
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific C-terminal dinitrophenylation to reconstitute the antibody Fc functions for nanobodies†
Haofei
Hong
a,
Zhifang
Zhou
 a,
Kun
Zhou
a,
Shaozhong
Liu
a,
Zhongwu
Guo
*b and
Zhimeng
Wu
*a
a,
Kun
Zhou
a,
Shaozhong
Liu
a,
Zhongwu
Guo
*b and
Zhimeng
Wu
*a
aKey Laboratory of Carbohydrate Chemistry & Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi, 214122, China. E-mail: zwu@jiangnan.edu.cn
bDepartment of Chemistry, University of Florida, 214 Leigh Hall, Gainesville, Florida 32611, USA. E-mail: zguo@chem.ufl.edu
First published on 28th August 2019
Abstract
Nanobodies are a class of camelid-derived single-domain antibodies that have many potential advantages over conventional antibodies and have been utilized to develop new therapeutic strategies for cancer and other diseases. However, nanobodies lack the Fc region of a conventional antibody, which possesses many functions, e.g., eliciting antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), essential for effective immunotherapy. The small molecular size of nanobodies also leads to poor pharmacokinetics, such as short in vivo half-life. To address these deficiencies, an endogenous antibody-based strategy to reconstitute the Fc functions for nanobodies was developed. As a proof-of-principle, an anti-human EGFR nanobody, 7D12, was selected to conduct C-terminal modification with the dinitrophenyl (DNP) hapten through Sortase A-mediated site-specific ligation. It was expected that the DNP motif would recruit endogenous human anti-DNP antibodies to indirectly reinstate the Fc functions. The resultant nanobody-DNP conjugates were shown to exhibit specific and high affinity binding to human EGFR expressed on target cancer cells. It was further proved that in the presence of anti-DNP antibody, these conjugates could mediate potent ADCC and CDC in vitro and exhibit significantly elongated half-life in vivo. Ultimately, it was proven in severe combined immunodeficiency (SCID) mice that treatment with the nanobody 7D12-DNP conjugate and anti-DNP mouse serum could inhibit xenograft tumor growth efficiently. In view of the abundance of anti-DNP and other endogenous antibodies in the human blood system, this could be a generally applicable approach employed to reconstitute the Fc functions for nanobodies and develop nanobody-based cancer immunotherapy and other therapies.
Introduction
Monoclonal antibodies (mAbs) are widely used for cancer immunotherapy.1 The anticancer effects of mAbs involve a variety of mechanisms.2 Among them, the antibody Fc region-invoked innate immunity plays a crucial role in killing cancer cells. The Fc region of an antibody can trigger both complement-dependent cytotoxicity (CDC) by eliciting the complement reaction cascade and antibody-dependent cellular cytotoxicity (ADCC) by activating immune cells such as natural killer cells, δγT cells and macrophages.3 For example, Rituximab, a mAb approved for the treatment of B-cell malignancies, has been demonstrated to realize its anticancer efficacy via Fc-mediated CDC and ADCC. However, mAbs are large and complex biomolecules (150 kDa), which makes their production and application challenging. Moreover, potential immune responses to mAbs are still a concern for mAb-based cancer therapy in the clinic.4Nanobodies are unique antibodies derived from camelids, which contain only the heavy chain variable domain.5 Compared to conventional antibodies, nanobodies exhibit some special and useful properties, such as much smaller size (15 kDa, 2.5 nm diameter and 4 nm height), easy production, good stability and solubility, low immunogenicity, and high specificity and affinity for their antigen.6 Consequently, nanobody-based medicines have been developed to combat infectious, autoimmune, inflammatory and neurological diseases.7–10 For cancer treatment, nanobodies and their multivalent constructs have been developed as antagonists to target extracellular proteins expressed on cancer cell surfaces for the prevention of tumor progression.11,12 In addition, many efforts are focused on conjugating nanobodies with effector molecules, such as toxins, radionuclides and photosensitizers, for cancer therapy.13–18 However, these conjugates usually display poor pharmacokinetics as they can be rapidly cleared from the circulation system via renal excretion. Recently, a nanobody-Fc fusion protein was explored to reconstitute the biological functions of Fc, and subsequent in vitro studies revealed that this fusion protein was able to kill target cancer cells via CDC.19,20 Nonetheless, Fc-fused nanobodies may have similar intrinsic problems to typical mAbs. Moreover, different allotypes of the Fc portion may have potential immunogenicity and trigger anti-Fc antibodies, resulting in an adverse acute immune response.21–23 Furthermore, complicated Fc N-glycan engineering is needed to gain enhanced binding activity with the FcγIIIa receptor on immune cells for improved ADCC and CDC.24–26 Therefore, for cancer therapy, it is crucial to reconstitute the Fc functions of nanobodies without the disadvantages of the aforementioned technologies.
Endogenous antibodies refer to naturally occurring antibodies in the human circulatory system, such as those against the dinitrophenyl (DNP) group,27 galactose-α-(1,3)-galactose (αGal)28 and rhamnose (Rha).29,30 They are abundant in human serum. For example, about 1% of circulating endogenous antibodies are against DNP. Redirecting these antibodies to target specific disease cells could lead to adaptive immunities for selective and effective therapy.31–37 For instance, a pioneering work from McNaughton's group demonstrated that DNP-modified nanobody 5F7 could recruit anti-DNP antibodies to HER2+ cancer cells and elicit a specific ADCC effect in vitro. However, no in vivo studies have been reported yet. Furthermore, the pharmacokinetic properties of nanobody-DNP conjugates, which will be key to determine the in vivo efficacy, are largely unknown.33
Inspired by the above studies, we report here the design and chemo-enzymatic synthesis of site-specifically modified nanobody-DNP conjugates through Sortase A-mediated ligation. Linkers containing various lengths of polyethylene glycol (PEG) between the nanobody and the DNP group were designed to explore the structure–activity relationship. It is expected that the conjugates would direct DNP antibodies to cancer cells to trigger CDC and ADCC via the engagement of DNP antibodies (Fig. 1). Moreover, the conjugates might also form large immune complexes with DNP antibodies in situ to avoid their rapid renal clearance and rescue them from lysosome degradation via the classical neonatal Fc receptor-mediated recycling mechanism to improve their pharmacokinetic profile. In addition, this type of conjugate may avoid the immunogenicity elicited by different allotypes of Fc in Fc-nanobody design, since anti-DNP antibodies are a natural, endogenous antibody. Here, we also disclose the in vitro antitumor activity and in vivo antitumor efficacy of an optimized nanobody-DNP conjugate in mouse xenograft models. The pharmacokinetic properties of these nanobody-DNP conjugates were evaluated in vivo as well.
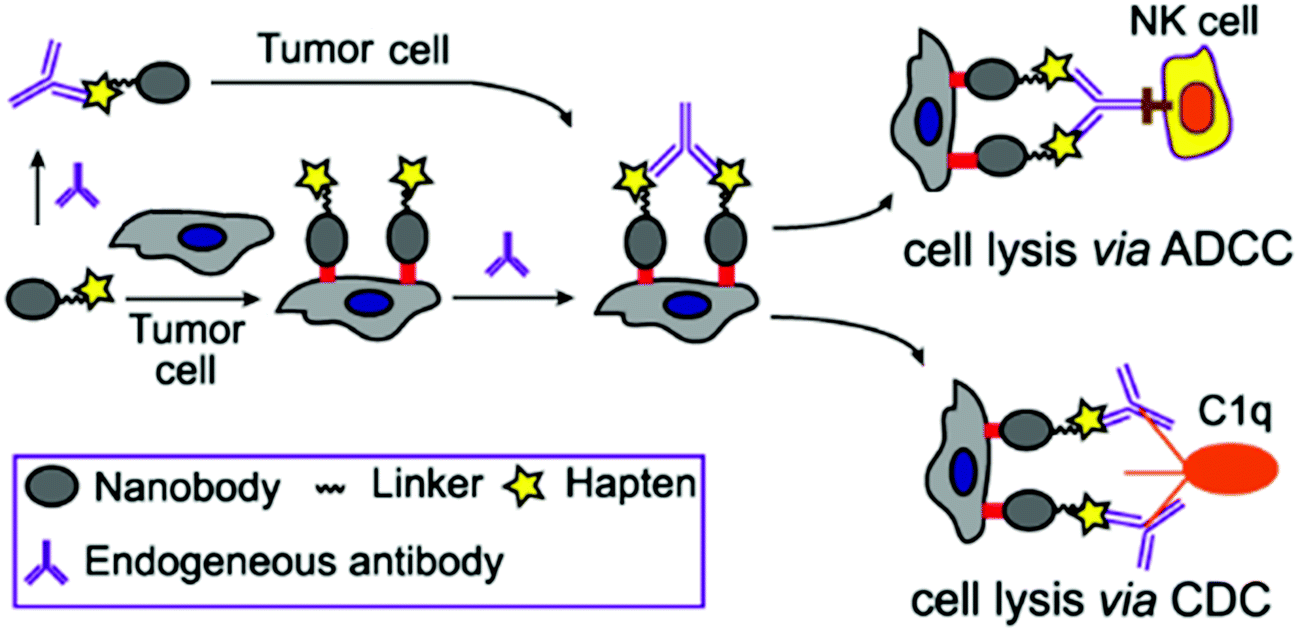 | ||
| Fig. 1 Nanobody-DNP conjugate-mediated targeting of cancer cells and recruitment of endogenous antibodies to mediate ADCC, CDC and other immune reactions against cancer. | ||
Results and discussion
Design, synthesis and characterization of nanobody 7D12-DNP conjugates
Nanobody 7D12, which recognizes human epidermal growth factor receptor (EGFR) expressed by many tumors, such as A431 cells,38 was utilized as a model to prove our concept. The well-established Sortase-A (SrtA)-mediated ligation (SML) method39–44 was used for the site-specific modification of nanobody 7D12 with DNP. SrtA is a bacterial enzyme that recognizes a specific peptide motif, LPXTG (X can be any amino acid except for cysteine), known as the sorting signal, at the protein C-terminus and cleaves the peptide bond between threonine and glycine to form a thioester intermediate, which is followed by reacting with substrates containing oligoglycine to provide the ligation product.45,46 Therefore, to be compatible with SML for site-specific conjugation and product purification, the nanobody 7D12 sequence was genetically engineered to contain at its C-terminus a cmyc tag (for immunoassay), a sorting signal (LPETGG) and a 6-His tag (Fig. 2). The nanobody 7D12 was expressed successfully in E. coli as a soluble protein with a yield of 60 mg L−1. The purified 7D12 was characterized by reduced SDS-PAGE, which displayed a single protein band with a molecular weight of approximately 16 kDa (Fig. S1a†). It was further characterized by mass spectrometry (MS) (Fig. S1b†). | ||
| Fig. 2 The structure of nanobody 7D12 containing cmyc, sorting signal, and His tags at the C-terminus. | ||
We intended to couple DNP to nanobody 7D12 via a hydrophilic and biocompatible PEG linker commonly used in biotherapeutics. To probe the impact of the linker on the therapeutic efficacies of the nanobody-DNP conjugates, four conjugates (1, 2, 3 and 4) which contained different lengths of PEG were designed. Their synthesis is outlined in Scheme 1. As SrtA accepts oligoglycine as its substrate, DNP-triglycine derivatives (13 to 16) are required. Accordingly, t-butoxycarbonyl (Boc)-protected amino derivatives of DNP (5 to 8) were prepared via a series of established transformations, respectively (the detailed synthetic procedure can be found in the ESI†). They were subjected to trifluoroacetic acid (TFA) treatment in dichloromethane (DCM, 50%) to give free amino derivatives, followed by coupling with Boc protected triglycine using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDCI) as the condensation reagent in the presence of N,N-diisopropylethylamine (DIPEA) to provide 9 to 12, which were deprotected subsequently with 50% TFA in DCM to afford NH2-GGG-(PEG)n-DNP conjugates 13 to 16 (n = 1, 3, 6 and 12, respectively). These compounds, as well as all synthetic intermediates involved, were fully characterized with 1H- and 13C-NMR and HR MS (refer to the ESI†).
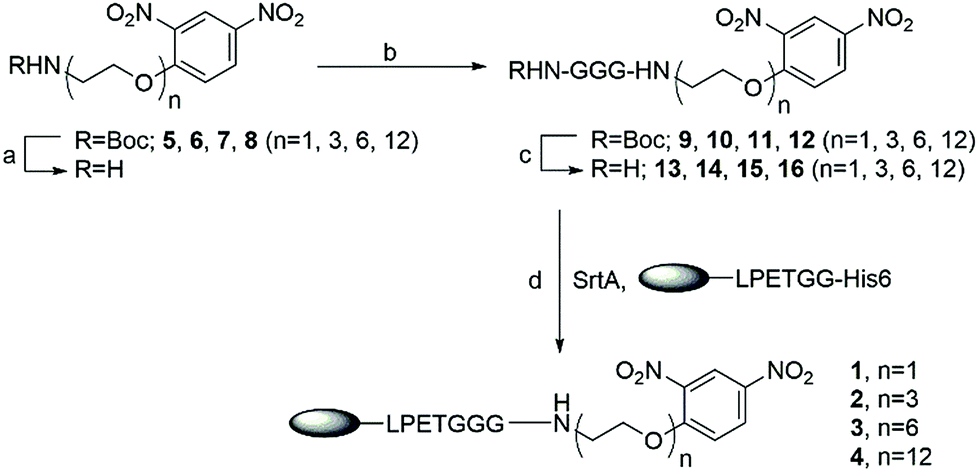 | ||
Scheme 1 Chemoenzymatic synthesis of nanobody 7D12-DNP conjugates 1, 2, 3 and 4. Reagents and conditions: (a) 50% TFA in DCM, rt, 2 h; (b) EDCI, DIPEA, DCM/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), rt, 62% for 9; 60% for 10, 50% for 11 and 16% for 12; (c) 50% TFA in DCM, rt, 2 h, 95% for 13; 95% for 14, 95% for 15, and 95% for 16; (d) SrtA, Tris buffer (pH = 7.4), 16 °C, 2–4 h, 85% for 1; 85% for 2, 75% for 3 and 75% for 4. :2), rt, 62% for 9; 60% for 10, 50% for 11 and 16% for 12; (c) 50% TFA in DCM, rt, 2 h, 95% for 13; 95% for 14, 95% for 15, and 95% for 16; (d) SrtA, Tris buffer (pH = 7.4), 16 °C, 2–4 h, 85% for 1; 85% for 2, 75% for 3 and 75% for 4. | ||
SrtA-mediated site-specific enzymatic conjugation of nanobody 7D12 with DNP derivatives 13, 14, 15 or 16 was accomplished in Tris buffer (pH = 7.4) following a previously reported protocol.47 After the reaction was completed, the enzyme and any unreacted nanobody potentially remaining, both of which contained the His6 tag, were removed with magnetic nickel beads. Both the eluent and captured proteins on the beads were analyzed with SDS-PAGE. The results (Fig. 3a) showed that nearly no engineered 7D12 remained in the reaction mixture (lane 2, 4, 6 and 8), indicating its complete conversion into the conjugation products 1, 2, 3 and 4. The resulting conjugation products were finally purified with a G25 column and lyophilized to produce nanobody 7D12-DNP conjugates (1 to 4) as white powders in a yield ranging from 75% to 85%. The purified conjugates were characterized by both SDS-PAGE and western blot using a commercial anti-DNP antibody (Fig. 3b). Clearly, the western blot results verified that conjugates 1 to 4 contained the DNP hapten, whereas the nanobody 7D12 did not. The conjugates were further confirmed by MS, the results of which were consistent with the calculated molecular weight (Fig. S8†).
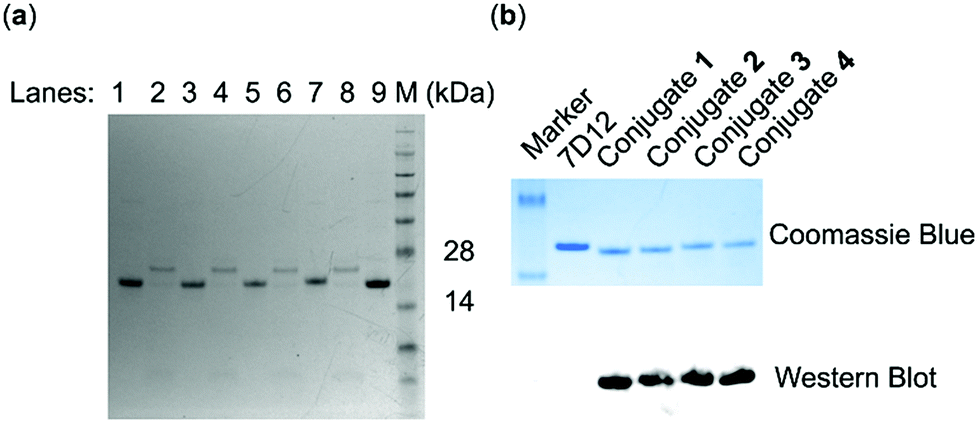 | ||
| Fig. 3 Characterization of nanobody 7D12-DNP conjugates 1, 2, 3 and 4 by (a) SDS-PAGE analysis (lanes 1, 3, 5 and 7: crude conjugation products 1, 2, 3 and 4, respectively; lanes 2, 4, 6 and 8: proteins captured by magnetic nickel beads from the four SML reactions; lane 9: nanobody 7D12; lane M: molecular markers) and (b) SDS-PAGE and western blot (nanobody 7D12, purified conjugates 1, 2, 3 and 4). | ||
Evaluation of the binding affinity and specificity of nanobody 7D12-DNP conjugates with cancer cells
The A431 cancer cell line with a high expression level of human EGFR, the MCF-7 cancer cell line with a low expression level of human EGFR, and the 4T1 cell line with a high expression level of murine EGFR were used to assess the binding affinity and specificity of the nanobody 7D12-DNP conjugates (1, 2, 3 and 4) with target cancer cells. After these cells were incubated with 100 nM of 7D12-DNP conjugates and Alexa Fluor 488-tagged anti-DNP IgG antibody, they were then imaged using a fluorescence microscope. As shown in Fig. 4a, A431 cells showed very strong fluorescence signals, whilst for the other two cell lines fluorescence signals were hardly observable under the same conditions (Fig. S2 and 3†). These results suggested that all of the nanobody 7D12-DNP conjugates (1 to 4) could specifically recognize and bind to the human EGFR expressed on A431 cells.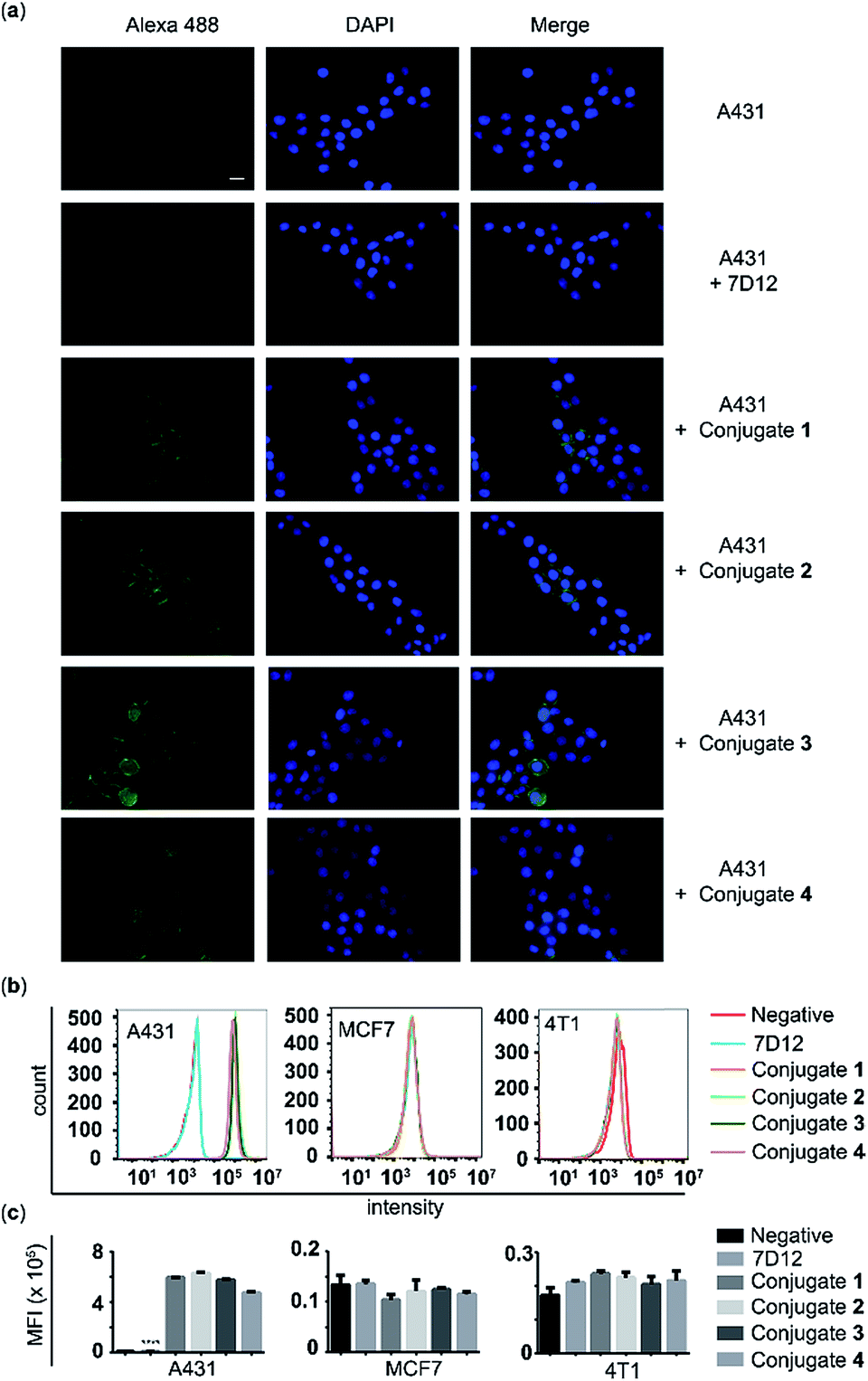 | ||
| Fig. 4 Cell-binding assay results. (a) Immunofluorescence images of A431 cells treated with nanobody 7D12, or conjugate 1, 2, 3 or 4 (scale bar = 20 μm). (b) Flow cytometry images and (c) MFI of various cell lines treated with nanobody 7D12, or conjugate 1, 2, 3 or 4. Error bars show the SD of three parallel experiments. ****: p < 0.0001. | ||
The above conclusion was further verified by the results of flow cytometry analysis. The histograms (Fig. 4b) and the corresponding mean fluorescence intensities (MFIs) (Fig. 4c) of these cells after treatment with conjugate 1, 2, 3 or 4 in the presence of Alexa 488-tagged anti-DNP IgG antibody clearly indicated that only A431 cells incubated with conjugates had significantly (P < 0.0001) increased MFI (5.94, 6.30, 5.75 and 4.73 × 105), by as high as 76.7, 82.4, 74.2 and 60.9 fold, respectively, as compared to A431 cells treated with nanobody 7D12. On the other hand, no obvious difference in fluorescence intensity was detected with MCF-7 and 4T1 cells as compared to the control groups. To further reveal the binding effect of the linker length in the conjugates to target cells, A431 cells treated with different concentrations of conjugates (1, 5, 20 and 50 nM) were analyzed using flow cytometry. It is observed that conjugates 1, 2 or 3 displayed better binding profiles to target A431 cancer cells than conjugate 4. In addition, for conjugates 1, 2 and 3, the fluorescence intensities were comparable, indicating that a linker length from PEG-1 to PEG-6 in the nanobody-DNP conjugates is suitable to obtain a good binding ability (ESI, Fig. S4†).
In vitro evaluation of ADCC and CDC induced by nanobody 7D12-DNP conjugates
As discussed above, the Fc region of conventional antibodies plays an important role in cancer immunotherapy because it can trigger ADCC and CDC by activating immune cells and eliciting a complement reaction cascade.2 Accordingly, we expected that the DNP-modified nanobody could trigger Fc-mediated ADCC and CDC via recruiting endogenous DNP antibodies to bind with the target cancer cells. To verify this concept, we evaluated nanobody 7D12-DNP conjugate-induced ADCC in vitro in the presence of anti-DNP antibodies. In these experiments, A431 cells were incubated first with conjugate 1, 2, 3 or 4 and anti-DNP IgG antibody and then co-cultured with freshly isolated healthy human peripheral blood mononuclear cells (PBMC), which served as the source of immune cells, such as natural killer cells, δγT cells and macrophages. Finally, cancer cell death was determined by the lactate dehydrogenase (LDH) test using a commercial kit. As shown in Fig. 5a, significant cancer cell lysis was observed in the groups treated with nanobody 7D12-DNP conjugates (1 to 4) and it was concentration-dependent, whereas under the same conditions there was no obvious cell lysis in the group treated with nanobody 7D12 at a concentration up to 100 nM. These results indicated clearly that nanobody 7D12-DNP conjugates 1 to 4 could successfully bind with target cancer cells and recruit anti-DNP IgG antibodies to trigger ADCC. Among them, 7D12-DNP conjugate 4 exhibited an inferior ADCC activity, proving again that a longer linker (PEG-12) could decrease the cell binding ability and result in less potent ADCC activity.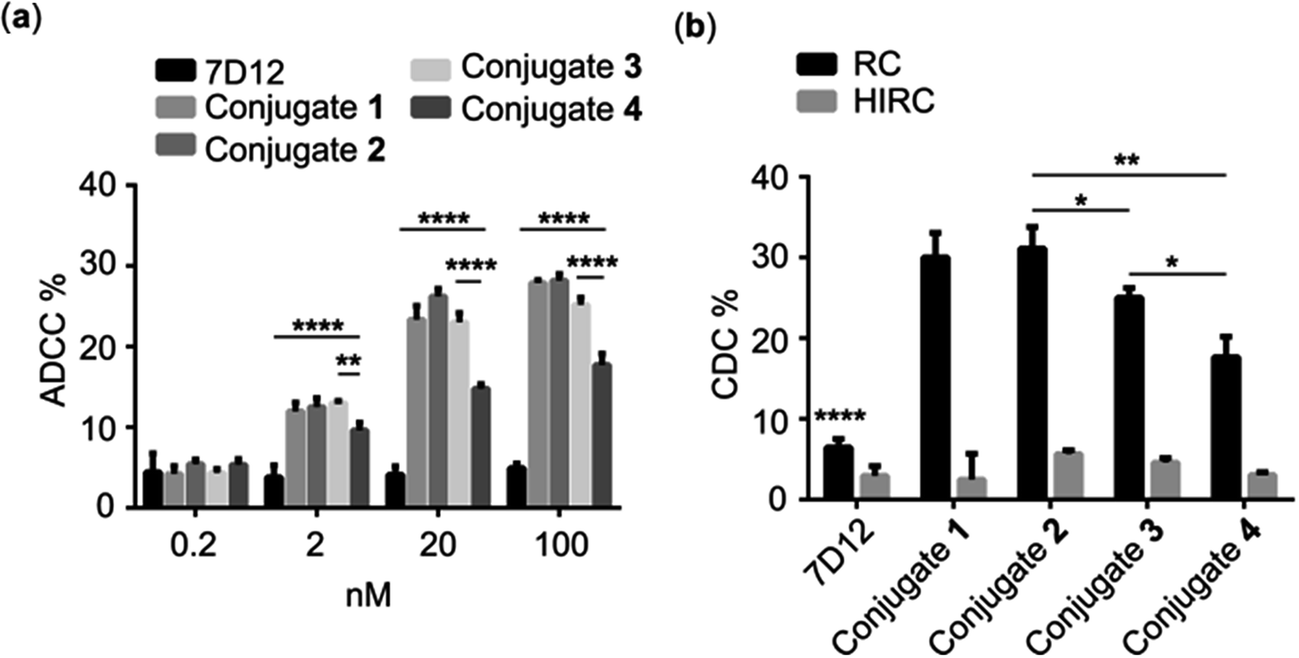 | ||
| Fig. 5 In vitro assays of (a) ADCC mediated by different concentrations of nanobody 7D12, and conjugates 1, 2, 3 and 4, and (b) CDC mediated by nanobody 7D12, and conjugates 1, 2, 3 and 4 in the presence of rabbit complement (RC) or heat inactive rabbit complement (HIRC). Error bars represent the SD of six parallel experiments. *: p < 0.05; **: p < 0.01; ****: p < 0.0001. | ||
We also examined the activities of DNP-modified nanobodies to mediate CDC. For this purpose, A431 cells were first treated with conjugate 1, 2, 3 or 4 and anti-DNP IgG antibody and then incubated with 1:100 diluted rabbit complement at 37 °C for 4 h. Thereafter, cell lysis was measured with a commercial CCK8 kit. As presented in Fig. 5b, the cell lysis percentages in the groups treated with 100 nM of conjugates 1 to 4 were 30.0, 31.7 25.0 and 17.7%, respectively, and only low (<8%) cell lysis percentages were observed in the control groups treated with nanobody 7D12 or heat inactive rabbit complement. Furthermore, conjugates 1 and 2 displayed better CDC efficacy than conjugates 3 and 4. As for conjugates 1 and 2, they exhibited a very similar ability to recruit anti-DNP antibodies to mediate effective CDC against the target cancer cells.
CDC is one of the most powerful complement-dependent mechanisms to kill cancer cells, and the formation of an antibody-complement component 1q (C1q) complex is required for activating the classic complement pathway. It seems that endogenous anti-DNP antibodies may be able to serve this purpose. However, conventional anti-EGFR antibodies, such as Cetuximab, Matuzumab and Panitumumab, failed to induce CDC because of their low affinity to C1q.48 Reconstitution of these mAbs to improve their affinity to C1q and their CDC activity required complicated antibody engineering. Thus, our new strategy would provide a very simple and effective approach to solve this problem via modification of nanobodies with DNP.
Evaluation of the in vivo pharmacokinetic properties of nanobody 7D12-DNP conjugates
The half-lives of nanobody 7D12-DNP conjugates 1 to 4 were evaluated in mice and compared to that of nanobody 7D12. In this regard, nanobody 7D12 and the 7D12-DNP conjugates were pre-mixed with naive mouse serum or anti-DNP mouse serum (obtained by immunization of mice with DNP-OVA conjugate) and the mixtures were i.v. injected into Balb/c mice. Subsequently, mouse blood samples were collected at different time points, and nanobody concentrations in the blood samples were determined by enzyme-linked immunosorbent assay (ELISA) according to the standard curves (Fig. S5†). As shown in Fig. 6, nanobody 7D12 and its DNP conjugates (1–4) with naive mouse serum treatment exhibited similar and very short half-lives (t1/2 = 9.4, 10.5, 12.8, 12.5 and 12.5 min, respectively). As expected, anti-DNP mouse serum-treated nanobody 7D12 also had a very short half-life (t1/2 = 9.5 min). In contrast, significantly improved half-lives were observed for nanobody-DNP conjugates 1–4 treated with anti-DNP mouse serum. The observed half-life was 285.1, 298.6, 273.3 and 270.3 min, respectively, which was a 26.1, 23.2, 20.8 and 20.6 fold increase. Clearly, the interaction of the DNP hapten in conjugates 1–4 with the endogenous anti-DNP antibodies could significantly improve the pharmacokinetics profiles of the conjugates and extend their circulation time in mice. However, the half-lives of the anti-DNP serum-treated nanobody-DNP conjugates were still shorter than that of IgG antibody, and the reason for this may be the same as for therapeutic mAbs and needs further exploration in the future.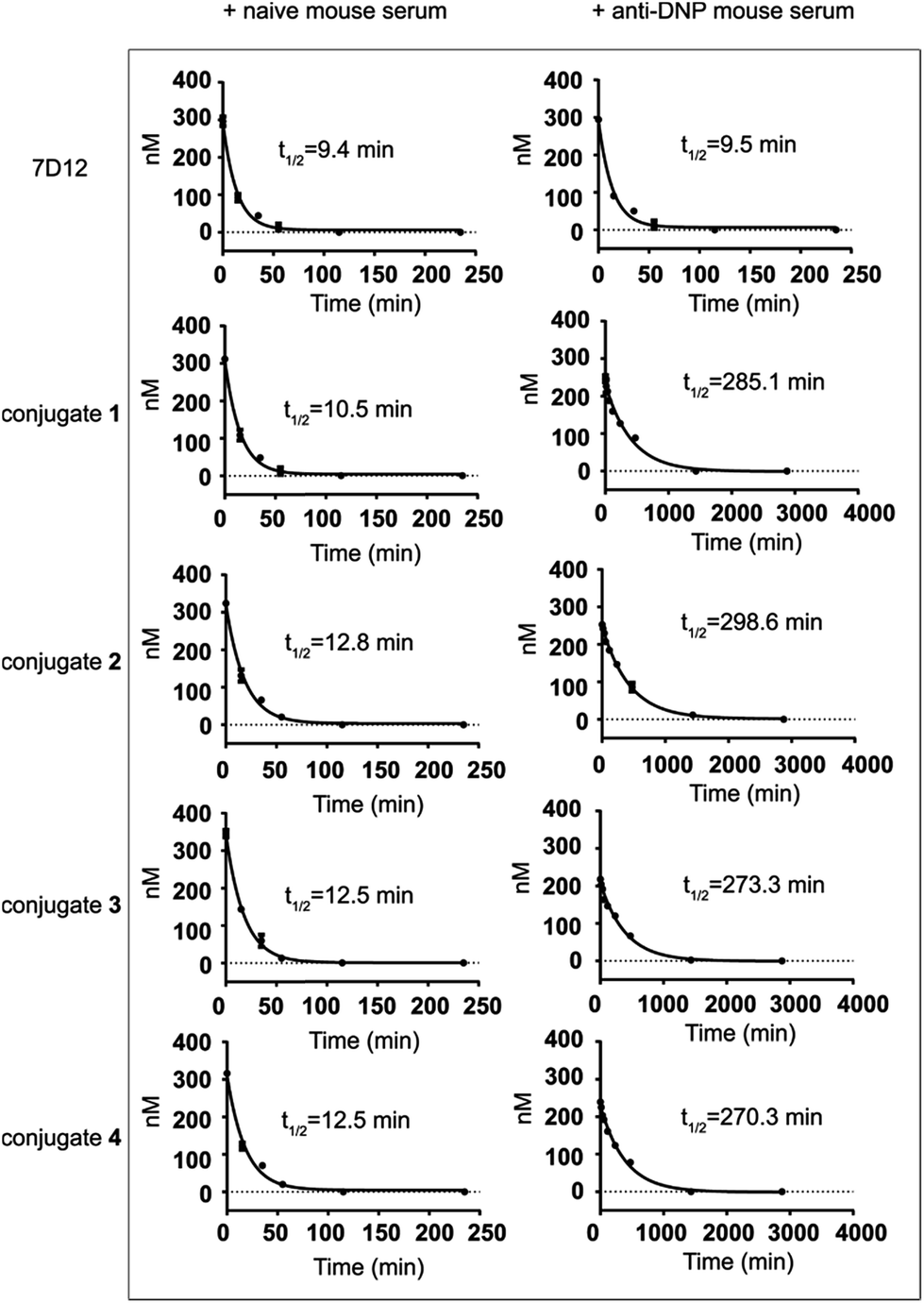 | ||
| Fig. 6 Serum concentrations and half-lives of nanobody 7D12, and 7D12-DNP conjugates 1, 2, 3 and 4 after their i.v. injection into Balb/c mice. Nanobodies were pre-treated with naive mouse serum or anti-DNP mouse serum. Error bars represent the SD of five parallel experiments in each group. | ||
One of the major issues of nanobody-based therapeutics is their relatively short half-life in vivo, which can be attributed to their low molecular weight and the lack of Fc region of the nanobody.49 This has limited the practical applications of nanobodies in cancer treatment. Traditional technologies used to improve the pharmacokinetics profiles of nanobodies include PEGylation,50 multivalency38 and fusion of nanobodies with other proteins.51–53 DNP modification of the nanobody as an alternative and convenient approach to address the clearance problem may improve the half-life of the nanobody in two ways. Firstly, DNP-modified nanobodies can form in situ immunocomplexes with endogenous anti-DNP antibodies to increase the molecular size to a range above the renal clearance threshold so as to prevent rapid renal clearance.54 Secondly, the Fc fragment of the anti-DNP antibodies in the immunocomplexes may help rescue the nanobodies from intracellular degradation and recycle them back into the circulation system through the classical neonatal Fc-receptor (FcRn)-mediated recycling mechanism,55 thereby resulting in elongated half-life.
Evaluation of the in vivo antitumor efficacy of nanobody 7D12-DNP conjugate 2
Because conjugate 2 displayed better ADCC and CDC in vitro and pharmacokinetics profiles in vivo than the other conjugates, we chose it to perform further in vivo evaluations of its antitumor efficacy employing a tumor model established in severe combined immunodeficiency (SCID) mice. As depicted in Fig. 7a, first, a group of five normal Balb/c mice were immunized four times with the DNP-ovalbumin (DNP-OVA) conjugate to generate anti-DNP antisera, which were proved to contain high titers of anti-DNP antibodies by ELISA (Fig. S6†). The mouse sera were pooled and used to provide “endogenous” anti-DNP antibodies for cancer immunotherapy. In the other three groups, SCID mice (n = 5) were challenged with A431 cells to create the cancer xenograft mouse model. Then, the xenograft mice received treatment with i.p. injection of PBS + pooled anti-DNP serum, nanobody 7D12 + pooled anti-DNP serum or conjugate 2 + the pooled anti-DNP serum obtained above, respectively, three times per week for 2 weeks. The dosages were 40 μg of nanobody 7D12 or conjugate 2/mouse/injection plus 50 μL of anti-DNP serum/mouse/injection, respectively. The size of the tumor in each mouse was individually measured three times a week using a caliper to evaluate the relative tumor volume (RTV), and the weight of the tumor was evaluated at the experimental endpoint.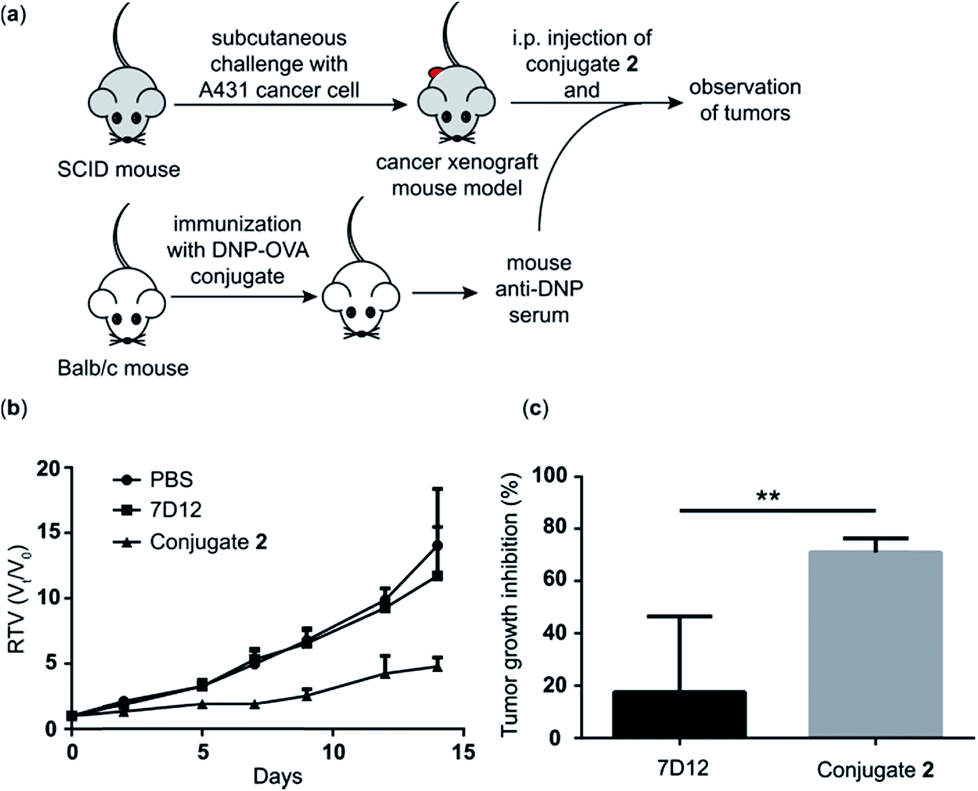 | ||
| Fig. 7 Evaluation of in vivo antitumor activities. (a) Experimental design for immunotherapy of xenograft A431 tumors in SCID mice using conjugate 2. (b) RTVs of mice treated with PBS + anti-DNP mouse serum, nanobody 7D12 + anti-DNP mouse serum or conjugate 2 + anti-DNP mouse serum. (c) Tumor growth inhibition of the treated mice at the experimental endpoint. Error bars represent the SEM (n = 5). **: p < 0.01. | ||
The RTV results shown in Fig. 7b clearly demonstrate that treatment with nanobody 7D12-DNP conjugate 2 + anti-DNP mouse serum could significantly inhibit tumor growth. The tumor growth inhibition rate of the treatment group was 71%, whereas only 17.5% inhibition was observed by using nanobody 7D12 (Fig. 7c). Nanobody 7D12 is anti-human EGFR and theoretically it can have antitumor activities through inhibition of the EGFR signaling pathway. However, treatment with nanobody 7D12 alone did not show any significant inhibition of tumor growth in mice. This was probably because of its lack of Fc region to mediate effective ADCC and CDC and its poor pharmacokinetics properties. On the other hand, 7D12-DNP conjugate 2 could reconstitute the Fc functions and had prolonged half-life in the blood system, which in combination may be responsible for its efficacious anti-tumor activities. It is also worth noting that no significant weight loss was observed in the mice treated with either nanobody 7D12 or conjugate 2 (Fig. S7†), indicating that both were essentially nontoxic.
Conclusions
A SrtA-based site-specific ligation approach was developed for the synthesis of DNP-nanobody conjugates. It was demonstrated that attaching DNP to the C-terminus of nanobody 7D12 did not adversely affect its specific and high-affinity binding to the human EGFR expressed on tumor cells. In vitro studies of these conjugates further revealed that the missing Fc biological functions, such as mediation of ADCC and CDC, in the nanobody were reconstituted with DNP-nanobody conjugates through formation of immunocomplexes with anti-DNP antibodies. In the meantime, it was shown that in the presence of anti-DNP antibodies, DNP-modified nanobody 7D12 exhibited significantly extended in vivo half-life presumably also through formation of immunocomplexes with anti-DNP antibodies in situ. Ultimately, the anticancer immunotherapeutic efficacy of nanobody 7D12-DNP conjugate 2 was confirmed in mice using a xenograft tumor model. These results have proved not only the feasibility of the new strategy to reconstitute the Fc functions for nanobodies through the incorporation of a DNP motif, but also the promise of conjugate 2 for cancer immunotherapy.Considering the abundance of endogenous anti-DNP antibodies as well as many other antibodies in the human circulatory system, we anticipated that this strategy to reconstitute nanobody Fc functions could be generally applicable to novel cancer immunotherapy development. Compared to existing technologies, such as complicated antibody engineering and nanobody-Fc fusion proteins, the current strategy to conjugate DNP with nanobodies via SrtA-based site-specific ligation should be more convenient and efficient to implement and the resultant conjugates exhibited desirable in vitro and in vivo activity and pharmacokinetics properties. In addition, DNP-nanobody conjugates may also help avoid the adverse immunogenicity and other problems associated with conventional antibodies and nanobody-Fc fusion proteins, since nanobodies have low immunogenicity and anti-DNP antibodies are natural and endogenous in the human system.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21472070 and No. 21602084), and the project was partly funded by the 111 Project (No. 111-2-06), the China Postdoctoral Science Foundation Grant 2018M632227, and the fund from KLCCB (No. KLCCB-KF201804) and supported by the Collaborative Innovation Center of Jiangsu Modern Industrial Fermentation. We appreciated the help of Professor Wei Huang in Shanghai Institute of Materia Medica for the MS spectrum of the nanobody and Professor Xiaodong Gao in Jiangnan University for the flow cytometry analysis.Notes and references
- A. M. Scott, J. D. Wolchok and L. J. Old, Nat. Rev. Cancer, 2012, 12, 278–287 CrossRef CAS PubMed.
- G. J. Weiner, Immunol. Res., 2007, 39, 271–278 CrossRef CAS PubMed.
- M. D. Pescovitz, Am. J. Transplant., 2006, 6, 859–866 CrossRef CAS PubMed.
- A. Beck, T. Wurch, C. Bailly and N. Corvaia, Nat. Rev. Immunol., 2010, 10, 345–352 CrossRef CAS PubMed.
- C. Hamerscasterman, T. Atarhouch, S. Muyldermans, G. Robinson, C. Hamers, E. B. Songa, N. Bendahman and R. Hamers, Nature, 1993, 363, 446–448 CrossRef CAS PubMed.
- S. Muyldermans, Annu. Rev. Biochem., 2013, 82, 775–797 CrossRef CAS PubMed.
- J. Wang, H. Mukhtar, L. Ma, Q. Pang and X. Wang, Sensors, 2018, 18, 485 CrossRef CAS PubMed.
- E. Beirnaert, A. Desmyter, S. Spinelli, M. Lauwereys, L. Aarden, T. Dreier, R. Loris, K. Silence, C. Pollet, C. Cambillau and H. de Haard, Front. Immunol., 2017, 8, 867 CrossRef PubMed.
- K. Manoutcharian, R. Perez-Garmendia and G. Gevorkian, Curr. Neuropharmacol., 2017, 15, 779–788 CrossRef CAS PubMed.
- J. C. Fernandes, Drug Discovery Today, 2018, 23, 1996–2002 CrossRef CAS PubMed.
- X. Peyrassol, T. Laeremans, M. Gouwy, V. Lahura, M. Debulpaep, J. Van Damme, J. Steyaert, M. Parmentier and I. Langer, J. Immunol., 2016, 196, 2893–2901 CrossRef CAS PubMed.
- F. Kazemi-Lomedasht, M. Behdani, K. P. Bagheri, M. Habibi-Anbouhi, M. Abolhassani, R. Arezumand, D. Shahbazzadeh and H. Mirzahoseini, Mol. Immunol., 2015, 65, 58–67 CrossRef CAS PubMed.
- D. Schumacher, J. Helma, A. F. L. Schneider, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed. Engl., 2018, 57, 2314–2333 CrossRef CAS PubMed.
- T. Pirzer, K. S. Becher, M. Rieker, T. Meckel, H. D. Mootz and H. Kolmar, ACS Chem. Biol., 2018, 13, 2058–2066 CrossRef CAS PubMed.
- K. T. Xenaki, S. Oliveira and P. M. P. van Bergen En Henegouwen, Front. Immunol., 2017, 8, 1287 CrossRef PubMed.
- A. Alibakhshi, F. A. Kahaki, S. Ahangarzadeh, H. Yaghoobi, F. Yarian, R. Arezumand, J. Ranjbari, A. Mokhtarzadeh and M. de la Guardia, J. Controlled Release, 2017, 268, 323–334 CrossRef CAS PubMed.
- M. Kijanka, B. Dorresteijn, S. Oliveira and P. M. van Bergen en Henegouwen, Nanomedicine, 2015, 10, 161–174 CrossRef CAS PubMed.
- W. M. Yin, X. L. Yu, X. J. Kang, Y. G. Zhao, P. F. Zhao, H. Y. Jin, X. H. Fu, Y. K. Wan, C. Y. Peng and Y. Z. Huang, Small, 2018, 14, 1802372 CrossRef PubMed.
- S. De Buck, J. Nolf, T. De Meyer, V. Virdi, K. De Wilde, E. Van Lerberge, B. Van Droogenbroeck and A. Depicker, Plant Biotechnol. J., 2013, 11, 1006–1016 CrossRef CAS PubMed.
- V. Bobkov, A. M. Zarca, A. Van Hout, M. Arimont, J. Doijen, M. Bialkowska, E. Toffoli, A. Klarenbeek, B. van der Woning, H. J. van der Vliet, T. Van Loy, H. de Haard, D. Schols, R. Heukers and M. J. Smit, Biochem. Pharmacol., 2018, 158, 413–424 CrossRef CAS PubMed.
- R. Jefferis and M. P. Lefranc, mAbs, 2009, 1, 332–338 CrossRef PubMed.
- K. A. van Schie, G. J. Wolbink and T. Rispens, mAbs, 2015, 7, 662–671 CrossRef CAS PubMed.
- D. Levin, B. Golding, S. E. Strome and Z. E. Sauna, Trends Biotechnol., 2015, 33, 27–34 CrossRef CAS PubMed.
- D. J. Falconer, G. P. Subedi, A. M. Marcella and A. W. Barb, ACS Chem. Biol., 2018, 13, 2179–2189 CrossRef CAS PubMed.
- F. Tang, L. X. Wang and W. Huang, Nat. Protoc., 2017, 12, 1702–1721 CrossRef CAS PubMed.
- A. Loos and H. Steinkellner, Arch. Biochem. Biophys., 2012, 526, 167–173 CrossRef CAS PubMed.
- K. M. Shokat and P. G. Schultz, J. Am. Chem. Soc., 1991, 113, 1861–1862 CrossRef CAS.
- O. Oyelaran, L. M. McShane, L. Dodd and J. C. Gildersleeve, J. Proteome Res., 2009, 8, 4301–4310 CrossRef CAS PubMed.
- C. E. Jakobsche, P. J. McEnaney, A. X. Zhang and D. A. Spiegel, ACS Chem. Biol., 2012, 7, 315–320 CrossRef PubMed.
- R. T. C. Sheridan, J. Hudon, J. A. Hank, P. M. Sondel and L. L. Kiessling, ChemBioChem, 2014, 15, 1393–1398 CrossRef CAS PubMed.
- X. X. Li, X. J. Rao, L. Cai, X. L. Liu, H. X. Wang, W. N. Wu, C. G. Zhu, M. Chen, P. G. Wang and W. Yi, ACS Chem. Biol., 2016, 11, 1205–1209 CrossRef CAS PubMed.
- S. Li, B. Yu, J. Wang, Y. Zheng, H. Zhang, M. J. Walker, Z. Yuan, H. Zhu, J. Zhang, P. G. Wang and B. Wang, ACS Chem. Biol., 2018, 13, 1686–1694 CrossRef CAS PubMed.
- M. A. Gray, R. N. Tao, S. M. DePorter, D. A. Spiegel and B. R. McNaughton, ChemBioChem, 2016, 17, 155–158 CrossRef CAS PubMed.
- P. J. McEnaney, C. G. Parker, A. X. Zhang and D. A. Spiegel, ACS Chem. Biol., 2012, 7, 1139–1151 CrossRef CAS PubMed.
- R. J. Amato, A. Shetty, Y. Lu, R. Ellis and P. S. Low, J. Immunother., 2013, 36, 268–275 CrossRef CAS PubMed.
- T. M. Doran, M. Sarkar and T. Kodadek, J. Am. Chem. Soc., 2016, 138, 6076–6094 CrossRef CAS PubMed.
- M. J. S. Feigman and M. M. Pires, ACS Infect. Dis., 2018, 4, 664–672 CrossRef CAS PubMed.
- R. C. Roovers, T. Laeremans, L. Huang, S. De Taeye, A. J. Verkleij, H. Revets, H. J. de Haard and P. M. P. V. Henegouwen, Cancer Immunol. Immunother., 2007, 56, 303–317 CrossRef CAS PubMed.
- Z. M. Wu and Z. W. Guo, J. Carbohydr. Chem., 2012, 31, 48–66 CrossRef CAS PubMed.
- Y. Zhang, K. Y. Park, K. F. Suazo and M. D. Distefano, Chem. Soc. Rev., 2018, 47, 9106–9136 RSC.
- J. M. Antos, M. C. Truttmann and H. L. Ploegh, Curr. Opin. Struct. Biol., 2016, 38, 111–118 CrossRef CAS PubMed.
- S. Tsukiji and T. Nagamune, ChemBioChem, 2009, 10, 787–798 CrossRef CAS PubMed.
- L. Q. Pan, W. B. Zhao, J. Lai, D. Ding, Q. Zhang, X. Y. Yang, M. M. Huang, S. J. Jin, Y. C. Xu, S. Zeng, J. J. Chou and S. Q. Chen, Small, 2017, 13, 1602267 CrossRef PubMed.
- P. Yousefpour, L. Ahn, J. Tewksbury, S. Saha, S. A. Costa, J. J. Bellucci, X. Li and A. Chilkoti, Small, 2019, e1804452, DOI:10.1002/smll.201804452.
- S. K. Mazmanian, G. Liu, T. T. Hung and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS PubMed.
- H. Ton-That, G. Liu, S. K. Mazmanian, K. F. Faull and O. Schneewind, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12424–12429 CrossRef CAS PubMed.
- Z. M. Wu, X. Q. Guo and Z. W. Guo, Chem. Commun., 2011, 47, 9218–9220 RSC.
- M. Dechant, W. Weisner, S. Berger, M. Peipp, T. Beyer, T. Schneider-Merck, J. J. L. van Bueren, W. K. Bleeker, P. W. H. I. Parren, J. G. J. van de Winkel and T. Valerius, Cancer Res., 2008, 68, 4998–5003 CrossRef CAS PubMed.
- G. Hassanzadeh-Ghassabeh, N. Devoogdt, P. De Pauw, C. Vincke and S. Muyldermans, Nanomedicine, 2013, 8, 1013–1026 CrossRef CAS PubMed.
- P. Bailon and C. Y. Won, Expert Opin. Drug Delivery, 2009, 6, 1–16 CrossRef CAS PubMed.
- J. T. Andersen, R. Pehrson, V. Tolmachev, M. B. Daba, L. Abrahmsen and C. Ekblad, J. Biol. Chem., 2011, 286, 5234–5241 CrossRef CAS PubMed.
- M. R. Muller, K. Saunders, C. Grace, M. Jin, N. Piche-Nicholas, J. Steven, R. O'Dwyer, L. Wu, L. Khetemenee, Y. Vugmeyster, T. P. Hickling, L. Tchistiakova, S. Olland, D. Gill, A. Jensen and C. J. Barelle, mAbs, 2012, 4, 673–685 CrossRef PubMed.
- B. M. Tijink, T. Laeremans, M. Budde, M. S. V. Walsum, T. Dreier, H. J. de Haard, C. R. Leemans and G. A. M. S. van Dongen, Mol. Cancer Ther., 2008, 7, 2288–2297 CrossRef CAS PubMed.
- J. H. Lin, Curr. Drug Metab., 2009, 10, 661–691 CrossRef CAS PubMed.
- T. T. Kuo, K. Baker, M. Yoshida, S. W. Qiao, V. G. Aveson, W. I. Lencer and R. S. Blumberg, J. Clin. Immunol., 2010, 30, 777–789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Enzyme and nanobody expression, experimental methods, supporting figures, and synthetic details. See DOI: 10.1039/c9sc03840j |
| This journal is © The Royal Society of Chemistry 2019 |