
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormamide catalyzed activation of carboxylic acids – versatile and cost-efficient amidation and esterification†
Peter H.
Huy
 * and
Christelle
Mbouhom
* and
Christelle
Mbouhom
Saarland University, Institute of Organic Chemistry, P. O. Box 151150, D-66041 Saarbruecken, Germany. Web: https://www.peterhuylab.de/E-mail: peter.huy@uni-saarland.de
First published on 17th June 2019
Abstract
A novel, broadly applicable method for amide C–N and ester C–O bond formation is presented based on formylpyrrolidine (FPyr) as a Lewis base catalyst. Herein, trichlorotriazine (TCT), which is the most cost-efficient reagent for OH-group activation, was employed in amounts of ≤40 mol% with respect to the starting material (100 mol%). The new approach is distinguished by excellent cost-efficiency, waste-balance (E-factor down to 3) and scalability (up to >80 g). Moreover, high levels of functional group compatibility, which includes acid-labile acetals and silyl ethers, are demonstrated and even peptide C–N bonds can be formed. In comparison to reported amidation procedures using TCT, yields are considerably improved (for instance from 26 to 91%) and esterification is facilitated for the first time in synthetically useful yields. These significant enhancements are rationalized by activation by means of acid chlorides instead of less electrophilic acid anhydride intermediates.
Introduction
Amide and ester C–N and C–O bond formations account for the most important and frequently performed transformations in chemistry.1,2 Both functional groups are ubiquitous as in peptides and proteins, synthetic polymers and pharmaceuticals, for instance. The majority of all amides 2 and esters 3 are synthesized by the straightforward condensation of carboxylic acids 1 with N- and O-nucleophiles (Scheme 1A), which is referred to as amidation and esterification, respectively.1,2 Here, conventional reagents are employed to convert the hydroxy group of 1 into a decent leaving group, which facilitates subsequent nucleophilic substitution. As a consequence, waste by-products are formed, which typically results in poor sustainability and cost-efficiency.3 | ||
| Scheme 1 Previous and this work. | ||
 | ||
| Fig. 1 (A) Amidation and esterification of carboxylic acids and (B) comparison of costs for various reagents for this transformation with respect to the converted substrate (prices were obtained from Sigma Aldrich, see Chapter 1.2 in the ESI† for more details). | ||
Intriguing protocols for amidation without the necessity of reagents have been recently established based on boron,4–7 hafnium and zirconium8 Lewis acid catalysts, for example.1e,g–k,m The latest generation of catalysts such as the remarkable dioxoazatriborinane of Shibasaki and Kumagai, which is prepared in five steps, allows even for the synthesis of amides derived from challenging aromatic and sterically encumbered aliphatic carboxylic acids.6,7
Despite the significant progress accomplished with regard to catalytic C–N and C–O1e bond formation, not all esters and amides are accessible by means of catalysis with high levels of efficiency. In fact, a united appeal of several leading pharmaceutical companies evidences a highly urgent demand for the development of novel methods for amidation.9 A comparison of some of the most common reagents for stoichiometric OH group activation of carboxylic acids and alcohols, for instance, is compiled in Scheme 1B.10 Indeed, trichlorotriazine (TCT),11 which is also called cyanuric chloride, emerges as the least expensive reagent besides phosgene (COCl2).12 When TCT is used in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry with regard to the hydroxyl group bearing starting material, the costs are even lower than those when using common coupling reagents such as thionyl chloride (SOCl2), iso-butyl chloroformate (IBCF), oxalyl chloride (C2O2Cl2) and dicyclohexylcarbodiimide (DCC). Other frequently used coupling reagents like Ghosez's chloro enamine13 and T3P14 are associated unavoidably with significantly higher costs. In contrast to oxalyl and thionyl chloride and phosgene, the application of TCT in addition prevents the formation of HCl as the by-product, which should enable an enhanced functional group compatibility.
:1 stoichiometry with regard to the hydroxyl group bearing starting material, the costs are even lower than those when using common coupling reagents such as thionyl chloride (SOCl2), iso-butyl chloroformate (IBCF), oxalyl chloride (C2O2Cl2) and dicyclohexylcarbodiimide (DCC). Other frequently used coupling reagents like Ghosez's chloro enamine13 and T3P14 are associated unavoidably with significantly higher costs. In contrast to oxalyl and thionyl chloride and phosgene, the application of TCT in addition prevents the formation of HCl as the by-product, which should enable an enhanced functional group compatibility.
In fact, TCT (33–40 mol%) in conjunction with NMM (N-methylmorpholine) has been exploited for the synthesis of secondary amides 2, diazoketones 4, acyl azides 5, and hydroxamic acids 2 (R2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OH, Scheme 1A).15,16 However, the yields are often moderate (e.g. for secondary amides 65–75%)15a and esterification has rarely been reported.16 Thereby, activation of 1 is not realized through conversion to acid chlorides 6,15a,c which could also be confirmed by experiments in this study (vide infra). Instead, mixed anhydrides of type I have been proposed as intermediates.15
OH, Scheme 1A).15,16 However, the yields are often moderate (e.g. for secondary amides 65–75%)15a and esterification has rarely been reported.16 Thereby, activation of 1 is not realized through conversion to acid chlorides 6,15a,c which could also be confirmed by experiments in this study (vide infra). Instead, mixed anhydrides of type I have been proposed as intermediates.15
Recently, we discovered that N-formylpyrrolidine (FPyr) is a potent Lewis base catalyst17 for the transformation of alcohols 7 into alkyl chlorides 8, which is mediated by either benzoyl chloride or TCT (Scheme 1B).18,19
Furthermore, substituted cyclopropenones and tropone, the latter even in catalytic quantities, have been employed as Lewis bases by the groups of Lambert and Nguyen in the production of acid chlorides, amides and esters with C2O2Cl2.20
Against this background, considering the excellent cost-efficiency of TCT, we envisioned the opportunity to develop a novel method for the synthesis of esters and amides via formamide catalyzed acid chloride generation (Scheme 1C). Since chlorides of type 6 are more reactive towards nucleophiles than anhydrides such as I, we expected a much broader applicability and significantly enhanced yields. Herein, a novel method for low-cost chlorination, amidation and esterification of carboxylic acids based on FPyr and TCT is disclosed.
Results and discussion
Optimization of reaction conditions
While the full details of the optimization of the reaction conditions are provided in the ESI† (Chapter 3), only the essential findings are discussed in the following based on the synthesis of a model amide 21a (Table 1), which is a frequently used test substrate for CH-activation.21 In terms of the solvent, MeCN was identified as optimal (entry 1). Nevertheless, the utilization of more environment-friendly EtOAc furnished amide 21a in a similar yield (entry 2). With respect to the transformation of 1 → 6, heating to 80 °C results in conveniently short reaction durations of ≤4 h (entries 1 + 2). However, the reaction temperature can be decreased to 40 °C, when 20 mol% FPyr is employed instead of 10 mol% (entry 3).
As an important finding, the activation of aromatic acids 1 can also be achieved at room temperature, when MeCN is used as the solvent (entry 4). Moreover, the catalyst loading can be minimized to 5 mol% and instead of FPyr a two-fold amount of DMF is also feasible (entries 5 + 6).
In general, aliphatic acids are more reactive than aromatic ones, which enables lower reaction temperatures in the former case (rt to 40 °C). Indeed, a low TCT amount of around 40 mol% is pivotal for one-pot condensation in high yield. For instance, an increase to 60 mol% resulted in a decrease of the yield in the preparation of 2-naphthyl phenyl acetate from 93 to 68%.
Screening of many Lewis bases for the transformation of acids 1 into acid chlorides 6 including OPPh3, DMSO, tropone and cyclopropenone derivatives revealed FPyr as the most effective catalyst by far (Table S5, ESI†). This is surprising because some of the probed Lewis bases have been employed in related chlorination of carboxylic acids with oxalyl chloride.20 In comparison to alcohols,18b the conversion of carboxylic acids requires higher TCT amounts and increased reaction times. This is rationalized probably by a lower thermodynamic driving force, because the conjugation energy between the hydroxyl substituent and the carbonyl group in 1 is lost.
Pleasingly, for the formation of amides 2 and esters 3 from acid chloride intermediates 6, one equivalent of base is adequate, albeit in some cases yields are improved by using two. Even inexpensive K2CO3 is suitable for the synthesis of amides (entry 7), although the utilization of NMM or NEt3 sometimes causes enhanced yields (entries 1 + 8). For the synthesis of esters 3 derived from aromatic alcohols, the aforementioned amine bases are recommended (Tables S13–S16, ESI†). For the condensation of weakly nucleophilic aliphatic alcohols with substrates 1 in good yields, the addition of catalytic amounts of DMAP22 proved to be crucial (4-dimethylaminopyridine, Tables S17 and S18, ESI†). Finally, in the absence of FPyr, the product 21a was obtained in a very low yield of 11%, which proves the crucial role of FPyr as the catalyst (entry 9).
In fact, the solvent volume has a major impact on both scalability and sustainability.3 Fortunately, chlorination 1 → 6 can be performed utilizing very low solvent amounts ([1] = 2–4 M), which also results in shorter reaction durations. Due to the successive addition of nucleophiles and bases, the solvent amount has to be increased. However, in most cases a concentration of 1 M with respect to the starting material 1 is sufficient to ensure stirrability. Recommendations for reaction conditions depending on the starting materials are concluded in Scheme 3A for the chlorination step 1 → 6 and in Scheme 5A for the subsequent condensation 6 → 2/3. As a support for the practitioner, we also included a more detailed reaction condition guide in Chapter 5.2 in the ESI.†
Synthesis of acid chlorides
In clear contrast to preceding literature protocols,15 carboxylic acid chlorides of type 6 are generated as intermediates, which can be monitored by NMR spectroscopy (Chapter 3.1.6, ESI†). Simultaneously, reversible formation of the respective acid anhydride was observed. In order to unambiguously verify that the activation of 1 is accomplished by means of chlorination, four different acid chlorides were synthesized on a multigram scale and isolated after distillation in 79–90% yield (Scheme 2). | ||
| Scheme 2 Synthesis of carboxylic acid chlorides 6. | ||
Therefore, it is important to heat the reaction mixture to 80 °C in order to decompose FPyr entirely, which prevents codistillation with the product. During the reactions, a solid precipitates. Isolation through filtration and drying delivered a residue with a mass of ca. 80% of the theoretical yield of cyanuric acid (CA). Indeed, 13C-NMR confirmed CA as the major component, which proves its role as the main by-product. In Scheme 2, the yields for CA are provided after purification by means of precipitation with hydrochloric acid from a basic solution.
As an important aspect, a good waste balance is attested through E-factors as low as 2.8, which is the ratio of the mass of waste with respect to the mass of isolated products according to Sheldon.3 Remarkably, this beneficial E-factor is in a range typical for the production of bulk chemicals (1.5–5),3 which is explained by the low solvent and reagent amounts necessary. It is worth noting that in the absence of FPyr, even heating to 80 °C did basically not induce the formation of acid chlorides, which emphasises the importance of formamide catalysis. The addition of Brønsted bases has a deteriorative impact on the yield, in particular when acids bearing α-H-atoms are used (see Table S7, ESI.†)
Synthesis of amides
Initially, we investigated the substrate scope in terms of the synthesis of amides 2 (Scheme 3). A variety of aliphatic carboxylic acids bearing a primary (1°), secondary (2°) and tertiary (3°) α-carbon atom were transformed into the respective amides (Scheme 3B). Among them even dimethylmalonic acid could be coupled with two equivalents of phenyl glycinol to afford diamide 29i. In addition, amides deduced from electron-rich and poor (hetero)aromatic carboxylic acids are accessible in good to excellent yields (Scheme 3C). The method is also applicable to α,β-unsaturated substrates, wherein in example 216b only little isomerisation occurred (dr = 97:3, Scheme 3D). In cases where the nucleophile is considered as the more valuable building block, the carboxylic acid 1 can be used in excess. For instance, the amide 216b was synthesized from 1.3 equiv. of crotonic acid and 1.0 equiv. of ephedrine.
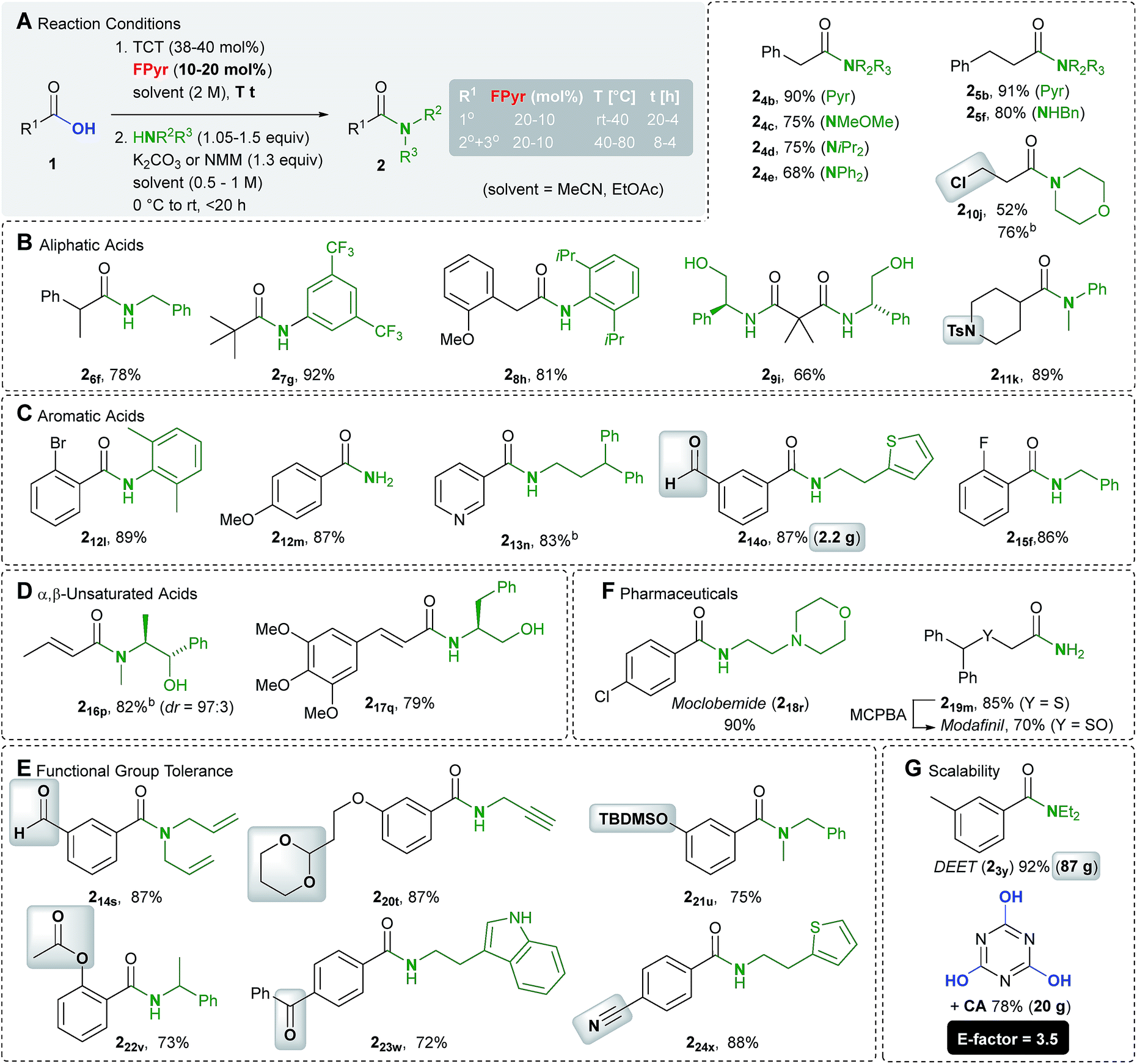 | ||
| Scheme 3 Synthesis of amides of type 2. aIsolated yield after chromatography or distillation if not mentioned otherwise. bYield determined by means of an internal NMR standard. cWith 1.3 equiv. 1, 49 mol% TCT, 1.7 equiv. NMM and 1.0 equiv. nucleophile. Pyr = N-pyrrolidyl, Ts = para-tolylsulfonyl, and TBDMS = tert-butyldimethylsilyl. | ||
Of particular significance is the functional group compatibility in the initial transformation of acids 1 into chlorides 6 (Scheme 3F). Since HCl is not released in stoichiometric quantities, even acid sensitive cyclic acetal and silyl ether functional groups are tolerated (examples 220t and 221u). Moreover, cyano and ester groups, ketones, aldehydes and chloro alkanes (example 210j) are compatible. Examples of functional groups for the production of esters also include carbamates (Scheme 5D).
To point out the synthetic value of our approach, the drugs moclobemide and modafinil have been prepared (Scheme 3F). Eventually, significant scalability is proven by the synthesis of the insect repellent DEET on a 500 mmol scale, for which standard glassware ≤1 L is sufficient (Scheme 3G). An E-factor of 3.5 denotes a reasonable waste balance for the determination of which, isolated cyanuric acid was also considered. In fact, in all other examples (214a, 330c and 331d) prepared on a multigram scale, environmentally benign EtOAc was employed as the solvent, too.
The scope of amine nucleophiles is diverse as well. Scheme 3 includes examples derived from various primary and secondary amines, electron poor and sterically encumbered, weakly nucleophilic aniline derivatives (examples 27g and 28h). In addition, the application of a plain aqueous ammonia solution enabled the synthesis of primary amides (examples 212m and 219m). As documented in the synthesis of the Weinreb amide 24c, hydrochloride salts of amines can be used as nucleophiles, which requires one additional equivalent of base.
Although β-amino acids are suitable starting materials (Scheme 5D, example 335j), the reaction of carbamate protected α-amino acid derivative 125 with TCT in the presence of FPyr yielded carboxamide 1025 (Scheme 4A). This outcome is most likely reasoned by the formation of the cyclic intermediate II25 after the activation of the acid functionality and subsequent nucleophilic substitution in the benzylic position by the chloride counterion.23
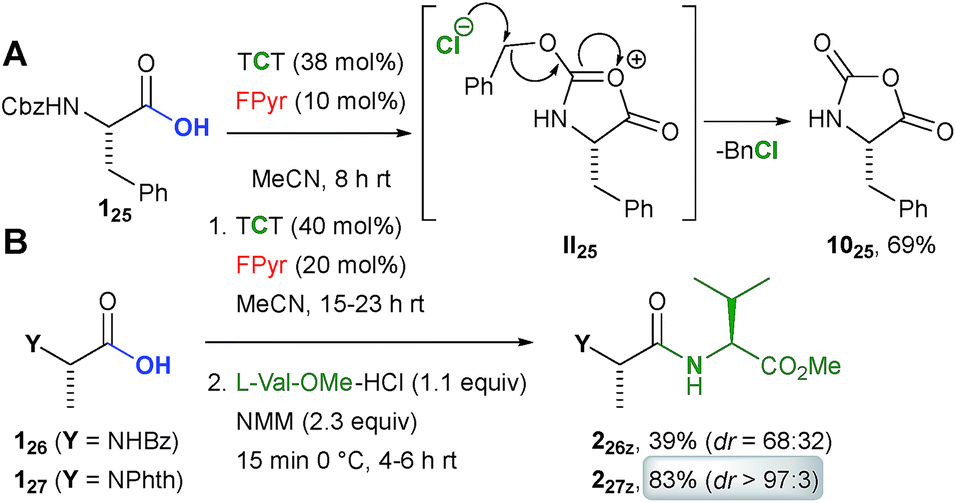 | ||
| Scheme 4 α-Amino acid derivatives. Phth = phthaloyl. | ||
An amide protected alanine derivative was converted into the dipeptide 226z in moderate yield under epimerization (Scheme 4B). Variation of the reaction conditions could neither enhance the yield nor the dr to a significant extent (see the ESI†). Gratifyingly, the dipeptide 227z was obtained from condensation with phthaloyl protected alanine 126 in an excellent yield as a virtually pure diastereomer.
Thus, the present protocol even allows for peptide C–N bond formation, when phthaloyl protected amino acids are employed. In comparison to other amino acid chlorides,1 phthaloyl protected analogues are less prone to racemisation.24 Against this background, we would also like to highlight recent contributions to boron Lewis acid catalyzed synthesis of peptides.5–7
Synthesis of esters
Due to the lower nucleophilicity of alcohols, the synthesis of esters of type 3 is more challenging. This is most probably also the reason why esters have been rarely prepared by using TCT.16 Fortunately, the present catalytic method facilitates the synthesis of esters derived from both aromatic and aliphatic alcohols, in 73–89% yield (Scheme 5B–D).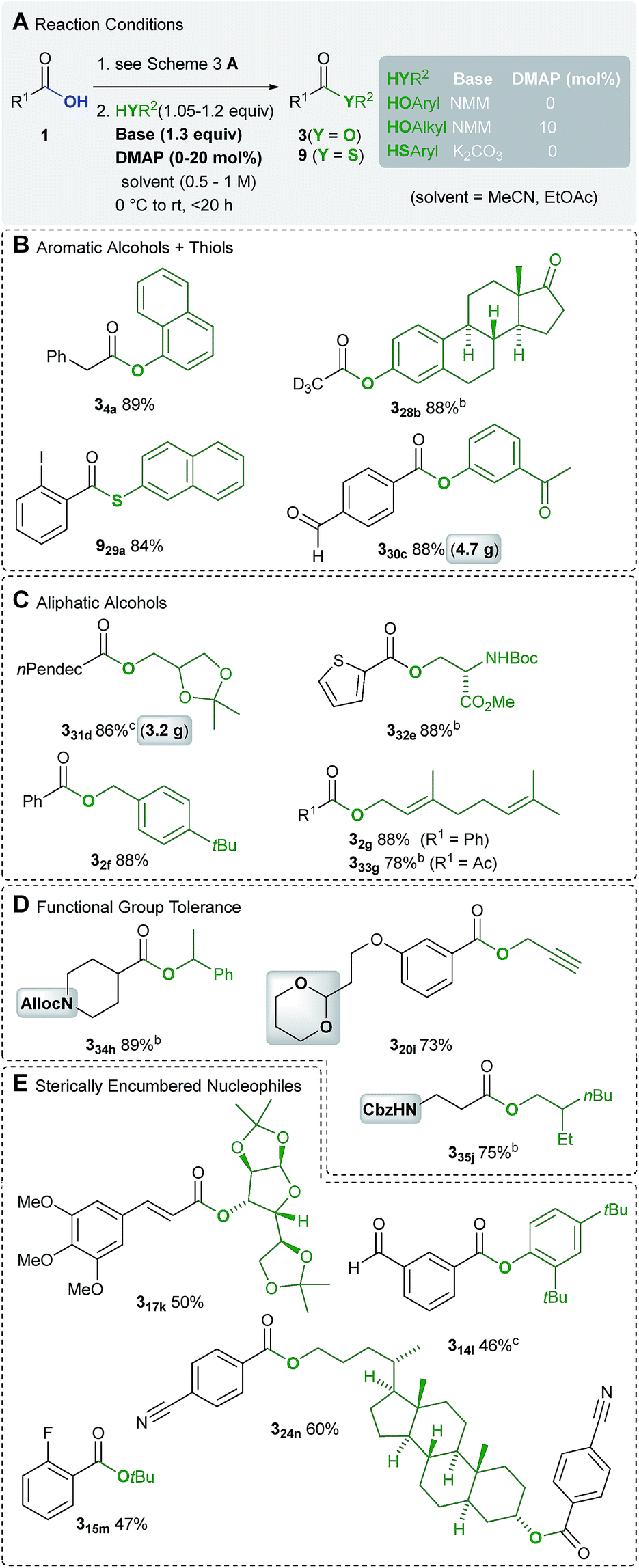 | ||
| Scheme 5 Synthesis of esters. aIsolated yield after chromatography. bWith 1.3 equiv. 1, 49 mol% TCT, 1.7 equiv. NMM and 1.0 equiv. nucleophile. cWith 1.1 equiv. 1, 44 mol% TCT, 2.3 equiv. NMM and 1.0 equiv. nucleophile. | ||
This also includes two examples on a 3–5 g scale, which certifies a high degree of practicability (examples 330c and 331d). Since acid chlorides are not isolated, the estrone derivative 328b could be obtained via volatile trideutero acetyl chloride. Because of higher nucleophilicity, 2-naphthalene thiol could be condensed with 2-iodobenzoic acid using K2CO3 instead of NMM (example 929a). Esterification of sterically demanding alcohols with acid chlorides is usually accomplished using strong bases such as nBuLi.25 Hence, the current method provides esters derived from sterically encumbered alcohols in moderate yields of 47–60% (Scheme 5D).
Comparison experiments
In order to compare our approach with a literature protocol using TCT and NMM (Scheme 1A),15a several amides and esters were prepared accordingly (also see Chapter 4, ESI†). Remarkably, in the event of four exemplary amides, the differences between the yield of the reported protocol and the current catalytic method are in the range of 21 and 65% (Scheme 6A). When K2CO3 was used as the base instead of NMM, the amides 24b and 21a were obtained in even more decreased yields ≤17% (88–90% in this method). | ||
| Scheme 6 Comparison of the present method with the literature method15a employing TCT and NMM. aFor reaction conditions see Schemes 3 and 5; yields refer to the isolated material after chromatographic purification or distillation. bFor reaction conditions see Scheme 1A and Chapter 4, ESI;† yield determined with an internal NMR standard. | ||
With 8–58% the yields for the preparation of esters based on ordinary TCT/NMM-activation15a must be classified as synthetically non-useful (current method, 86–89%, Scheme 6B). Utilization of DMAP, which has not been described in the literature, allows improvement of the yield of the ester 331d (8 → 50%), whereas for the example 330c a slightly decreased yield was obtained (42 → 36%). It should be emphasized that we optimized the literature conditions thoroughly in order to allow a meaningful comparison (see Tables S19–S22, ESI†). The significantly improved yields testify our concept that acid chloride activation is superior to anhydride formation.
Proposed mechanism
In agreement with our previous work on catalytic chlorination of alcohols,18 the related transformation of carboxylic acids 1 into acid chlorides 6 is most likely initiated by nucleophilic aromatic substitution (SNAr) using TCT and FPyr (Scheme 7A). The fact that chloride substitution on a triazine scaffold proceeds via a stepwise SNAr mechanism has been demonstrated previously.26,27 Next, the plausible intermediate III, which shows structural similarities to the Vilsmeier–Haack reagent, should undergo replacement of the triazine moiety through the substrate 1. In the following, the resulting intermediate IV would surpass substitution with the chloride counterion to afford the product 6 and regenerate FPyr. Dichlorohydroxytriazine (V with X = Cl) results from the conversion of III with 1 to IV. As verified through the formation of CA and the low stoichiometry of TCT (40 mol% → 1.2 equiv., vide supra), V (X = Cl) undergoes two consecutive reactions with FPyr until all chlorine atoms are replaced.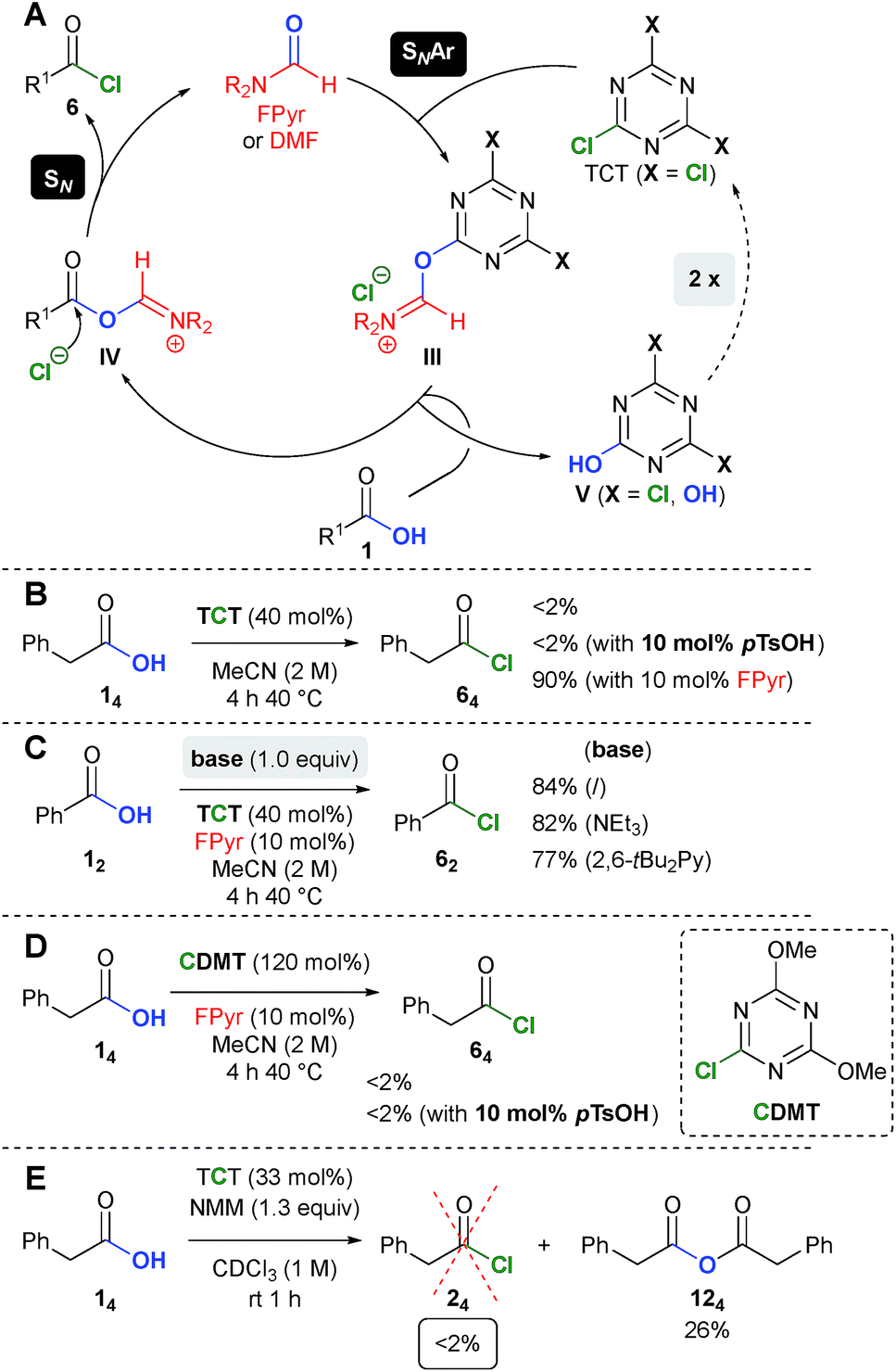 | ||
| Scheme 7 (A) Proposed mechanism and (B–E) comparison experiments. Yields were determined using an internal NMR standard. | ||
The fact that alkoxy iminium salts derived from alcohols have been previously proven by us,18a supports related carboxyl iminium salts of type IV as key intermediates. Preliminary mechanistic elucidations according to the normalized time scale method of Burés28 revealed a reaction order of 1 in FPyr (Chp. 3.1.6, ESI†), which is in agreement with the proposed mechanism.
Remarkably, in the absence of FPyr, no phenylacetyl chloride (64) is formed at all (Scheme 7B). As an important finding, para-tolylsulfonic acid (pTsOH) does not promote the production of 64, which rules out Brønsted acid catalysis. Furthermore, Brønsted acid cocatalysis can also be excluded, since the conversion of benzoic acid into benzoyl chloride is virtually not inhibited by bases such as NEt3 and 2,6-bis-tert-butylpyridine (Scheme 7C, Table S7, ESI†).
The reaction of phenyl acetic acid (14) with CDMT, which is closely related to intermediate V (X = OH), does not give rise to the respective acid chloride 64 (Scheme 7C, also see Table S8, ESI†). Thus, the reason for the replacement of the last chlorine atom in V (X = OH) is currently unclear (Scheme 7D).
The fact that activation of carboxylic acids with NMM and TCT does not yield acid chlorides of type 615a,c was confirmed experimentally (Scheme 7E, see Table S23, ESI†). The proposed intermediate I (see Scheme 1), which might be insoluble in CDCl3, was also not observed. Instead, anhydride 124 was obtained in 26% yield, which provides an additional explanation for the low yields in the comparison experiments (vide supra).
Conclusions
In summary, a versatile formamide catalyzed method for the transformation of carboxylic acids into acid chlorides, amides, esters and thioesters has been developed. For this, cyanuric chloride (TCT), which is the cheapest reagent for OH group activation (alongside phosgene), is used as a promoter in quantities ≤40 mol%. As a consequence, the presented protocol exhibits (1) an exceptional cost-efficiency. Additional important features are high levels of (2) scalability (>80 g) and (3) functional group compatibility including acid labile acetals and silyl ethers; (4) a reasonable waste-balance (E-factor down to 3) and (5) operational simplicity, since non-dry reaction conditions are viable. Remarkably, the formation of peptidic C–N bonds is possible when phthaloyl protected amino acids are employed.In comparison to ordinary protocols using TCT, not only the yields of amidation are significantly improved (e.g. 91% instead of 26%), but also esterification is enabled in synthetically useful yields for the first time. The significant advancements are explained by the formation of carboxylic acid chlorides as intermediates instead of less electrophilic anhydrides. Finally, high levels of applicability were testified by the preparation of the pharmaceuticals moclobemide and modafinil and the insecticide DEET. Considering the manifold enhancements, rapid application of the current method in academic and industrial laboratories is expected.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the German Research Foundation (DFG) and the Fonds of the Chemical Industry (Liebig fellowship for P. H.) for generous support. In addition, we would like to thank Rudolf Thomes and Dr Klaus Hollemeyer for HR-MS measurements and Dr Josef Zapp for the measurement of high temperature NMR spectra.Notes and references
- Reviews on amidation:
(a)
T. Ziegler, in Science of Synthesis, ed. S. M. Weinreb, Georg Thieme, Stuttgart, 2005, vol. 21, pp. 43–77 Search PubMed
; (b) E. Haslam, Tetrahedron, 1980, 36, 2409 CrossRef CAS
- Reviews on esterification:
(a) E. Haslam, Tetrahedron, 1980, 36, 2409 CrossRef CAS
- Selected reviews regarding sustainability:
(a) R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC
- Reviews on boron Lewis acid catalysis:
(a) H. Charville, D. Jackson, G. Hodgesc and A. Whiting, Chem. Commun., 2010, 46, 1813 RSC
- Seminal publications:
(a) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196 CrossRef CAS
-
(a) H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Nat. Chem., 2017, 9, 471 CrossRef
- For other leading boron Lewis acid catalyzed methods for the synthesis of challenging amides see:
(a) K. Ishihara and Y. Lu, Chem. Sci., 2016, 7, 1276 RSC
- For example see:
(a) H. Lundberg, F. Tinnis and H. Adolfsson, Chem.–Eur. J., 2012, 18, 3822 CrossRef CAS
- A round-table of representatives of several pharmaceutical companies repeatedly requested more efficient methods for amidation as the primary target:
(a) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaksh and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC
- For the cost assessment for each reagent the lowest price from Sigma-Aldrich was selected. We are fully aware that the costs would drop significantly on an industrial scale. Nevertheless, we are convinced that the current study allows for a relative ranking with respect to cost-efficiency. For more details, see Chapter 1.2 in the ESI.†.
- Reviews on TCT:
(a) S. S. Voyutskii and V. L. Vakula, Russ. Chem. Rev., 1964, 92–103 Search PubMed
- At Sigma-Aldrich, phosgene is only available as a relatively costly solution and is therefore not included in Fig. 1. As carbonyl dichloride is readily prepared from CO and Cl2, the real costs are likely to be significantly lower..
-
(a) A. Devos, J. Remion, A. M. Frisque-Hesbain, A. Colens and L. Ghosez, J. Chem. Soc., Chem. Commun., 1979, 1180 RSC
-
(a) H. Wissmann and H.-J. Kleiner, Angew. Chem., Int. Ed. Engl., 1980, 19, 133 CrossRef
-
(a) H. L. Rayle and L. Fellmeth, Org. Process Res. Dev., 1999, 3, 172 CrossRef CAS
- Acid chlorides, amides and esters have been prepared using TCT (50 mol%) and NEt3: K. Venkataraman and D. R. Wagle, Tetrahedron Lett., 1979, 20, 3037 CrossRef
- Reviews on Lewis base catalysis in nucleophilic substitution:
(a) J. An, R. M. Denton, T. H. Lambert and E. D. Nacsa, Org. Biomol. Chem., 2014, 12, 2993 RSC
-
(a) P. H. Huy, S. Motsch and S. M. Kappler, Angew. Chem., Int. Ed., 2016, 55, 10145 (
Angew. Chem.
, 2016
, 128
, 10300
) CrossRef CAS PubMed
- In addition, formamides such as dimethylformamide (DMF) are known to catalyse the chlorination of carboxylic acids using thionyl and oxalyl chloride, respectively:
(a) R. Richter and B. Tucker, Helv. Chim. Acta, 1959, 1653 Search PubMed
-
(a) D. J. Hardee, L. Kovalchuke and T. H. Lambert, J. Am. Chem. Soc., 2010, 132, 5002 CrossRef CAS
- See for instance:
(a) Y. Ano, M. Tobisu and N. Chatani, Org. Lett., 2012, 14, 354 CrossRef CAS PubMed
- For DMAP catalyzed amidation and esterification using DCC and Boc2O, respectively, see:
(a) B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522 CrossRef
-
N-Boc-amino acids have been converted to carboxamides upon activation of the C-terminus:
(a) S. Mobashery and M. Johnston, J. Org. Chem., 1985, 50, 2200 CrossRef CAS
- J. C. Sheehan, D. W. Chapman and R. W. Roth, J. Am. Chem. Soc., 1952, 71, 3823 Search PubMed
-
(a) E. M. Kaiser and R. A. Woodruff, J. Org. Chem., 1970, 35, 1198 CrossRef CAS
- Z. J. Kamiki, P. Paneth and J. Rudzinski, J. Org. Chem., 1998, 63, 4248 CrossRef
- For the substitution of all chlorine atoms on the triazine core of TCT through O- and N-nucleophiles, respectively, the reaction temperature has to be gradually increased from 0 to 60 °C (see ref. 11). This experimental observation is most probably rationalized by a decreasing reaction rate for the consecutive SNAr-substitutions in the order 1st > 2nd > 3rd Cl-atom, since each replacement introduces a new electron donating substituent. In contrast, the present method allows quantitative conversion of sterically not hindered acids such as dodecanoic acid at room temperature (see Table S3, entries 1 + 2 + 12, ESI†). Since 40 mol% of TCT (=1.2 equiv.) have been used, even the 3rd chlorine atom of TCT must have been partially replaced.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02126d |
| This journal is © The Royal Society of Chemistry 2019 |