances of 5+ indicative of an agostic interaction were observed by 1H NMR spectroscopy at room temperature in CD2Cl2, cooling the sample to −20 °C resulted in sharper, more resolved aromatic resonances and the appearance of a new singlet at 4.01 ppm coupled to an aromatic carbon (δ13C = 97.1, 1JCH = 125 Hz). The upfield shifts in both the 1H and 13C resonances, as well as the substantially reduced 1JCH (typically 150–170 for aromatic C–H), are hallmarks of a C–H agostic interaction.22
Deprotonation of cationic iridium hydride complexes
With well-defined and electrochemically accessible cationic hydrides in hand, base-promoted C–H bond activation was targeted. Treating 2,6-lutidine complex 4+ with 1 equiv. tBuP1(pyrr) in 1,2-DFB resulted in full conversion within minutes at room temperature. Two sets of product signals with upfield hydride resonances indicative of five-coordinate Ir(III) complexes (−43.2, −46.3 ppm) are observed by 1H NMR spectroscopy. New 1H and 19F resonances confirm the presence of a fluoroaryl group bound to iridium.12 The combined spectroscopic data are consistent with clean conversion to (tBu4PCP)Ir(H)(2,3-C6F2H3) as a mixture of rotamers, 6a and 6b (Scheme 4). This structure was confirmed by addition of CO to produce the known complex (tBu4PCP)Ir(H)(CO)(2,3-C6F2H3).23 The high regioselectivity, with exclusive C–H bond activation at the 3-position, is expected based on the ortho-fluorine stabilizing effect.24–26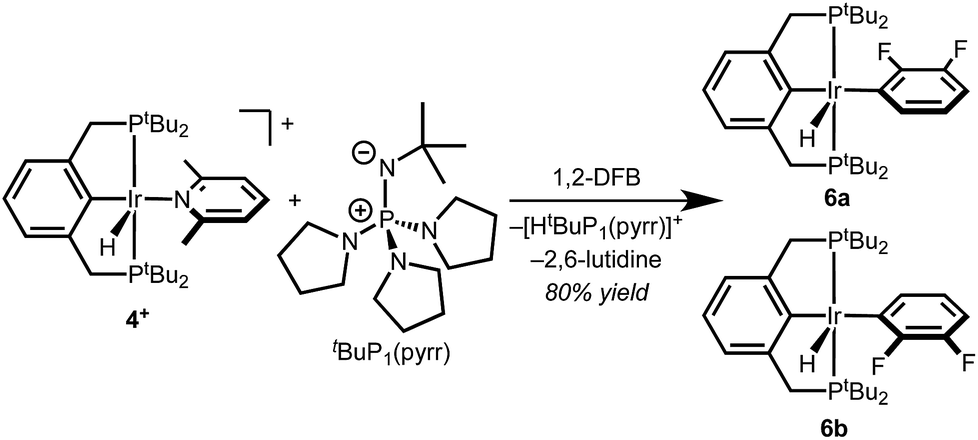 | ||
| Scheme 4 C–H activation of 1,2-DFB by deprotonation of 4+. | ||
While the agostic interaction of 5+ seemed likely to bias C–H activation reactivity towards cyclometallation, deprotonation of 5+ in 2-phenylpyridine solutions led to a mixture of several species at room temperature, only converging to the expected ortho-metallated phenylpyridine complexes upon heating (indicating that C–H addition occurs without prior pyridine coordination, as noted previously).12,27–29 Further studies focused on the well-defined reactivity of 4+ in 1,2-DFB.
Electrochemical C–H activation of 1,2-difluorobenzene
Having successfully demonstrated both key steps in the proposed C–H bond activation scheme, we next attempted a one-pot electrochemical C–H activation of 1,2-DFB starting from (tBu4PCP)IrH4, 20 equiv. lut, and 5 equiv. tBuP1(pyrr). Oxidation by CPE resulted in a color change from pale orange to dark orange-red, and passage of current amounting to 2.7e− per Ir. Analysis of the product mixture by 1H NMR spectroscopy showed that 1 indeed provided 6a and 6b, but with limited conversion (19% yield, Scheme 5). The reaction proceeds in higher yield (30%) when the 2,6-lutidine is left out of the reaction. The formation of the desired product is a promising indication of the viability of this electrochemical C–H activation strategy.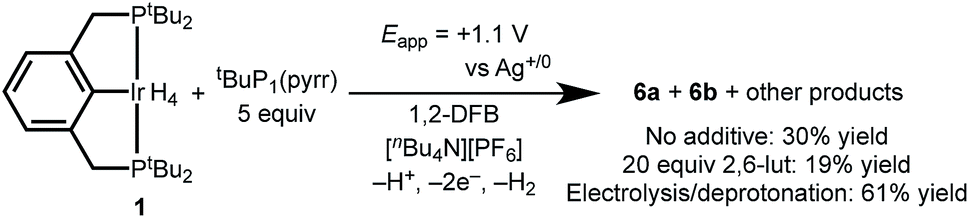 | ||
| Scheme 5 Electrochemical C–H activation of 1,2-DFB by 1. | ||
We hypothesized that incomplete conversion of 1 in the electrochemical process was due to complications arising from direct oxidation of the base, tBuP1(pyrr). Indeed, tBuP1(pyrr) is more easily oxidized than 1 (Fig. S33 in the ESI†) and we suspect that the oxidized tBuP1(pyrr) may lead to electrode fouling or other side reactions.
To circumvent direct oxidation of tBuP1(pyrr), a one-pot procedure was carried out. Electrochemical generation of 4+ was followed immediately by addition of tBuP1(pyrr), producing 6a and 6b in a significantly improved yield of 61%.
Comparison to sacrificial olefin-mediated process
To compare electrochemical and thermal C–H activation processes, the reactivity of (tBu4PCP)IrH4 in 1,2-DFB with the prototypical hydrogen acceptor tert-butylethylene (TBE) was evaluated. Allowing (tBu4PCP)IrH4 to react with 2 equiv. TBE in 1,2-DFB led to no observed reaction after 24 hours at 20 °C. Heating the mixture at 50 °C for 17 h resulted in full conversion to a mixture of products, of which 79% were C–H activation products of 1,2-DFB in a 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (6a:6b).
:1 ratio (6a:6b).
The thermal and electrochemical arene activation reactivity are notably distinct. The traditional thermal approach requires a sacrificial alkene (TBE) as a chemical hydrogen acceptor, producing an equivalent of 2,2-dimethylbutane as a byproduct. Given that both the chemical and electrochemical reactions are proposed to proceed through the same (tBu4PCP)Ir intermediate, the elevated temperatures required by the thermal C–H bond activation reaction are consistent with a rate-determining reaction with TBE, in accord with previous studies.5 The new electrochemical approach, in contrast, does not require an olefinic sacrificial hydrogen acceptor. The electrochemical method described here does require a chemical additive in the form of a phosphazene base. In an envisioned electrochemical application, the protonated base would migrate to the cathode and supply protons for H2 evolution, regenerating the free base. Furthermore, the low barrier to electrochemical activation to reach the (tBu4PCP)Ir intermediate allows the reaction to proceed at room temperature.
Conclusions
The need for alkene hydrogen acceptors has been a major limitation in (PCP)Ir-catalyzed C–H functionalization reactions. The electrochemical C–H activation conditions reported here lift the requirement for sacrificial hydrogen acceptor reagents. Furthermore, the electrochemical reaction proceeds at room temperature, whereas even the acceptor-driven reaction must be heated. Cationic iridium hydride complexes are likely intermediates in the electrochemical C–H bond activation pathway. Three such species were isolated and characterized, displaying a rich structural diversity that may be attributed to agostic interactions.This work illuminates some areas where modified catalysts or conditions could be beneficial. First, a solvent that can support electrochemical studies while withstanding reaction with the Ir complex is needed. Second, the development of metal hydride complexes that are easier to oxidize than the base present in solution would improve efficiency.30 Ongoing efforts to develop new electrochemical activation methodologies will expand future opportunities for acceptorless C–H functionalization of substrates lacking directing groups.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The synthesis and cyclic voltammetry were supported through funding from NSF Center for Enabling New Technologies through Catalysis (CENTC), CHE-1205189. Controlled potential electrolysis and thermal C–H activation studies were supported through the NSF Chemical Catalysis program under Grant No. CHE-1665135 and CHE-1665146.Notes and references
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed.
- J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed.
- T. J. Hebden, K. I. Goldberg, D. M. Heinekey, X. Zhang, T. J. Emge, A. S. Goldman and K. Krogh-Jespersen, Inorg. Chem., 2010, 49, 1733–1742 CrossRef CAS PubMed.
- K. B. Renkema, Y. V Kissin and A. S. Goldman, J. Am. Chem. Soc., 2003, 125, 7770–7771 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS.
- C. Ma, P. Fang and T. S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS.
- A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed.
- F. Novak, B. Speiser, H. A. Mohammad and H. A. Mayer, Electrochim. Acta, 2004, 49, 3841–3853 CrossRef CAS.
- D. A. Laviska, PhD thesis, Rutgers University, 2013.
- T. R. O'Toole, J. N. Younathan, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1989, 28, 3923–3926 CrossRef.
- S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC.
- V. M. Iluc, A. J. M. Miller, J. S. Anderson, M. J. Monreal, M. P. Mehn and G. L. Hillhouse, J. Am. Chem. Soc., 2011, 133, 13055–13063 CrossRef CAS PubMed.
- A. G. Walden, A. Kumar, N. Lease, A. S. Goldman and A. J. M. Miller, Dalton Trans., 2016, 45, 9766–9769 RSC.
- O. B. Ryan, M. Tilset and V. D. Parker, J. Am. Chem. Soc., 1990, 112, 2618–2626 CrossRef CAS.
- J. D. Hackenberg, S. Kundu, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2014, 136, 8891–8894 CrossRef CAS PubMed.
- G. P. Connor, N. Lease, A. Casuras, A. S. Goldman, P. L. Holland and J. M. Mayer, Dalton Trans., 2017, 46, 14325–14330 RSC.
- B. Olgemoeller and W. Beck, Inorg. Chem., 1983, 22, 997–998 CrossRef CAS.
- A. M. Camp, M. R. Kita, J. Grajeda, P. S. White, D. A. Dickie and A. J. M. Miller, Inorg. Chem., 2017, 56, 11141–11150 CrossRef CAS PubMed.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- S. A. Hauser, J. Emerson-King, S. Habershon and A. B. Chaplin, Chem. Commun., 2017, 53, 3634–3636 RSC.
- E. Clot, M. Besora, F. Maseras, C. Mégret, O. Eisenstein, B. Oelckers and R. N. Perutz, Chem. Commun., 2003, 98, 490–491 RSC.
- E. Clot, C. Mégret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2009, 131, 7817–7827 CrossRef CAS PubMed.
- M. E. Evans, C. L. Burke, S. Yaibuathes, E. Clot, O. Eisenstein and W. D. Jones, J. Am. Chem. Soc., 2009, 131, 13464–13473 CrossRef CAS PubMed.
- X. Zhang, M. Kanzelberger, T. J. Emge and A. S. Goldman, J. Am. Chem. Soc., 2004, 126, 13192–13193 CrossRef CAS PubMed.
- A. Casuras, PhD thesis, Rutgers University, 2019.
- D. A. Ahlstrand, A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Chem.–Eur. J., 2017, 23, 1748–1751 CrossRef CAS PubMed.
- C. R. Waidmann, A. J. M. Miller, C.-W. A. Ng, M. L. Scheuermann, T. R. Porter, T. A. Tronic and J. M. Mayer, Energy Environ. Sci., 2012, 5, 7771–7780 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1935784, 1935785 and 1935786. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03076j |
| ‡ These co-authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |