
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThymine DNA glycosylase recognizes the geometry alteration of minor grooves induced by 5-formylcytosine and 5-carboxylcytosine†
Tianran
Fu‡
ab,
Liping
Liu‡
cd,
Qing-Lin
Yang‡
e,
Yuxin
Wang
 ab,
Pan
Xu
cd,
Lin
Zhang
ab,
Shien
Liu
cd,
Qing
Dai
f,
Quanjiang
Ji
g,
Guo-Liang
Xu
e,
Chuan
He
f,
Cheng
Luo
*cd and
Liang
Zhang
*ab
ab,
Pan
Xu
cd,
Lin
Zhang
ab,
Shien
Liu
cd,
Qing
Dai
f,
Quanjiang
Ji
g,
Guo-Liang
Xu
e,
Chuan
He
f,
Cheng
Luo
*cd and
Liang
Zhang
*ab
aDepartment of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, P. R. China. E-mail: liangzhang2014@sjtu.edu.cn; cluo@simm.ac.cn
bShanghai Universities Collaborative Innovation Center for Translational Medicine, Shanghai, P. R. China
cCAS Key Laboratory of Receptor Research, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
eState Key Laboratory of Molecular Biology, Chinese Academy of Sciences Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, Shanghai, China
fDepartment of Chemistry, Department of Biochemistry and Molecular Biology, Institute for Biophysical Dynamics, The University of Chicago, Chicago, Illinois, USA
gSchool of Physical Science and Technology, ShanghaiTech University, Shanghai, China
First published on 27th June 2019
Abstract
The dynamic DNA methylation–demethylation process plays critical roles in gene expression control and cell development. The oxidation derivatives of 5-methylcytosine (5mC) generated by Tet dioxygenases in the demethylation pathway, namely 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), could impact biological functions by altering DNA properties or recognition by potential reader proteins. Hence, in addition to the fifth base 5mC, 5hmC, 5fC, and 5caC have been considered as the sixth, seventh, and eighth bases of the genome. How these modifications would alter DNA and be specifically recognized remain unclear, however. Here we report that formyl- and carboxyl-modifications on cytosine induce the geometry alteration of the DNA minor groove by solving two high-resolution structures of a dsDNA decamer containing fully symmetric 5fC and 5caC. The alterations are recognized distinctively by thymine DNA glycosylase TDG via its finger residue R275, followed by subsequent preferential base excision and DNA repair. These observations suggest a mechanism by which reader proteins distinguish highly similar cytosine modifications for potential differential demethylation in order to achieve downstream biological functions.
Introduction
In mammals DNA methylation and demethylation at the C5 position of cytosine is a dynamic process which is critical for cell fate reprogramming and development.1,2 Aberrant methylation occurring in the human genome leads to numerous diseases and cancers, such as myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).3 Thus, this dynamic process has been considered as an important factor for studies of tumorigenesis mechanisms and the discovery of therapeutic targets. In the methylation pathway, cytosine (C) is converted to 5-methylcytosine (5mC) by DNA methyltransferases (DNMTs), which interferes with the recognition of transcriptional factors and silences gene expression.4 While in the demethylation pathway, 5mC is oxidized to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) in a stepwise manner by ten-eleven-translocation proteins (Tets).3,5 Subsequently, the last two oxidization pyrimidine products 5fC and 5caC, but not 5hmC, are recognized and excised by thymine DNA glycosylase (TDG) to form an apyrimidinic site (AP), followed by base-excision repair (BER) to revert to unmodified cytosine.6 Researchers established the DNA methylation pathway several decades ago. The indispensable biological roles of 5mC in fundamental processes such as genomic imprinting, X chromosome inactivation, suppression of transposable elements, and tumorigenesis have been broadly discussed.1,4 In contrast, the biological function of the active DNA demethylation pathway has remained unclear.Among the four cytosine modifications (5mC, 5hmC, 5fC, and 5caC) involved in the demethylation pathway, the latter three were newly discovered in the human genome in 2009 (5hmC) and 2011 (5fC and 5caC).7,8 In contrast to the distinguished gene suppression effect of 5mC in the genome, 5hmC, 5fC, and 5caC appeared to be intermediate products without any clear independent biological function. Along with the rapid development of sequencing and nucleic acid modification detection technologies against cytosine modifications, these products are found to have tissue-specific distributions and may play functional roles in regulating the different stages of embryonic development or other potential biological processes.9–14 For instance, 5hmC was found to be highly enriched in the genome of the brain and central nervous system, at a level nearly equal to that of 5mC.15 5hmC recruits specific reader proteins such as MeCP2 to activate neuronal function-related gene expression by inducing the loss of H3K27me3.16–19 Compared with numerous studies on the distribution and functions of 5hmC, there are few reports on the biological function of 5fC and 5caC. Unlike the stable geometry of 5hmC in dsDNA, 5fC was reported to alter the tertiary structure of dsDNA and facilitate its transformation from B-form to Z-form, or even to F-form.18,20,21 Recent research further showed that 5fC is enriched in early embryos at approximately double the level in the parental genome after fertilization, while 5caC is increased in breast cancers and gliomas and affects the transcriptional rate and specificity of RNA polymerase II.14,22,23 These discoveries suggest additional roles of 5fC and 5caC in altering duplex DNA properties, in addition to functioning as intermediates of the oxidative demethylation pathway.
To date, thymine DNA glycosylase (TDG) has been the only functional “reader” protein to recognize 5fC and 5caC modifications and achieve downstream biological functions.6,24 TDG belongs to the uracil DNA glycosylase (UDG) superfamily, and is thought to be involved in the dsDNA repair of G·T and G·U mismatch in the mammalian genome for decades.25 When repairing, TDG uses a “finger residue” Arg275 to recognize and flip out the damaged/aberrant base into the active site from mismatch-containing dsDNA, and performs subsequent cleavage of the glycosylic bond of these base lesions.26–28 The mutagenesis of R275 (R275L) significantly reduces the glycosylase activity of TDG,29 which may be related to lung adenocarcinoma tumorigenesis (TCGA library data). In 2011, He et al. showed that TDG catalyzed the base excision of 5fC and 5caC, thus acknowledging TDG as a key factor to complete the DNA demethylation pathway.6,27 Furthermore, TDG shows distinctive substrate preference for 5fC compared with 5caC with an unknown mechanism.30,31 Here we have solved the crystal structures of a dsDNA decamer containing fully symmetric 5mC, 5hmC, 5fC or 5caC. Structural analysis suggests that 5fC and 5caC induce distinct geometry alteration of the dsDNA minor groove, which is subsequently recognized by the finger residue Arg275 of TDG. The mutagenesis of Arg275 to alanine (R275A) abolishes the excision of 5fC with a minor effect on the 5caC excision. Our findings thus reveal the mechanism of substrate preference of TDG with regard to C, 5mC, 5hmC, 5fC, and 5caC in the demethylation pathway, providing insights into the principle on how reader proteins distinguish these highly similar cytosine modifications in order to achieve downstream biological functions.
Results
Overall structures of 5mC, 5hmC, 5fC and 5caC containing duplex DNA
In order to investigate the structural characteristics of dsDNA modified by 5mC, 5hmC, 5fC and 5caC, a 10-bp self-complementary DNA decamer (5′-CCAGXGCTGG-3′, X refers to 5mC, 5hmC, 5fC or 5caC) was synthesized by using solid-phase synthesis. Since +4 to −4 base pairs aside from the central substrate modification are required for TDG recognition,30,31 the modified base was set at the central position of the decamer. The oligo was further annealed, generating a 10bp decamer dsDNA containing a fully symmetric 5mC·G, 5hmC·G, 5fC·G or 5caC·G modification at the central CpG site. The crystal screening was performed using the hanging drop method at 4 °C, and square-like crystals appeared under acidic conditions in 4 to 6 days. The 5mC-dsDNA, 5hmC-dsDNA, 5fC-dsDNA and 5caC-dsDNA structures were determined under resolution 1.40 Å, 2.85 Å, 1.56 Å and 1.06 Å with the space group C2, C2, P61 and P21, respectively. The structures of 5mC-dsDNA and 5hmC-dsDNA were solved by using the molecular replacement method (MR) with a published dsDNA structure (pdb code: 1EN9; sequence: 5′-CCAGCGCTGG-3′) as the search model,32 while the structures of 5fC-dsDNA and 5caC-dsDNA were solved by using a direct method due to their extremely high resolutions.33 All the structures were subsequently refined by using the maximum-likelihood refinement method carried out with the Phenix software package.34Table 1 and ESI Table S1† summarize the data collection and structure refinement statistics.| 5fC-dsDNA | 5caC-dsDNA | ||
|---|---|---|---|
| a Highest-resolution shell is shown in parentheses. | |||
| Data collection | |||
| Space group | P61 | P21 | |
| Cell dimensions | a, b, c (Å) | 54.548, 54.548, 45.819 | 22.465, 36.333, 30.215 |
| α, β, γ (°) | 90.00, 90.00, 120.00 | 90.00, 100.175, 90.00 | |
| Wavelength (Å) | 0.9795 | 0.9795 | |
| Resolution (Å)a | 50–1.56 (1.62–1.56) | 50–1.06 (1.10–1.06) | |
| R merge (%) | 6.1 (21.3) | 4.3 (8.2) | |
| I/σI | 37.6 (11.6) | 26.1 (11.2) | |
| Completeness (%) | 99.5 (98.6) | 98.0 (99.4) | |
| Redundancy | 9.8 (9.0) | 3.2(3.1) | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Refinement | |||
| Resolution (Å) | 47.2–1.56 | 29.7–1.06 | |
| No. of reflections | 10569 |
21838 |
|
| R work/Rfree | 0.147/0.162 | 0.156/0.181 | |
| No. of atoms | Nucleic acids | 410 | 410 |
| Water | 118 | 174 | |
| Ligand/ion | 8 | N/A | |
| B-factors | Nucleic acids | 21.006 | 8.761 |
| Water | 36.410 | 22.506 | |
| Ligand/ion | 32.764 | N/A | |
| R.m.s deviations | Bond lengths (Å) | 0.007 | 0.010 |
| Bond angles (°) | 1.338 | 1.802 | |
Structural analysis suggests that the 5mC-dsDNA, 5fC-dsDNA and 5caC-dsDNA structures exhibit a single non-canonical right-hand double helix pattern. Interestingly, 5hmC-dsDNA displays two non-canonical right-hand double helixes in an asymmetric unit, where one strand is complemented with half of the other two stands at the same time, exhibiting a non-standard “X” base paring pattern. Within the double stranded helix of the structures, 5mC-dsDNA, 5hmC-dsDNA and 5caC-dsDNA helixes exhibit B-form dsDNA patterns, which are similar to the published B-form 5C-dsDNA structure (pdb code: 1EN9),32 while the 5fC-dsDNA helix exhibits an A-form pattern. All the modification groups point towards the major groove as expected. The C5 atoms from the modification groups in each structures are 7.4 Å (5mC), 7.7 Å/7.0 Å (5hmC), 10.4 Å (5fC) or 7.2 Å (5caC) away from each other (distance between C5m atoms) (Fig. 1 and ESI Fig. S1 and S2†).
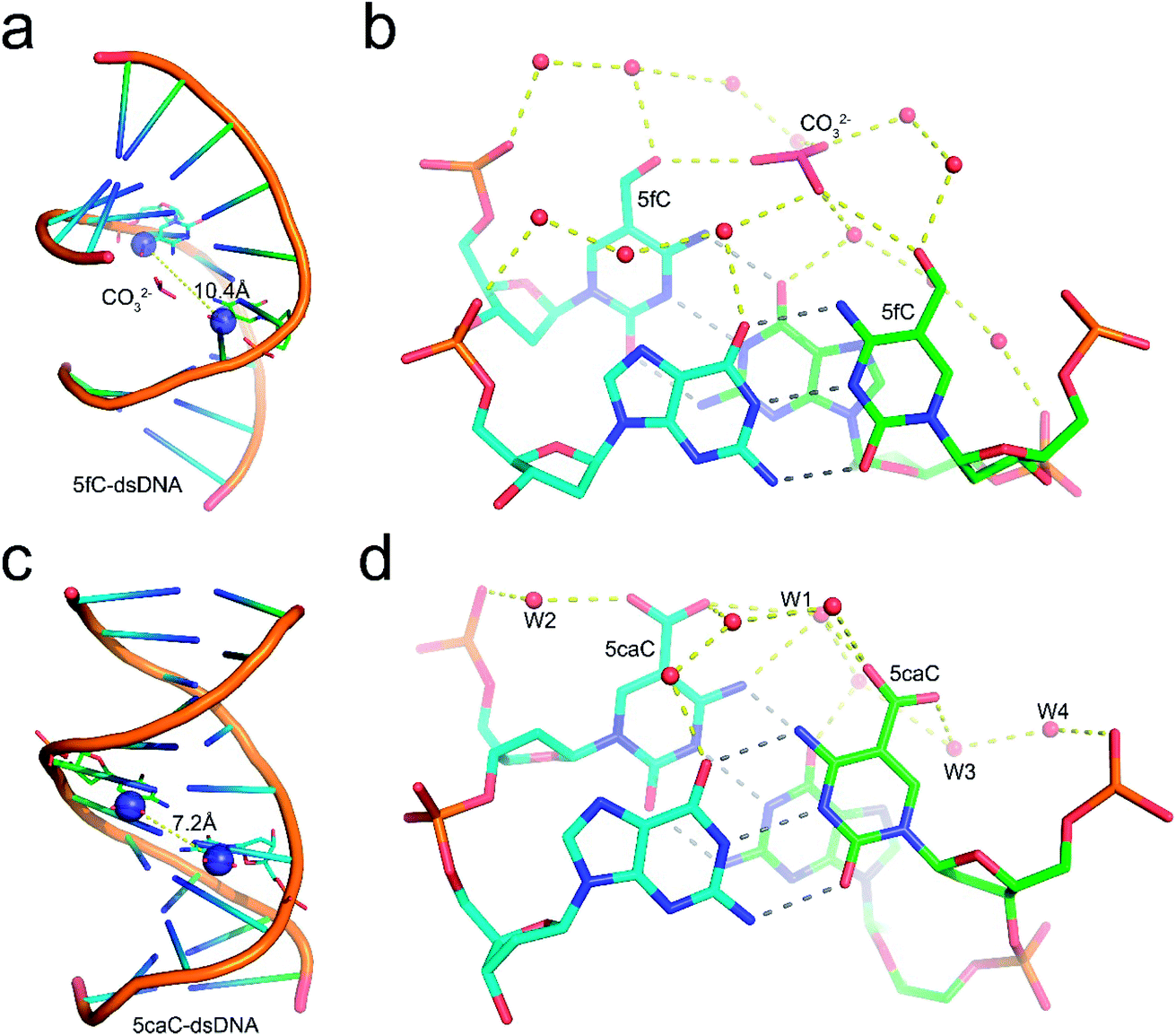 | ||
| Fig. 1 Schematic diagram of 5fC-dsDNA and 5caC-dsDNA crystal structures. (a) Overall structure of 5fC-dsDNA. Two complementary strands are colored in green and cyan, respectively. The fully symmetric 5fC bases are shown as sticks and their C5m atoms are shown as purple spheres. The distance between two C5m atoms is shown as yellow dashes. (b) The hydrogen bond networks around 5fC base pairing. The water molecules are shown as red dots, and the carbonate ions are shown as magenta sticks. The hydrogen bonds are shown as yellow dashes. (c) The overall structure of 5caC-dsDNA. (d) The hydrogen bond networks around 5caC base pairing. | ||
Further local rotational and translational base-step parameter analysis including the slide displacement and roll angle indicates structural variations among these crystal structures (Fig. 2a and c). Remarkably, we observed distinct structural alteration in 5fC and 5caC containing dsDNA. Regarding 5fC containing dsDNA, ∼2 Å shift displacement alteration at the 5fC·G site occurs compared with the adjacent base pairs, leading to a 3–5 Å opening in the major groove width and 2–3 Å narrowing in the minor groove (Fig. 2b, d and e). It is noteworthy that such alterations have been observed in previously reported 5fC-containing DNA structures under different crystallization conditions suggesting that 5fC modification specifically contributes to DNA structure conformational alteration through inducing groove geometry alteration in the helix.21 Within the 5fC·G base pair, the formyl-group does not interfere with the regular G·C base paring sterically, and the sp2 hybridization plane of the formyl group lies in the same plane as the pyrimidine ring (Fig. 1b). The O5 atom of the formyl group forms an intramolecular hydrogen bond with the exocyclic N4 amino group of the pyrimidine, and locks the rotation of the O5-C5m-C5-C6 dihedral angle of the formyl group (Fig. 3c). Moreover, the formyl groups are stabilized at the right place via hydrogen bond networks in the major groove. In the center of the networks, a carbonate ion source from the DNA purification reagent triethylammonium bicarbonate (TEAB) mediates the crosstalk of the two fully symmetric formyl groups as well as the interactions among the formyl groups, water molecules, and the phosphodiester backbones as a hub.
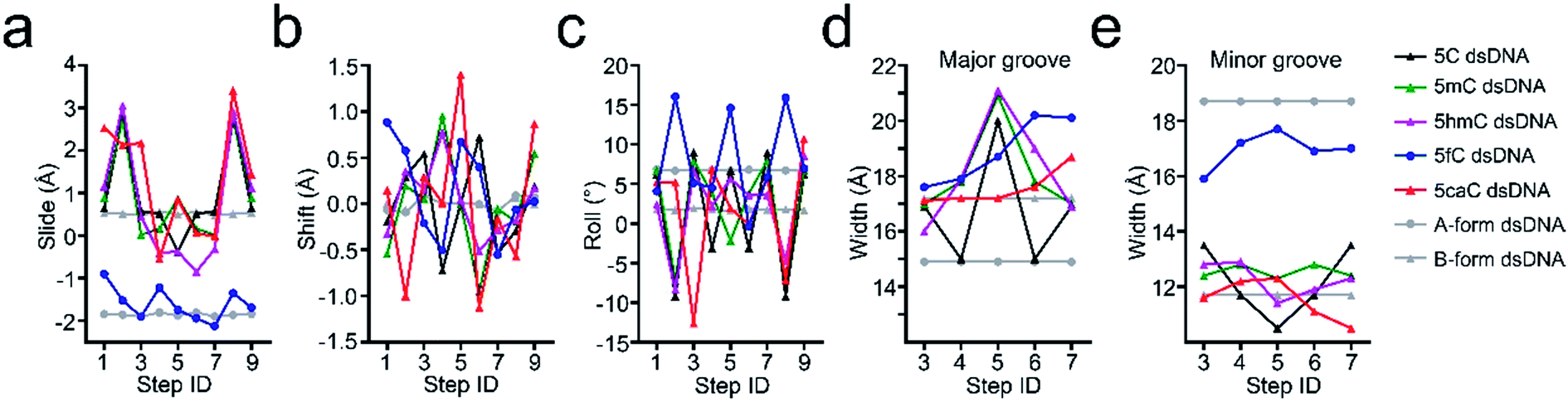 | ||
| Fig. 2 Comparison of the base-step and groove rigid body parameters of 5C-, 5mC-, 5hmC-, 5fC- and 5caC-dsDNA with canonical A-form and B-form dsDNA. (a–c) The comparison of slide, shift, and roll base pair parameters. (d, e) The major and minor groove width comparison. | ||
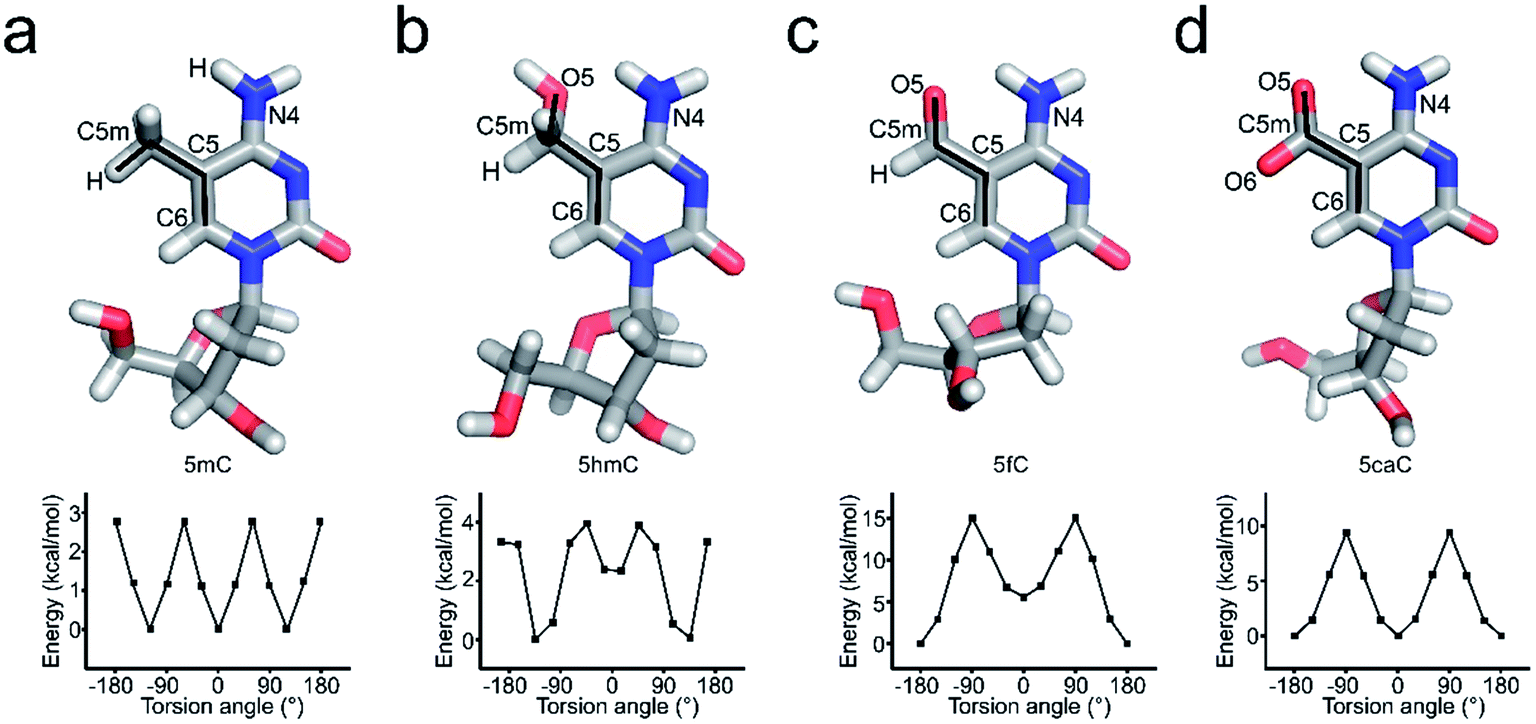 | ||
| Fig. 3 The dihedral angle rotation analysis against 5mC (a), 5hmC (b), 5fC (c), and 5caC (d). The modified bases are shown as sticks, and atoms in the modified group are labeled. The potential energy calculated during O5/H-C5m-C5-C6 dihedral angle rotation is shown as curves. | ||
Similar to 5fC containing dsDNA, a ∼2 Å shift displacement alteration occurs at the 5caC·G site compared with the adjacent base pairs in 5caC containing dsDNA, suggesting that the negative charges at the C5m position of the cytosine pyrimidine ring could induce similar geometry alteration of dsDNA (Fig. 2b). Unexpectedly, further structural alteration in the major and minor grooves is not observed, thus indicating that 5caC has distinctive contribution to dsDNA structural alteration compared with 5fC (Fig. 2d and e). Within the 5caC·G base paring, the carboxyl group of 5caC does not interfere with the regular G·C base paring sterically, and the sp2 hybridization plane of the carboxyl group lies in the same plane as the pyrimidine ring as well (Fig. 1d). Due to the significantly shorter distance between two fully symmetric carboxyl groups in dsDNA, a bridging water molecule (W1) directly connects the two carboxyl groups, mediating the crosstalk between two 5caC instead of the carbonate ion in the 5fC-DNA structure. What is more, the carboxyl group is further locked by the intramolecular hydrogen bond with the exocyclic N4 amino group of the pyrimidine, and hydrogen bonds with the phosphodiester backbone are mediated by one (W2) or two (W3 and W4) water molecules (Fig. 1d).
Effects of C, 5mC, 5hmC, 5fC, and 5caC in inducing the geometry alteration of dsDNA
5hmC, 5fC, and 5caC generated by TET dioxygenases from 5mC share highly similar chemical structures. However, TDG accurately distinguishes these derivatives by excluding C, 5mC, and 5hmC and catalyzing the removal of 5fC significantly faster than that of 5caC.30,31 Density functional theory (DFT) analysis against the O5/H-C5m-C5-C6 dihedral angle suggests that the methyl group in the 5mC-dsDNA structures adopts the most stable conformation in the minimum energy state at −120°, 0° or 120°, while formyl and carboxyl groups adopt the most stable conformation in the minimum energy state at −180° or 180° and −180°, 0° or 180°, respectively (Fig. 3). Dihedral angle rotation of formyl and carboxyl groups along the C5m-C5 bond from 0° to 90° leads to an increase of potential energy and instability of the structures. The maximum energy state is reached at 90°, and subsequently decreases from 90° to 180°. At 180°, the carboxyl group returns to the most stable conformation in the minimum energy state. The energy of the formyl group at 0° is significantly higher than that at 180° due to the loss of the intramolecular hydrogen bond with the exocyclic N4 amino group of the pyrimidine, suggesting a critical role of the intramolecular hydrogen bonding in stabilizing the formyl- and carboxyl-group conformation in the structure. What is more, the energy state landscape of 5hmC is totally different from that of 5mC, 5fC and 5caC. The minimum energy state of the 5hmC dihedral angle is around 136° or −133° rather than 0° or 180° in 5fC and 5caC structures and −120°, 0° or 120° in the 5mC structure. This is most likely caused by the sp3 hybridization of the hydroxymethyl group, and such a distinctive orientation and energy state of 5hmC potentially leads to the unrecognition of TDG on 5hmC for catalysis.In order to investigate the effects of the cytosine modifications on dsDNA conformation, we employed a scrupulous method to define the force field and perform further molecular dynamic simulation according to our previous study35,36 (see the method section), which will eliminate the interference from the DNA sequence and crystallization condition variations. As shown in Fig. 4, after 400 ns simulation, the overall structures of 5C-, 5mC-, 5hmC-, 5fC- and 5caC-dsDNA tend to be canonical B-form dsDNA, confirming that the A-form conformation observed from the 5fC containing structure was caused by crystal packing rather than the variation of the cytosine modification.
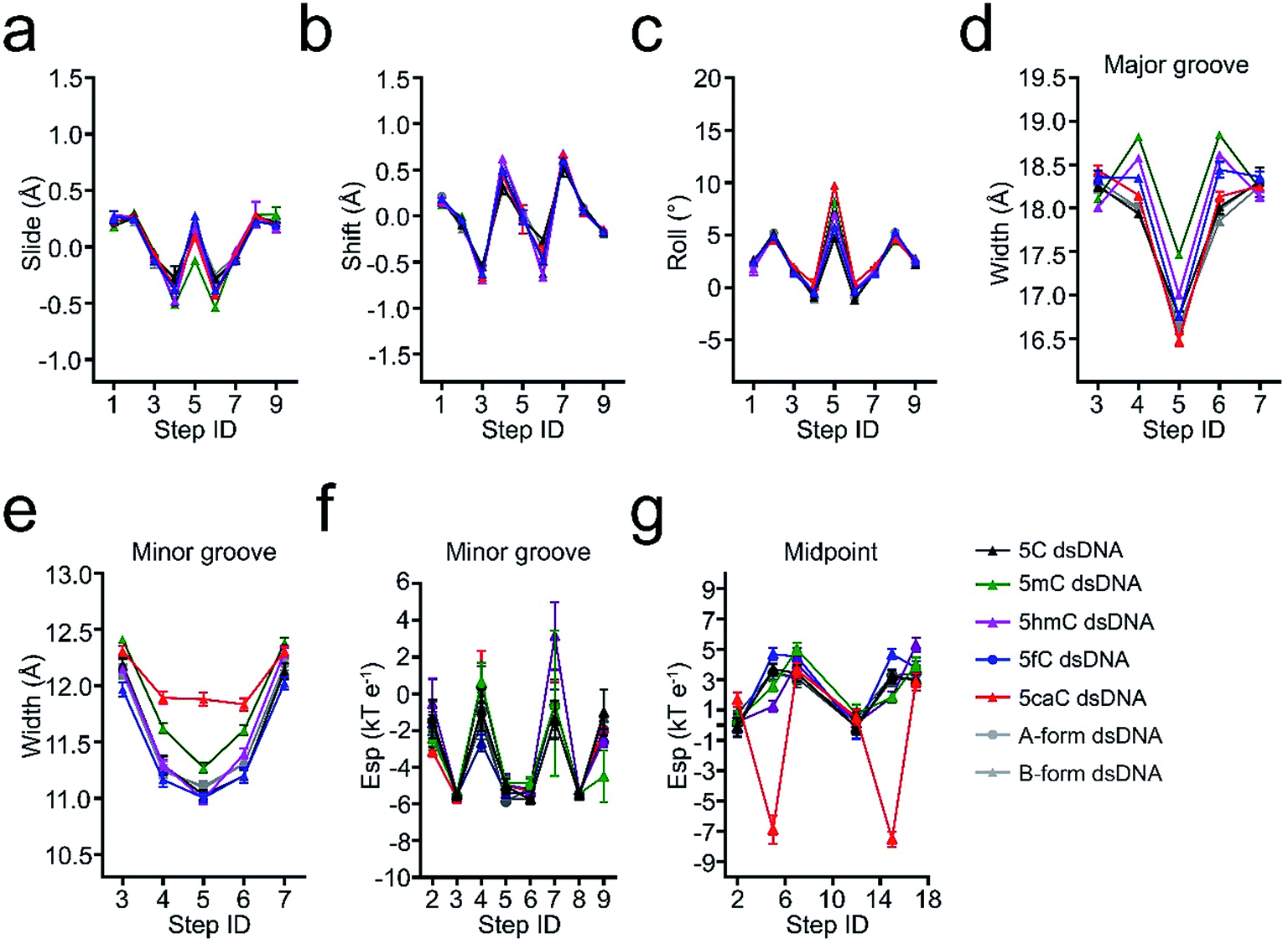 | ||
| Fig. 4 Comparison of the structural parameters of 5C-, 5mC-, 5hmC-, 5fC- and 5caC-dsDNA with canonical A-form and B-form dsDNA from molecular dynamics simulation. (a–c) The comparison of slide, shift, and roll base pair parameters. (d and e) The major and minor groove width comparison. (f and g) The electric potential energy of the minor groove and the midpoint of C5-P (phosphate in the backbone). | ||
The slide displacement analysis showed that all the modifications have similar effects except that the slide displacements of 5mC containing dsDNA slightly decrease by 0.4 Å, while the roll angle of 5caC increases remarkably by 5°, leading to more bended structural conformations of 5caC-dsDNA, especially at the modification site compared with that of the canonical B-form dsDNA (Fig. 4a–c).
Further geometry analysis against major and minor grooves shows that the major grooves in 5mC- and 5hmC-dsDNA are significantly opened by ∼1 Å and ∼0.7 Å compared to that of 5fC- and 5caC-dsDNA respectively, leading to a relatively narrower major groove in 5fC- and 5caC-dsDNA structures, and mildly looser major grooves in 5mC- and 5hmC-dsDNA structures (Fig. 4d). Moreover, the minor groove of 5caC-dsDNA is significantly enlarged by ∼1 Å compared to other modification containing structures, which has not been observed previously, suggesting distinctive contribution of 5caC to minor groove geometry alteration (Fig. 4e).
By analyzing the electrostatic potential energy of the minor groove, 5fC and 5caC are found to be in a less stable state in base pairing due to the repulsive force produced by the same negative charges of the carbonyl oxygen atom in 5fC or the carboxyl group in 5caC and the phosphodiester backbone (Fig. 4f). However, the repulsive force in 5caC-dsDNA squeezes the 5caC base aside from the phosphate backbone, resulting in an opposite and more stable electric potential energy at the midpoint of C5-P (phosphate in the backbone) with 5fC (Fig. 4g). Together with these observations, the 5fC and 5caC modifications are found to variously influence the geometry alteration of the dsDNA major and minor grooves. The various conformational alterations are predominantly induced by the distinctive repulsive force between the formyl or carboxyl group and the phosphodiester backbone, which would benefit the preferential recognition and catalysis of 5fC and 5caC by various reader proteins, such as TDG.
TDG selectively recognizes 5fC and 5caC through the finger residue Arg275
To date, TDG has been the only enzyme discovered for the enzymatic removal of 5fC and 5caC from dsDNA with marked catalytic specificity. Structural analysis of the published TDG-dsDNA structure (PDB code: 3UO7) indicates that TDG recognizes DNA predominantly via two loops (Fig. S2†).30,31 Loop1 between residues 199–202 (GSKD) inserts into the major groove of the dsDNA. While loop2 between residues 274–277 (ARCA) inserts into the minor groove of the dsDNA, where the finger residue Arg275 penetrates into the DNA helix and pushes the modified base into the active pocket of TDG for catalysis. Considering the eminent influences of 5fC and 5caC on dsDNA groove geometry, polar residues in these two loops were mutated (S200A, K201A, and D202A in loop1, and R275A in loop2) in order to evaluate their potential roles in recognizing the conformational alteration of dsDNA induced by 5fC and 5caC. Surprisingly, only the mutation of Arg275 (R275A) on loop2 exhibits significant influence on 5fC/5caC selection through glycosylase activity assay. As shown in Fig. 5a, the wildtype enzyme catalyzed 5fC and 5caC excision completely in a 30 min reaction, while the R275A mutation preferentially abolished the excision of 5fC compared with the partial excision of 5caC in dsDNA. A single turnover kinetics assay further confirms this result. As shown in Table 2, mutagenesis of residues on loop1 (S200A, K201A, and D202A) reduces the maximal catalytic activity (Kmax) in catalyzing 5fC and 5caC excision by 1.1 to 4.7 fold as expected,28,29 suggesting that loop1 unlikely contributes to selective recognition and catalysis of substrates. However, the R275A mutation in loop2 dramatically decreases the catalysis activity against 5fC by 47 fold versus to 4.8 fold in catalyzing 5caC, resulting in a nearly ten times slower catalysis rate of TDG for 5fC removal compared with that of 5caC (Fig. 5b). These observations indicate that TDG makes substrate selection against 5fC and 5caC by recognizing the geometry alteration of the dsDNA minor groove by its finger residue Arg275 on loop2. Notably, a sharp activity decrease of TDG R275A in catalyzing uracil was also observed, suggesting that Arg275 can specifically recognize uracil besides 5fC (Fig. 5a, ESI Table S2†). As the uracil does not have a positive charge modification on the C5 position of the pyrimidine ring, such recognition could be due to the wobble guanine–uracil base pairing, which does not follow Watson–Crick base pairing rules.37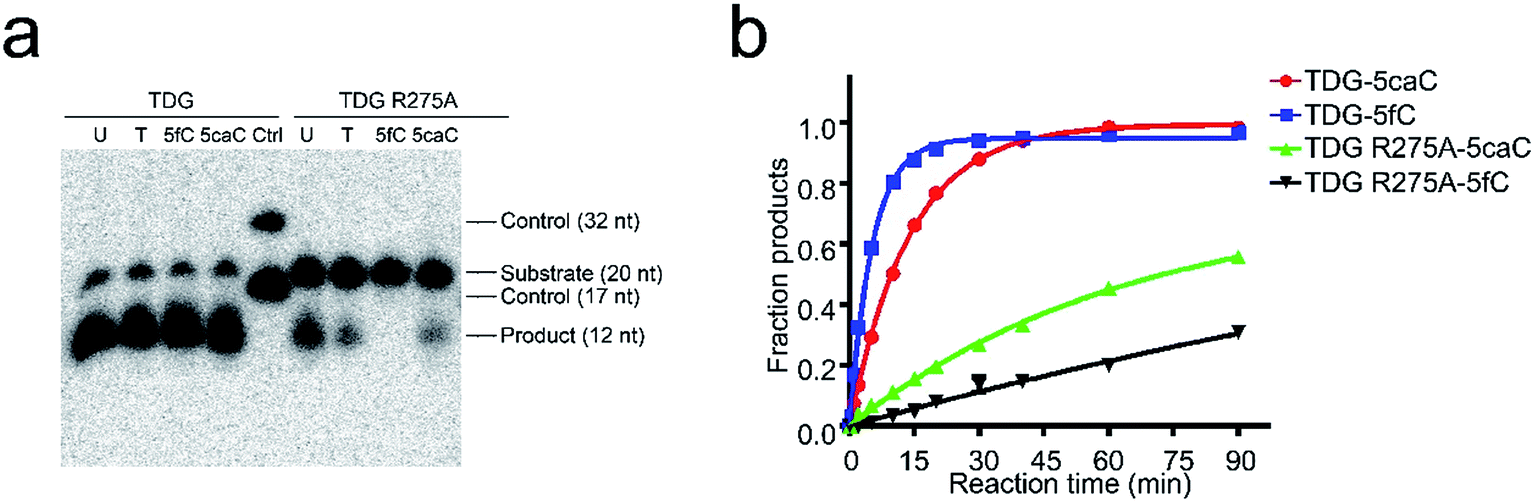 | ||
| Fig. 5 Schematic diagram of TDG glycosylase activity assay and single turnover assay results. (a) The glycosylase activity assay of wild type and R275A mutants against various substrates containing uracil, thymine, 5fC, and 5caC, respectively. (b) The single turnover assay against 5fC or 5caC containing substrates (representative of three independent experiments). | ||
| Substrate | Enzyme | K max (min−1) | K max(WT)/Kmax (Mut) |
|---|---|---|---|
| 5caC·G | TDG | 0.072 ± 0.001 | — |
| TDG S200A | 0.020 ± 0.001 | 3.6 | |
| TDG K201A | 0.068 ± 0.001 | 1.1 | |
| TDG D202A | 0.026 ± 0.002 | 2.8 | |
| TDG R275A | 0.015 ± 0.001 | 4.8 | |
| 5fC·G | TDG | 0.189 ± 0.004 | — |
| TDG S200A | 0.079 ± 0.003 | 2.4 | |
| TDG K201A | 0.106 ± 0.010 | 1.8 | |
| TDG D202A | 0.040 ± 0.008 | 4.7 | |
| TDG R275A | 0.004 ± 0.001 | 47.3 |
Our results here show that TDG R275 is a key residue that recognizes the distinctive geometry alterations in the minor groove of dsDNA induced by modified bases such as 5fC and 5caC, and flips the selected substrate base into the active pocket for catalysis to achieve independent downstream biological functions. Our results provide a potential mechanism by which reader proteins distinguish these highly similar 5-substituents of cytosine. This selective recognition mechanism further provides novel insights into the biological functions of these epigenetic modifications in the active DNA demethylation pathway, as well as in development and tumorigenesis.
Discussion
Our studies have revealed that methyl-, hydroxyl-, formyl-, and carboxyl-group modifications on cytosine have distinctive influences on the geometry of dsDNA which potentially allow for the preferential recognition and catalysis among these highly similar derivatives by various reader proteins, such as TDG. Since full methylation on a chromosome has been well known as a regular epigenetic event, all the oxidative modifications (5hmC, 5fC and 5caC) generated from the oxidation of fully methylated cytosine have been detected by numerous whole genome sequencing studies (e.g. TAB seq for 5hmC and fCAB-seq for 5fC).11,38 This indicated that fully symmetric 5hmC, 5fC or 5caC sites in the genome do have a significant independent biological meaning. We synthesized and crystallized 10-bp self-complementary DNA decamers containing a fully symmetric 5mC·G, 5hmC·G, 5fC·G or 5caC·G modified CpG site in the middle position, and analyzed the conformation alteration by DFT computation and molecular dynamics simulations. The sp2 hybridized formyl and carboxyl groups adopt plane conformations, and the ketone from the formyl and carboxyl groups forms an intramolecular hydrogen bond with the exocyclic N4 amino group of the pyrimidine, locking the sp2 hybridization plane to lie in the same plane as the cytosine pyrimidine ring. In contrast, the hydroxylmethyl group adopts a distinctive non-planar sp3 hybridization, and forms a ∼136° angle with the pyrimidine plane in base paring.18 Furthermore, the negative charge carried by 5caC generates a repulsive force with the phosphodiester backbone of dsDNA, which influences the geometry of dsDNA by predominantly widening the minor groove and loosening the dsDNA structures compared with canonical B-form dsDNA. Meanwhile, the repulsive force in 5caC-dsDNA squeezes the 5caC base aside from the phosphate backbone, causing more drastic geometry alteration in the minor groove of dsDNA and opposite electric potential energy at the midpoint of C5-P compared with 5fC.Then, using two techniques, we investigated the key residue which could sense the geometry alteration in the minor groove, and observed that the mutagenesis of Arg275 on the loop2 of TDG to alanine (R275A) sharply decreases the activity of TDG against 5fC by 47 fold, thus suggesting a specific role of Arg275 in 5fC recognition and catalysis. However, the sharp drop of activity does not occur in 5caC catalysis to a similar degree. The activity of R275A against 5caC only decreases by 4.8 fold, which is similar to that of mutations in loop1 (S200A, K201A, and D202A), thus suggesting a non-specific recognition of Arg275 on 5caC. This difference could be explained by the deflection of the 5caC base as well as the more stable status of 5caC compared to 5fC, which might weaken the role of Arg275 in 5caC recognition and flipping.
It should be noted that a previous study reported that the residue Asn157 inside the active pocket of TDG is crucial for selective 5caC excision. The mutagenesis of Asn157 to aspartic acid (N157D) significantly reduces such preference at acidic pH,31 while our results here suggest that the substrate preferential selection of TDG occurs prior to the flipping of the base from the dsDNA helix. Our results further suggest that TDG N157D and R275A double mutation would abolish the base excision of 5fC by either blocking the base flipping or blocking the catalysis of the base excision, which would cause the accumulation of 5fC in the genome. Hence, this TDG double mutant could be used as a chemical biology tool to further study the biological function of 5fC in the differentiation of embryo stem cells in vivo.
The mammalian DNA demethylation is a critical process for cell development, and how enzymes involved in this process recognize the highly similar chemical structures of the cytosine derivatives is one of the key scientific questions in understanding the biological function of this process. A recent study reported that ten-eleven-translocation proteins (Tets) and TDG physically interact with one another to avoid DNA double strand breaks (DSB) in the demethylation.39 Our current results support their observation as we show that the oxidative cytosine derivatives such as 5fC and 5caC induce significant geometry alteration in the DNA minor groove, which may decrease the stability of the genome, leading to DSB. The way to avoid such a possibility is that the two enzymes work together, and no free 5fC and 5caC site is exposed.
All these results provide novel insights into the mechanisms of cytosine modifications in achieving independent downstream biological functions, which will in turn facilitate the understanding of biological functions of these epigenetic modifications in active DNA demethylation pathways, as well as in development and tumorigenesis.
Experimental methods
Synthesis and purification of modified DNA oligonucleotides
The oligonucleotides with sequences (5′-CCA GXG CTG G-3′ and 5′-ATA GAA GAA TTC XGT TCC AG-3′, X refers to 5mC, 5hmC, 5fC or 5caC) used in crystallization and biochemistry studies were synthesized by using solid-phase synthesis. The synthesized oligonucleotides were purified by using high-performance liquid chromatography (reverse-phase C18 column) and lyophilized as previously reported.40 The oligonucleotides containing normal bases (5′-CTG GAA CGG AAT TCT TCT AT-3′) were purchased from Sangon Company. The oligonucleotides were subsequently dissolved in buffer containing 20 mM HEPES, pH 7.0, and 100 mM NaCl, and heated to 100 °C for slow annealing.DNA crystallization, diffraction data collection, and structure determination
Crystallization of 5mC, 5hmC, 5fC or 5caC containing DNA decamers was performed by using a sitting drop vapor diffusion method. 1 μL of 2 mM 5mC-, 5hmC-, 5fC- or 5caC-dsDNA sample was mixed with an equal volume of reservoir solution containing 200 mM potassium chloride, 10 mM magnesium sulfate hexahydrate, 50 mM MES monohydrate, pH 5.6, and 10% PEG400 (for 5mC-dsDNA); 80 mM sodium chloride, 12 mM potassium chloride, 20 mM magnesium chloride hexahydrate, 40 mM sodium cacodylate trihydrate, pH 6.0, 30% MPD, and 12 mM spermine tetrahydrochloride (for 5hmC-dsDNA); 50 mM cacodylate acid, pH 6.0, 20 mM magnesium acetate, 0.5 mM spermine, 100 mM NaCl, and 25% MPD (for 5fC-dsDNA); or 50 mM sodium succinate, pH 5.5, 20 mM MgCl2, 0.5 mM spermine, and 3.0 M ammonium sulfate (for 5caC-dsDNA), respectively. The mixture was then equilibrated against 100 μL of the reservoir solution at 277 K. Crystals appeared after about 4–6 days. The crystals were then flash-frozen in liquid nitrogen with 20% glycerol (v/v) as the cryoprotectant solution. The diffraction data were collected at BL18U/19U at a wavelength of 0.9795 Å at the Shanghai Synchrotron Radiation Facility (SSRF; Shanghai, People's Republic of China). The diffraction images collected were integrated and scaled to a resolution of 1.40 Å, 2.85 Å, 1.56 Å and 1.06 Å with the space group C2, C2, P61 and P21 by using HKL3000, respectively. The phases of 5mC-dsDNA and 5hmC-dsDNA were determined by using the molecular replacement method (MR) with a published dsDNA structure (pdb code: 1EN9; sequence: 5′-CCAGCGCTGG-3′) as the search model.32 The phases of 5fC-dsDNA and 5caC-dsDNA were determined by using a direct method with the CCP4 suite,33 followed by subsequent maximum-likelihood refinement with the Phenix software package.34 The electron density-based model building was performed using the computer graphics program Coot,41 and the final structures were visualized by using PyMol software.42Table 1 and ESI Table S1† summarize the data collection and structure refinement statistics.Modeling, structural parameter calculation, and MD simulation
Beside the determined crystal structures of 5mC-dsDNA, 5hmC-dsDNA, 5fC-dsDNA and 5caC-dsDNA, the 5C-dsDNA structure was obtained from the published crystal structure (PDB code: 1EN9), while canonical A-form and B-form dsDNA structures with the same sequence were modeled by using Maestro software.The DNA rigid-body parameters were calculated and analyzed by using the ensemble scripts provided by the 3DNA suit.43 The data were processed using g-analyze from the Gromacs analysis tool. The vertical bars represent the error estimate of the average values calculated using the blocking method. All relaxed potential energy surface scans were carried out with the Gaussian 09 software package at the M062x/6-31+g(d,p) level.44,45 The dihedral torsion angle of O5/H-C5m-C5-C6 in the modified deoxycytidines was scanned every 30 degrees with geometry optimization at each conformation. For MD simulation, the RESP charges of 5mC, 5hmC, 5fC and 5caC were calculated with Gaussian 09 software at the HF/6-31(d,p) level and the force field parameters were produced using an antechamber based on previous reports.35,36 The force field for DNA was ff-nucleic-OL15.46 The solvent effects were involved using the polarizable continuum model with water as the solvent.47 Tleap was used to model all simulation systems, namely 5C-dsDNA, 5mC-dsDNA, 5hmC-dsDNA, 5fC-dsDNA, 5caC-dsDNA, A-form dsDNA, and B-form dsDNA. In all simulation systems, the DNA structure was submerged in explicit TIP3P water in cubic boxes with an extra 10 Å extension along each axis of the DNA. The net charge of the system was neutralized by adding a suitable number of counterions. Amber topology and coordinates files were converted into Gromacs format by using Acpype. All MD simulations herein were performed with the Gromacs package (version 5.1). The particle mesh Ewald (PME) method was applied to handle the long-range electrostatics.48 Nonbonded van der Waals forces and short-range electrostatic interactions between atoms were truncated at 10 Å. Periodic boundary conditions (PBCs) were used during the MD simulations. All the MD simulations were performed at 300 K and 1 atm. The LINCS was used to constrain the length of the hydrogen bonds, allowing the movements integrated numerically with a time step of 2 fs algorithm.49 The starting structure of each model was energy-minimized by using the steepest-descent algorithm to remove unfavorable steric clashes.50 Coordinates of each model were saved every 10 ps throughout the 400 ns production runs. The DNA structures were extracted from MD trajectories every 200 ps for further calculations. The Delphi program was used for electrostatic potential calculations.51 The partial charge and atom radii were taken from the topology file used during MD simulations. The salt concentration was 0.145 M and a 1.4 Å probe sphere was applied for the calculation of the solute molecule surface. The interior of the solute molecule was assigned an internal dielectric constant of 2 whereas exterior regions were assigned a dielectric constant of 80. The size of the cubical grid was set to 165. The minor groove electrostatic potential and the electrostatic potential of the geometric midpoint between the C5 atom and P atom were calculated based on a previous report.52,53
TDG expression and purification
The catalytic domain of human TDG was expressed and purified as previously reported.30 Briefly, the cDNA for the catalytic domain of human TDG (residues 111–308) was subcloned into a pMCGS19 plasmid and expressed in BL21 (DE3) cells containing the vector pRK1037. The cultures were grown at 37 °C until OD600 reached 0.6, and then induced at 25 °C with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight. Subsequently, the cells were harvested and re-suspended with a lysis buffer containing 20 mM Tris, pH 7.4, 500 mM NaCl, 20 mM imidazole, 1 mM dithiothreitol (DTT) and 0.25 mM phenylmethylsulphonyl fluoride (PMSF), and lysed using a French press. The lysate was centrifuged at 13000g for 40 min, and the supernatant was used for further purification via loading onto an affinity column (Ni-NTA), ion-exchange column (HiTrap SP column), and gel-filtration column (16/60 Superdex 75). The purified protein was concentrated and quantified by using the Bradford reagent (Bio-Rad), flash-frozen, and stored at −80 °C.
DNA glycosylase activity assay
A 20-mer 5fC- or 5caC-containing strand was labeled with γ-32P-ATP by incubation with T4 DNA polynucleotide kinase (T4 PNK, NEB) at 37 °C for 1 h (A 17-mer labeled strand was used as a control). Subsequently, the labeled oligonucleotide was annealed with a complementary strand and the duplex was purified for the glycosylase activity assay. The reactions were performed with 100 nM TDG and 10 nM DNA substrates (32P-5′-ATA GAA GAA TTC C*GT TCC AG-3′ and 5′-CTG GAA CGG AAT TCT TCT AT-3′) at 22 °C in the reaction buffer containing 25 mM HEPES, pH 7.4, 0.5 mM EDTA, 0.5 mg mL−1 BSA, and 0.5 mM DTT. Reactions were quenched by adding 1 M NaOH and 100 mM EDTA and incubating at 100 °C for 5 min to break the DNA strand containing an abasic site. The samples were cooled down and loaded into a denaturing gel for electrophoresis.Single turnover kinetics assay
Single turnover kinetics assay under saturating enzyme conditions was carried out to determine the rate constant (Kmax) of TDG. The reaction was performed with 5 μM TDG and 0.5 μM DNA substrates at 22 °C in the reaction buffer (25 mM HEPES, pH 7.4, 0.5 mM EDTA, 0.5 mg mL−1 BSA, and 0.5 mM DTT) and was quenched at specific time points with 50% (v:v) 0.1 M NaOH and 0.01 M EDTA. The samples were further boiled for 15 min at 85 °C (5fC and U were quenched with 0.3 M piperidine and 0.03 M EDTA and incubated at 85 °C for 15 min) and then cooled down for HPLC analysis. The products and reactants from different time points were separated and quantified by anion-exchange HPLC using reported denaturing conditions with a DNAPac PA200 column (Dionex). The oligonucleotides are detected by absorbance (260 nm), and the fraction product (F) is determined from the integrated peak areas for the product strands (AP1 and AP2) and target strand (AS) using the equation: F = (AP1 + AP2)/(AP1 + AP2 + AS), and the single turnover rate constant Kmax is calculated by using the equation: F = A[1 − exp(−Kmaxt)], in which A is the fraction of the substrate converted to the product at completion and t is the reaction time.
Accession codes
PDB: the atomic coordinates and structure factors for the reported crystal structures are deposited under accession codes 6JV5 (5mC-dsDNA), 6JV3 (5hmC-dsDNA), 5ZAS (5fC-dsDNA) and 5ZAT (5caC-dsDNA).Author contributions
L. Z., C. L. and C. H. designed the experiments; T. R. F. performed the protein purification and the single turnover assay; Y. X. W. and L. Z. performed the nucleic acid crystallization and structure determination; Q. L. Y. performed the glycosylase activity assay; L. P. L., P. X. and S. E. L. preformed the computational analysis; Q. D. performed the DNA synthesis and purification; L. Z. and C. L. wrote the paper. All authors discussed and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This project was supported by grants from the National Natural Science Foundation of China (21572133, 21722802 and 91853118 to L. Z.; 81821005, 81625022, and 21820102008 to C. L.) and the National Institutes of Health HG006827 (C. H.). The Program for Professors of Special Appointment (Eastern Scholar) at the Shanghai Institutions of Higher Learning (L. Z.), and facilities including the beamline BL17U/19U at the National Center for Protein Sciences Shanghai (NCPSS) and the Shanghai Synchrotron Radiation Facility (SSRF) are acknowledged.References
- R. Jaenisch and A. Bird, Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals, Nat. Genet., 2003, 33, 245–254 CrossRef CAS PubMed.
- N. Bhutani, D. M. Burns and H. M. Blau, DNA demethylation dynamics, Cell, 2011, 146, 866–872 CrossRef CAS PubMed.
- Y. Huang and A. Rao, Connections between TET proteins and aberrant DNA modification in cancer, Trends Genet., 2014, 30, 464–474 CrossRef CAS PubMed.
- J. A. Law and S. E. Jacobsen, Establishing, maintaining and modifying DNA methylation patterns in plants and animals, Nat. Rev. Genet., 2010, 11, 204–220 CrossRef CAS PubMed.
- X. Wu and Y. Zhang, TET-mediated active DNA demethylation: mechanism, function and beyond, Nat. Rev. Genet., 2017, 18, 517–534 CrossRef CAS PubMed.
- Y. F. He, B. Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen, L. Li, Y. Sun, X. Li, Q. Dai, C. X. Song, K. Zhang, C. He and G. L. Xu, Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA, Science, 2011, 333, 1303–1307 CrossRef CAS PubMed.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine, Science, 2011, 333, 1300–1303 CrossRef CAS PubMed.
- L. Zhang, K. E. Szulwach, G. C. Hon, C. X. Song, B. Park, M. Yu, X. Lu, Q. Dai, X. Wang, C. R. Street, H. Tan, J. H. Min, B. Ren, P. Jin and C. He, Tet-mediated covalent labelling of 5-methylcytosine for its genome-wide detection and sequencing, Nat. Commun., 2013, 4, 1517–1527 CrossRef PubMed.
- C. X. Song, K. E. Szulwach, Y. Fu, Q. Dai, C. Yi, X. Li, Y. Li, C. H. Chen, W. Zhang, X. Jian, J. Wang, L. Zhang, T. J. Looney, B. Zhang, L. A. Godley, L. M. Hicks, B. T. Lahn, P. Jin and C. He, Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine, Nat. Biotechnol., 2011, 29, 68–72 CrossRef CAS PubMed.
- M. Yu, G. C. Hon, K. E. Szulwach, C. X. Song, L. Zhang, A. Kim, X. Li, Q. Dai, Y. Shen, B. Park, J. H. Min, P. Jin, B. Ren and C. He, Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome, Cell, 2012, 149, 1368–1380 CrossRef CAS PubMed.
- W. Li, X. Zhang, X. Lu, L. You, Y. Song, Z. Luo, J. Zhang, J. Nie, W. Zheng, D. Xu, Y. Wang, Y. Dong, S. Yu, J. Hong, J. Shi, H. Hao, F. Luo, L. Hua, P. Wang, X. Qian, F. Yuan, L. Wei, M. Cui, T. Zhang, Q. Liao, M. Dai, Z. Liu, G. Chen, K. Meckel, S. Adhikari, G. Jia, M. B. Bissonnette, X. Zhang, Y. Zhao, W. Zhang, C. He and J. Liu, 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers, Cell Res., 2017, 27, 1243–1257 CrossRef CAS PubMed.
- M. J. Booth, T. W. Ost, D. Beraldi, N. M. Bell, M. R. Branco, W. Reik and S. Balasubramanian, Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine, Nat. Protoc., 2013, 8, 1841–1851 CrossRef CAS PubMed.
- C. Zhu, Y. Gao, H. Guo, B. Xia, J. Song, X. Wu, H. Zeng, K. Kee, F. Tang and C. Yi, Single-Cell 5-Formylcytosine Landscapes of Mammalian Early Embryos and ESCs at Single-Base Resolution, Cell Stem Cell, 2017, 20, 720–731 CrossRef CAS PubMed.
- S. Kriaucionis and N. Heintz, The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain, Science, 2009, 324, 929–930 CrossRef CAS PubMed.
- M. Mellen, P. Ayata, S. Dewell, S. Kriaucionis and N. Heintz, MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system, Cell, 2012, 151, 1417–1430 CrossRef CAS PubMed.
- T. Zhou, J. Xiong, M. Wang, N. Yang, J. Wong, B. Zhu and R. M. Xu, Structural basis for hydroxymethylcytosine recognition by the SRA domain of UHRF2, Mol. Cell, 2014, 54, 879–886 CrossRef CAS PubMed.
- D. Renciuk, O. Blacque, M. Vorlickova and B. Spingler, Crystal structures of B-DNA dodecamer containing the epigenetic modifications 5-hydroxymethylcytosine or 5-methylcytosine, Nucleic Acids Res., 2013, 41, 9891–9900 CrossRef CAS PubMed.
- B. Xia, D. Han, X. Lu, Z. Sun, A. Zhou, Q. Yin, H. Zeng, M. Liu, X. Jiang, W. Xie, C. He and C. Yi, Bisulfite-free, base-resolution analysis of 5-formylcytosine at the genome scale, Nat. Methods, 2015, 12, 1047–1050 CrossRef CAS PubMed.
- S. Wang, Y. Long, J. Wang, Y. Ge, P. Guo, Y. Liu, T. Tian and X. Zhou, Systematic investigations of different cytosine modifications on CpG dinucleotide sequences: the effects on the B-Z transition, J. Am. Chem. Soc., 2014, 136, 56–59 CrossRef CAS PubMed.
- E. A. Raiber, P. Murat, D. Y. Chirgadze, D. Beraldi, B. F. Luisi and S. Balasubramanian, 5-Formylcytosine alters the structure of the DNA double helix, Nat. Struct. Biol., 2015, 22, 44–49 CrossRef CAS PubMed.
- M. Eleftheriou, A. J. Pascual, L. M. Wheldon, C. Perry, A. Abakir, A. Arora, A. D. Johnson, D. T. Auer, I. O. Ellis, S. Madhusudan and A. Ruzov, 5-Carboxylcytosine levels are elevated in human breast cancers and gliomas, Clin. Epigenet., 2015, 7, 88–93 CrossRef PubMed.
- L. Wang, Y. Zhou, L. Xu, R. Xiao, X. Lu, L. Chen, J. Chong, H. Li, C. He, X. D. Fu and D. Wang, Molecular basis for 5-carboxycytosine recognition by RNA polymerase II elongation complex, Nature, 2015, 523, 621–625 CrossRef CAS PubMed.
- A. Maiti and A. C. Drohat, Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites, J. Biol. Chem., 2011, 286, 35334–35338 CrossRef CAS PubMed.
- P. Neddermann, P. Gallinari, T. Lettieri, D. Schmid, O. Truong, J. J. Hsuan, K. Wiebauer and J. Jiricny, Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase, J. Biol. Chem., 1996, 271, 12767–12774 CrossRef CAS PubMed.
- A. Maiti, M. T. Morgan, E. Pozharski and A. C. Drohat, Crystal structure of human thymine DNA glycosylase bound to DNA elucidates sequence-specific mismatch recognition, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8890–8895 CrossRef CAS PubMed.
- S. Cortellino, J. Xu, M. Sannai, R. Moore, E. Caretti, A. Cigliano, M. Le Coz, K. Devarajan, A. Wessels, D. Soprano, L. K. Abramowitz, M. S. Bartolomei, F. Rambow, M. R. Bassi, T. Bruno, M. Fanciulli, C. Renner, A. J. Klein-Szanto, Y. Matsumoto, D. Kobi, I. Davidson, C. Alberti, L. Larue and A. Bellacosa, Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair, Cell, 2011, 146, 67–79 CrossRef CAS PubMed.
- H. Hashimoto, S. Hong, A. S. Bhagwat, X. Zhang and X. Cheng, Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation, Nucleic Acids Res., 2012, 40, 10203–10214 CrossRef CAS PubMed.
- A. Maiti, M. T. Morgan and A. C. Drohat, Role of two strictly conserved residues in nucleotide flipping and N-glycosylic bond cleavage by human thymine DNA glycosylase, J. Biol. Chem., 2009, 284, 36680–36688 CrossRef CAS PubMed.
- L. Zhang, X. Lu, J. Lu, H. Liang, Q. Dai, G. L. Xu, C. Luo, H. Jiang and C. He, Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA, Nat. Chem. Biol., 2012, 8, 328–330 CrossRef CAS PubMed.
- H. Hashimoto, X. Zhang and X. Cheng, Selective excision of 5-carboxylcytosine by a thymine DNA glycosylase mutant, J. Mol. Biol., 2013, 425, 971–976 CrossRef CAS PubMed.
- T. K. Chiu and R. E. Dickerson, 1 A crystal structures of B-DNA reveal sequence-specific binding and groove-specific bending of DNA by magnesium and calcium, J. Mol. Biol., 2000, 301, 915–945 CrossRef CAS PubMed.
- S. Fortier, Direct Methods for Solving Macromolecular Structures, Nato Asi, 2010, vol. 507 Search PubMed.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Towards automated crystallographic structure refinement with phenix.refine, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352–367 CrossRef CAS PubMed.
- L. Hu, J. Lu, J. Cheng, Q. Rao, Z. Li, H. Hou, Z. Lou, L. Zhang, W. Li, W. Gong, M. Liu, C. Sun, X. Yin, J. Li, X. Tan, P. Wang, Y. Wang, D. Fang, Q. Cui, P. Yang, C. He, H. Jiang, C. Luo and Y. Xu, Structural insight into substrate preference for TET-mediated oxidation, Nature, 2015, 527, 118–122 CrossRef CAS PubMed.
- L. T. Da, Y. Shi, G. Ning and J. Yu, Dynamics of the excised base release in thymine DNA glycosylase during DNA repair process, Nucleic Acids Res., 2018, 46, 568–581 CrossRef CAS PubMed.
- D. Xu, T. Landon, N. L. Greenbaum and M. O. Fenley, The electrostatic characteristics of G·U wobble base pairs, Nucleic Acids Res., 2007, 35, 3836–3847 CrossRef CAS PubMed.
- B. Xia, D. Han, X. Lu, Z. Sun, A. Zhou, Q. Yin, H. Zeng, M. Liu, X. Jiang, W. Xie, C. He and C. Yi, Bisulfite-free, base-resolution analysis of 5-formylcytosine at the genome scale, Nat. Methods, 2015, 12, 1047–1050 CrossRef CAS PubMed.
- A. R. Weber, C. Krawczyk, A. B. Robertson, A. Kuśnierczyk, C. B. Vågbø, D. Schuermann, A. Klungland and P. Schär, Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism, Nat. Commun., 2016, 7, 10806–10818 CrossRef CAS PubMed.
- Y. Mishina and C. He, Probing the structure and function of the Escherichia coli DNA alkylation repair AlkB protein through chemical cross-linking, J. Am. Chem. Soc., 2003, 125, 8730–8731 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Coot: model-building tools for molecular graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- W. L. DeLano, Use of PYMOL as a communications tool for molecular science, Am. Chem. Soc., 2004, 228, 313–314 Search PubMed.
- X. J. Lu and W. K. Olson, 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures, Nucleic Acids Res., 2003, 31, 5108–5121 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09, revision A. 2, 2009 Search PubMed.
- M. Zgarbova, J. Sponer, M. Otyepka, T. E. Cheatham 3rd, R. Galindo-Murillo and P. Jurecka, Refinement of the Sugar-Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA, J. Chem. Theory Comput., 2015, 11, 5723–5736 CrossRef CAS PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N log (N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, LINCS: a linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S.-e. Liu, J.-c. Hu, H. Zhang, P. Xu, W. Wan, M.-y. Zheng, K.-q. Yu, H. Ding, H.-l. Jiang, L. Zhou and C. Luo, Conformation and dynamics of the C-terminal region in human phosphoglycerate mutase 1, Acta Pharmacol. Sin., 2017, 38, 1673–1682 CrossRef CAS PubMed.
- L. Li, C. Li, S. Sarkar, J. Zhang, S. Witham, Z. Zhang, L. Wang, N. Smith, M. Petukh and E. Alexov, DelPhi: a comprehensive suite for DelPhi software and associated resources, BMC Biophys., 2012, 5, 9 CrossRef PubMed.
- R. Rohs, S. M. West, A. Sosinsky, P. Liu, R. S. Mann and B. Honig, The role of DNA shape in protein–DNA recognition, Nature, 2009, 461, 1248–1253 CrossRef CAS PubMed.
- R. Joshi, J. M. Passner, R. Rohs, R. Jain, A. Sosinsky, M. A. Crickmore, V. Jacob, A. K. Aggarwal, B. Honig and R. S. Mann, Cell, 2007, 131, 530–543 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02807b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |