
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective switching of multiple azobenzenes†
Andreas H.
Heindl
 ab,
Jonathan
Becker
c and
Hermann A.
Wegner
*ab
ab,
Jonathan
Becker
c and
Hermann A.
Wegner
*ab
aInstitute of Organic Chemistry, Justus Liebig University, Heinrich-Buff-Ring 17, 35392 Giessen, Germany. E-mail: hermann.a.wegner@org.chemie.uni-giessen.de
bCenter for Materials Research (LaMa), Justus-Liebig-University, Heinrich-Buff-Ring 16, 35392 Giessen, Germany
cInstitute of Inorganic and Analytical Chemistry, Justus Liebig University, Heinrich-Buff-Ring 17, 35392 Giessen, Germany
First published on 21st June 2019
Abstract
Multi-state photoswitchable compounds are highly attractive for application in data storage or multi-responsive materials. In this work, a trisazobenzene macrocycle capable of three-state isomerization is presented. The compound can be switched into each of the states with more than 70% of the isomer solely by light and heat as stimuli representing the first example for an oligo-azobenzene containing identical photochromic units which can be selectively adressed. Detailed spectroscopic, crystallographic, HPLC as well as computational investigations and the comparison to a less and a higher strained derivative revealed macrocyclic ring strain to be responsible for the compounds unique isomerization behavior.
Introduction
Photoswitchable compounds open a vast number of possibilities of influencing systems on the molecular level. Especially the intensively studied E–Z isomerization of azobenzene (AB)1,2 was utilized in many fields of research, reaching from photobiology3 to materials science4 or solar fuel applications.5 Thermally stable E-AB can be converted to the Z isomer by irradiation with UV light of ca. 350 nm, and the Z isomer converts back to the E conformation under irradiation at ca. 450 nm photochemically or thermally upon heating.1 Electronic effects are mainly responsible for the thermal half-lives of Z-ABs, which can be controlled by suitable functional group substitution of the phenyl rings. Moreover, subtle interactions, such as London dispersion in alkyl-substituted ABs, were also found to have a significant influence on the thermal Z-AB isomerization.6 Combining these approaches, ABs featuring thermal half-lives from milliseconds up to years can be designed.Integrating multiple chromophoric units in a single molecule opens the possibility of multistate switching with high information storage density. This approach drives photoswitches beyond a simple “on–off” behavior, which is especially interesting for the design of intelligent responsive materials. By combining two different chromophores with alternating properties, the complexity of the system rises, such as synthesis and characterization. Construction of multiswitches composed of equal photochromic units drastically increases design and application options relying, nevertheless, on the same principles. In the case of AB, the simplest approach is the linkage of two azo units at one shared phenyl ring to create AB dimers.7 Due to high electronic conjugation, ortho-bis(AB) features extremely high thermal Z → E relaxation (ms range). Furthermore, para-bis(AB) exhibits the highest conjugation and shows a dramatically lower excited state lifetime and low isomerization quantum yield. As a result, no (E,Z) or (Z,Z) isomers can be accumulated under standard conditions. In contrast, meta-bis(AB) features the lowest electronic coupling between the two azo groups and behaves almost like two isolated AB units.8 Several approaches of breaking the conjugation between p-bis-(AB) have been presented,9 resulting in multistate switchable AB compounds. However, in order to achieve multistate switching, differently substituted AB units must be incorporated, which results in higher complexity of the photoswitches.10 As a conclusion, multiphotochromic AB compounds, that can be addressed selectively are still exceptional.11
An alternative approach towards the control of the multiphotochromic behavior is the incorporation of AB units in macrocyclic arrangements, resulting in azobenzophanes.12 In these compounds, the isomer stability is significantly influenced by ring strain. Additionally, the incorporation of more than one AB unit leads to multiple isomerization states, which is especially interesting for information storage applications.13,14 The first azobenzophane featuring two AB units was already described by Rau in 1982.15 In further studies, a disulfide-bridged and electronically decoupled (Z,Z)-bisazobenzophane with remarkable high thermal stability of ∼400 days caused by macrocyclic strain is presented.16 The addition of even more ring strain reverses the thermodynamics of AB and causes the Z isomers to be more stable.17–20 Tamaoki and Norikane reported electronically decoupled, meta-methylene21 and para-ethylene22,23 linked azobenzophanes that showed full and stepwise isomerization. In the case of the meta-methyl bridged bisazobenzophane, all three possible states could be generated as predominant isomers, however, the (E,Z) state in rather moderate excess. Alternatively, adding chiral information into an azobenzophane also allowed triple state isomerization.24
Motivated by these results, we designed a series of symmetric bis-, tris- and tetraazobenzophanes 1–3, linked with dimethyl(methylene) spacers in para positions (Fig. 1). Herein, we present the synthesis and detailed investigation on the isomerization properties of these macrocycles. Additionally, trisazobenzophane 2 was found to feature reversible, triple-state isomerization with relative amounts of >75% of the corresponding isomers. Ring strain was revealed to be the origin of this unique multi-photochromic behavior, supported by X-ray crystallography, UV-vis and NMR spectroscopy, HPLC and density functional theory (DFT). The presented study contributes to a better understanding of azobenzophanes towards their applications as multistate photoswitches.
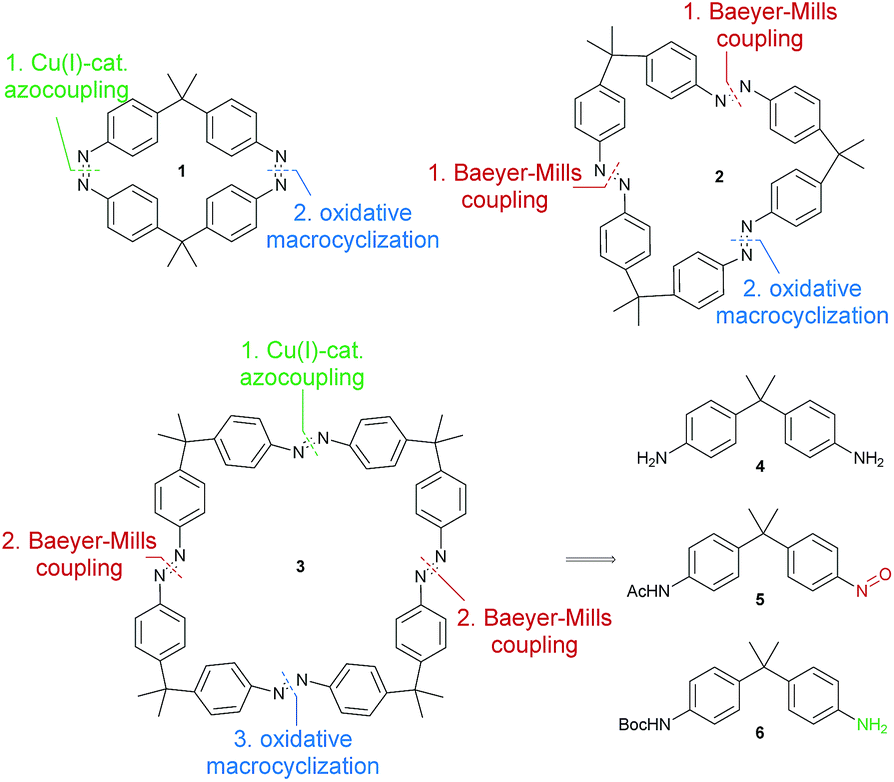 | ||
| Fig. 1 Retrosynthetic analysis of azobenzene macrocycles 1–3. | ||
Results and discussion
The syntheses of the azobenzene macrocycles were designed in a straight-forward manner relying on Baeyer–Mills reactions,25,26 Cu(I)-catalyzed oxidative couplings of anilines27 and oxidative macrocyclization reactions.19,28 The 2,2-bis(4-aminophenyl)propane starting material 4 can be readily obtained from aniline hydrochloride and acetone.29 After protection, the mono-nitroso compound 5 can be prepared via a convenient, mild oxidation method.30 For the symmetric Cu(I)-catalyzed oxidative azocoupling, the tert-butoxycarbonyl (Boc) protecting group has proven to be most effective in previous syntheses.19 Combining these general methods, all macrocycles with two, three and four azobenzene units can be accessed.With the general synthetic strategy in hand, (Z,Z)-cyclobisazobenzene 1 was prepared in four steps (Scheme 1). Starting from dianiline 4, which was previously synthesized on a multigram scale (see ESI†), the mono-Boc protected aniline 6 was obtained in 46% yield in the first step. After a Cu(I)-catalyzed oxidative azocoupling followed by deprotection, monoazo-dianiline 7 was synthesized in 83% yield over two steps. As previously reported,19 the yields of the macrocyclization of (Z,Z)-cyclobisazodiphenylmethane increased when the precursor solution is irradiated with UV light of 365 nm. Thereby, the Z-isomer of the precursor is accumulated and due to the closer proximity of the amino groups in the Z-form, the macrocyclization is favored. Using this approach, (Z,Z)-bisazo-macrocycle 1 was obtained in 19% yield.
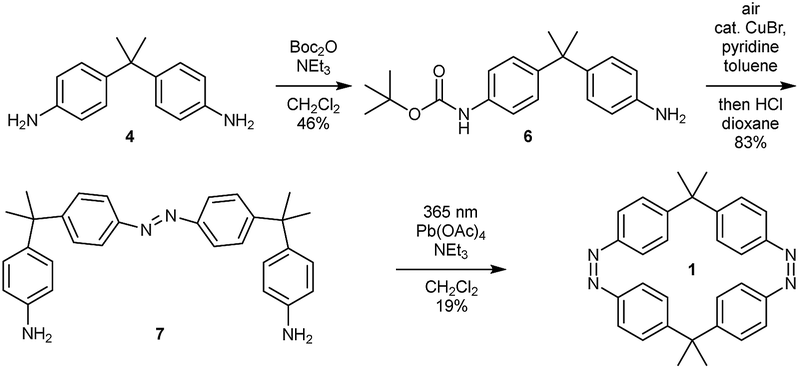 | ||
| Scheme 1 Synthesis of bisazo-macrocycle 1. | ||
For the synthesis of trisazomacrocycle 2, a double Baeyer–Mills coupling strategy was applied (Scheme 2). After mono-acetyl-protection of dianiline 4 and oxidation, nitroso compound 5 was obtained in good yield (79%). With mono-nitroso compound 5 in hand, and based on our results for one-pot preparations of bisazobenzenes,26 the same protocol was applied to synthesize bisazodianiline 9 with remarkably high efficiency on gram scale (97% yield over two steps). A one-pot double-azocoupling and deprotection reaction was applied, since the purification of the azocoupling product turned out to be ineffective due to its low solubility. The final macrocyclization step was challenging. Preliminary experiments showed that irradiation of the precursor solution gave slightly higher macrocyclization yields. Additionally, the reactions were carried out under pseudo-high dilution by the simultaneous dropwise addition of the precursor and the oxidant to a solution of base in dichloromethane. After the cyclization, workup and isolation was performed under ambient light in order to obtain the all-E isomer. Nevertheless, with these optimized conditions the macrocycle 2 could only be obtained in 3% yield. A rationale for this can be found in the high macrocyclic ring strain that has to be built up.
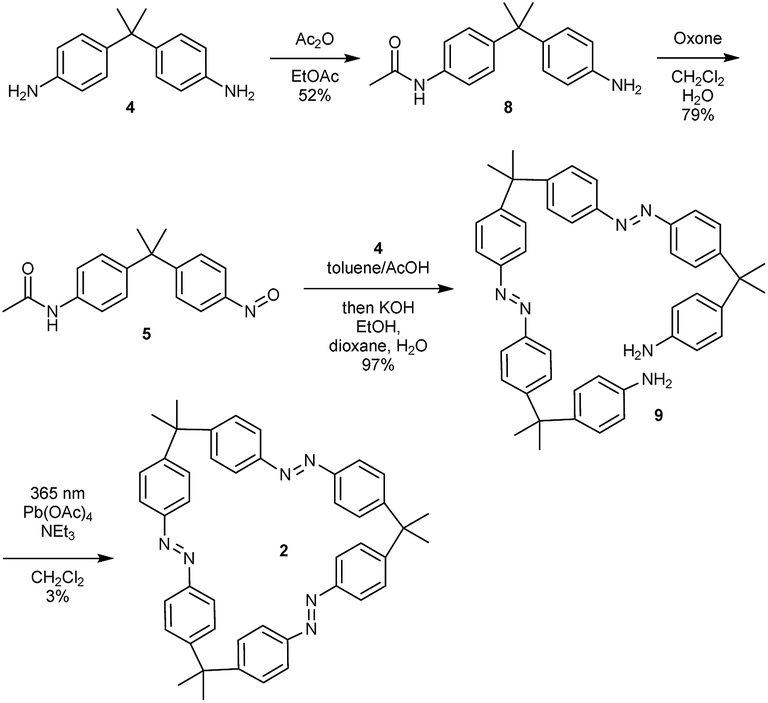 | ||
| Scheme 2 Synthesis of trisazo-macrocycle 2. | ||
Finally, the tetraazo-macrocycle 3 was synthesized via a double azocoupling–deprotection strategy, starting from the previously synthesized monoazo dianiline 7 and nitroso compound 5. In this reaction cascade, the macrocyclization precursor 10 was obtained in 49% yield over two steps. The final product was again obtained by an oxidative macrocyclization reaction under high dilution in 12% yield. After isolation and purification, it turned out that macrocycle 3 was insoluble in most solvents. It was slightly soluble in 1,1,2,2-tetrachloroethane (TCE, ∼1 mg mL−1) (Scheme 3).
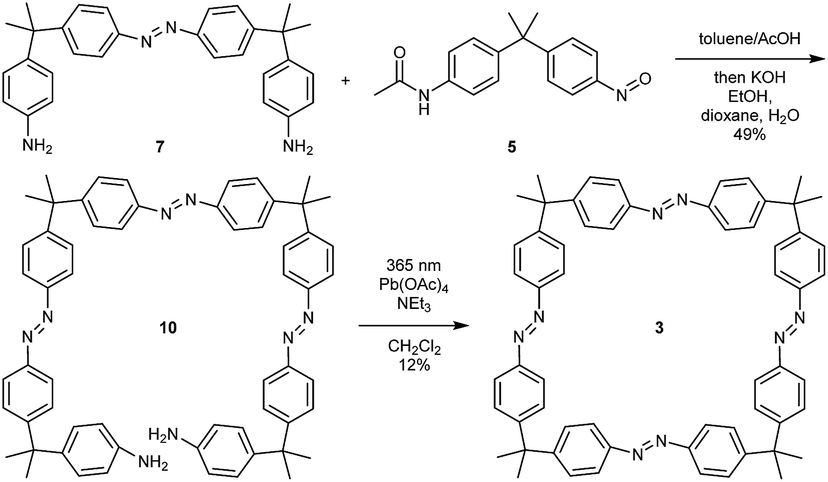 | ||
| Scheme 3 Synthesis of tetraazo-macrocycle 3. | ||
After the successful syntheses of compounds 1–3, single crystal X-ray diffraction was conducted for all macrocycles. In the case of bisazo-macrocycle 1, a very similar structure compared to cyclobisazodiphenylmethane,19 with almost ideal Z-azobenzene geometry, was obtained (see ESI, Fig. S11†). In the case of the trisazobenzophane 2, a distorted, triangular-shaped structure was found (Fig. 2). Furthermore, it became evident that the azobenzene units are bent out of the ideal planar geometry and thus feature considerable macrocyclic ring strain. On the other hand, the tetraazo-macrocycle 3 shows almost planar azobenzene units and thus features lower strain than the trisazo compound 2. In addition, the comparison of the sp3-carbon bridge angles mirrors the macrocyclic strain. Whereas the tetraazo-macrocycle 3 (aswell as the bisazobenzophane 1, see ESI†) features a value close to the ideal tetrahedral angle of 109.5°, the trisazo-macrocycle 2 deviates significantly with a smaller angle of 103°. Moreover, the higher distortion from the ideal geometry and, thus, higher ring strain in azobenzophane 2 is consistent with the lowest macrocyclization yield achieved.
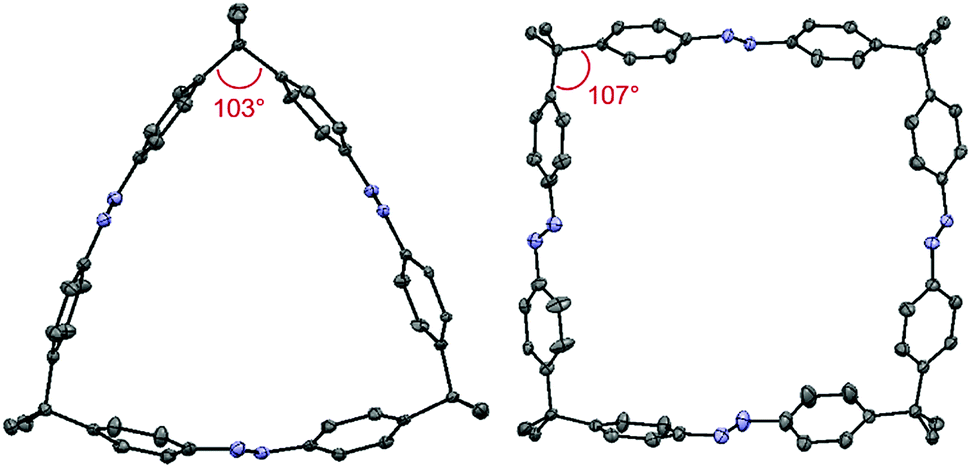 | ||
| Fig. 2 Molecular structures obtained by X-ray diffraction of macrocycles 2 (left) and 3 (right). Hydrogen atoms, disorder and solvent molecules are omitted for clarity. | ||
In order to get a deeper insight in the macrocyclic ring strain, isodesmic equations18,31 were used to compute ring strain energies of all possible isomers of the macrocycles (Table 1). As outlined in eqn (S1) (see ESI†), ΔEstrain is obtained by the difference of the electronic energies of the macrocycle and the sum of the corresponding numbers of 4-iPr-azobenzene and H2. As a result, the computations are consistent with the trend observed in the crystal structures: the highest ring strain of 18 kcal mol−1 per azo unit was found for (E,E,E)-2, in which the E-azobenzene moieties deviate most from their ideal geometry. Furthermore, the all-Z isomers of both macrocycles 2 and 3 featured less macrocyclic ring strain compared to their corresponding all-E isomers. These findings explain the higher macrocyclization yields when the staring materials were pre-irradiated, since less ring strain must be introduced during the reactions. Looking at all the possible isomers of macrocycles 1 and 3, it becomes obvious that the more azobenzene units exist in their E-conformation, the more macrocyclic ring strain is incorporated (see ESI, Tables S4–S6†).
| Compound | Isomer | ΔEstrain/kcal mol−1 | |
|---|---|---|---|
| Total | Per azo unit | ||
| 2 | (E,E,E) | 53 | 18 |
| (Z,Z,Z) | 40 | 14 | |
| 3 | (E,E,E,E) | 58 | 15 |
| (Z,Z,Z,Z) | 45 | 11 | |
In the most extreme case, cyclobisazobenzene 1, strain even reverses the thermodynamic stability of its isomers and forces the system to its (Z,Z) form. Analogously to cyclobisazodiphenylmethane, the (E,Z) and (E,E) isomers were not observed on the considered timescale (see following sections). The compound might be able to isomerize, but the relaxation to the thermodynamically most stable (Z,Z)-form is expected to occur on the millisecond timescale, as it was previously observed for cyclobisazodiphenylmethane.20
The photophysical properties of macrocycles 1–3 were investigated using UV-vis spectroscopy (Fig. 3a). While both the tris- and tetraazo-macrocycles featured strong π,π* transition bands with maxima at 341 nm, the bisazo-macrocycle 1 showed a spectrum typically observed for a Z-azobenzene with drastically reduced absorption in this region. Furthermore, characteristic n,π* bands around 440 nm were found for all three azobenzophanes.
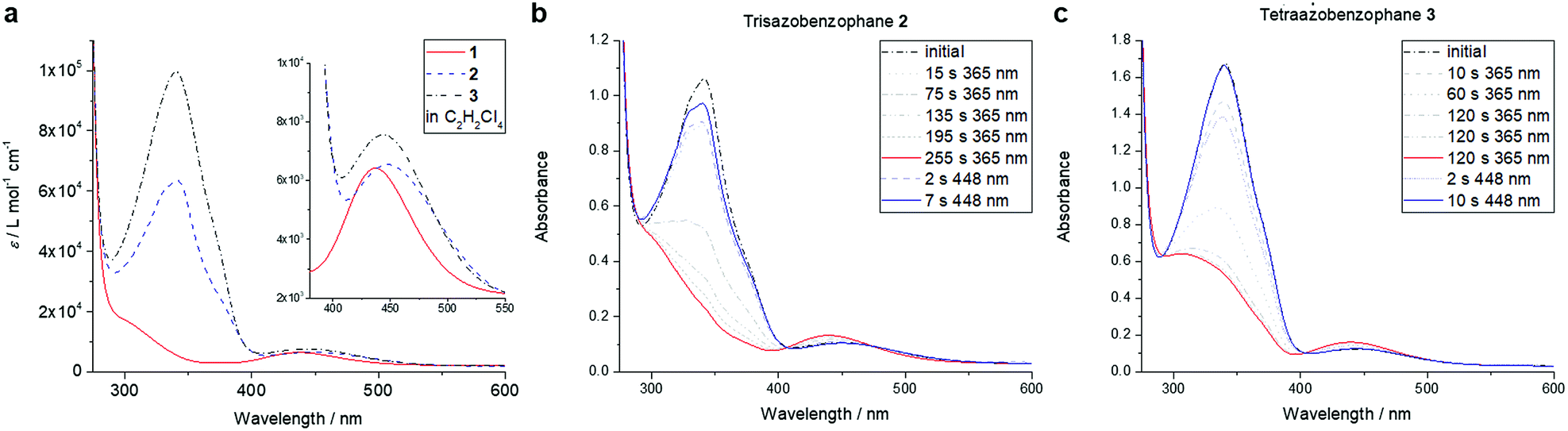 | ||
| Fig. 3 UV-vis spectra of azobenzophanes 1–3 in TCE (a). Photostationary states of macrocycles 2 (b) and 3 (c) in TCE (c = 1.6 × 10−5 mol L−1). | ||
Upon irradiation with high power LEDs at 365 nm, photostationary states (PSS) were reached after about 4 min in the case of 2 and after 7 min for 3, respectively (Fig. 3b and c). In this state, both macrocycles 2, 3 showed typical Z-azobenzene spectra with strongly decreased π,π* bands and increased, hypsochromically shifted n,π* absorptions. When irradiating the tetraazo-macrocycle 3 at 448 nm, the initial spectrum was restored within 10 s (Fig. 3c, blue spectrum). Interestingly, the initial spectrum of trisazobenzophane 2 could not be observed upon irradiation at this wavelength (Fig. 3b, blue spectrum). Furthermore, irradiation at 405, 425, 470 or 500 nm also could not restore the initial spectrum. As it was already expected, (Z,Z)-bisazobenzophane 1 did not isomerize upon irradiation with any wavelength at the observed timescale (see ESI, Fig. S7†).
1H NMR experiments were carried out assigning the isomeric ratio in the photostationary state (Fig. 4). At first, trisazo-macrocycle 2 was irradiated at different wavelengths and the resulting isomer mixtures were analyzed. By taking symmetry,14 COSY NMR spectroscopy and chemical shifts into consideration, the signals of all isomers were unambiguously assigned. As expected, the (E,E,E) as well as the (Z,Z,Z) isomers showed the most symmetric 1H NMR spectra with two signals in the aromatic region. Both the (E,E,Z) and (E,Z,Z) isomers showed six signals each due to their lower symmetry (Fig. 4a). Upon irradiation at 365 nm, a (Z,Z,Z) enriched state was obtained (73%, Fig. 4b). After heating to 60 °C for 23 h (exposed to ambient light), the (E,E,E) isomer predominated in the mixture, showing the reversibility of the isomerization process. However, still significant amounts (ca. 10%) of the (E,E,Z) and (E,Z,Z) isomers were detected. Interestingly, after irradiation at 385 nm, a PSS containing the (E,Z,Z) isomer as the main component in relative concentration of 47% was obtained. Upon irradiation at 448 nm, a PSS with roughly one third (E,E,E), (E,E,Z) and (E,Z,Z) of each isomer was generated. All in all, the NMR experiments showed that three different isomers of 2 could be generated selectively as the major components depending on the experimental conditions. Furthermore, the thermal isomerization of trisazobenzophane 2 was monitored by 1H NMR spectroscopy (Fig. 4c and d). Within the first three hours at 60 °C, the (Z,Z,Z) isomers concentration decreased almost completely, whereas the accumulation of the (E,Z,Z) isomer reached its maximum. Remarkably, this isomer showed the lowest isomerization rate (t1/2 = 10.7 h). In contrast, the (E,E,Z) → (E,E,E) conversion was found to be significantly faster than the other steps, as the quasistationary concentration of (E,Z,Z)-2 did not change during the experiment. All in all, the rate determining step of the complete process was observed for the (E,Z,Z) → (E,E,Z) isomerization.
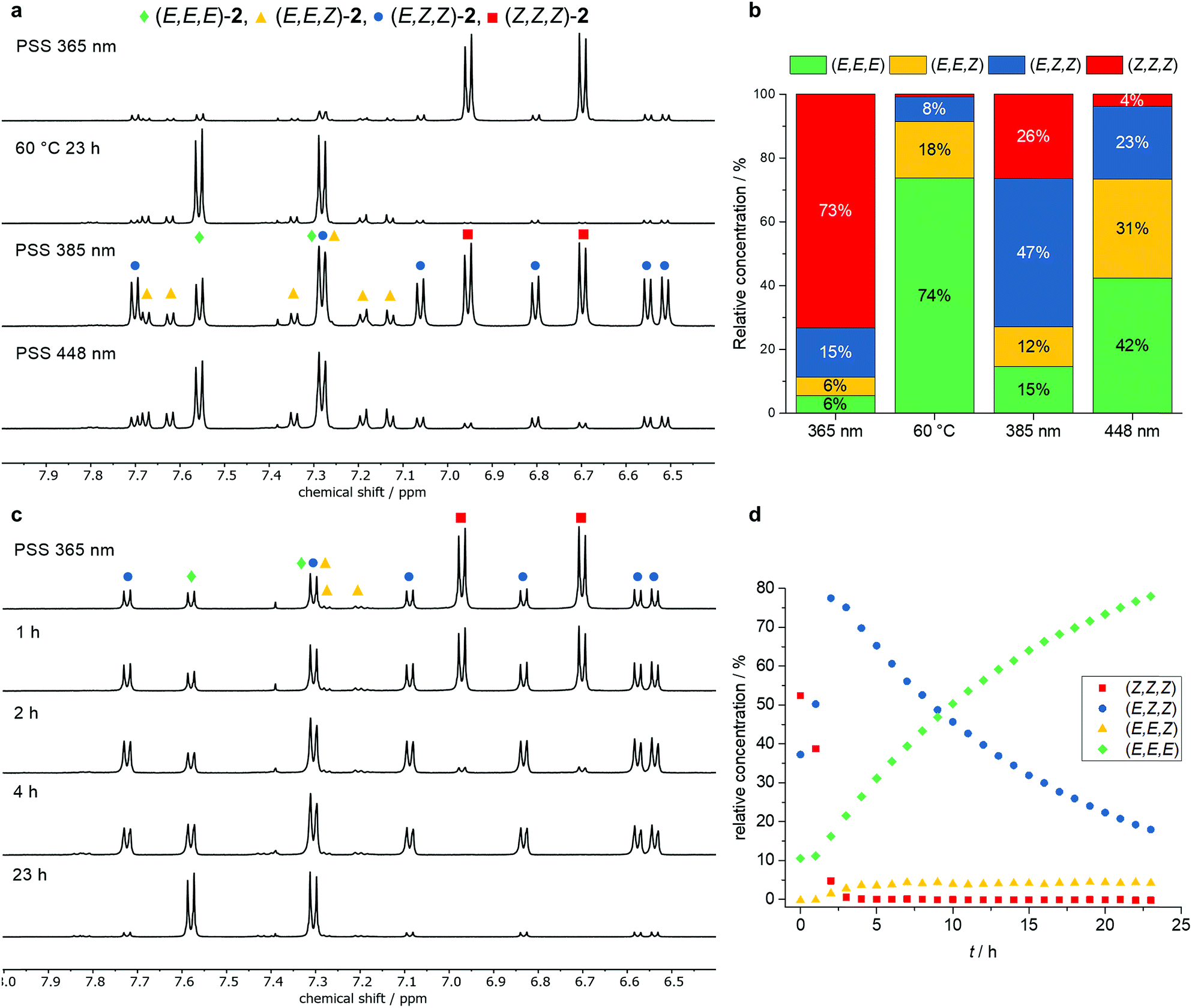 | ||
| Fig. 4 Aromatic region of the 1H NMR spectra of 2 in TCE-d2 at different PSSs (a). Relative concentrations of the isomers after irradiation at the corresponding wavelength (b). Aromatic region of the 1H NMR spectra of the thermal isomerization of 2 at 60 °C (c). Relative isomer concentrations during thermal isomerization at 60 °C in TCE-d2 (d). | ||
In the case of tetraazobenzophane 3, similar NMR isomerization experiments were carried out. However, it was not possible to assign all observed signals in the mixture. Nonetheless, the all-E, all-Z, (E,Z,E,Z) and the (E,Z,Z,Z) isomers were identified (see ESI†). As a result of the isomerization experiments, at least three different PSSs were also found for macrocycle 3. Taking a look at the overall all-Z → all-E thermal isomerization, the thermodynamically most stable all-E conformation could be fully generated after heating to 60 °C for 19 h. Furthermore, the total conversion occurred faster compared to the smaller analogue 2 (see ESI†). In order to support the experimental isomerization results, the thermal all-Z → all-E pathways of all macrocycles were computed using density functional theory (DFT) on the PBE0-D3(BJ)/def2-TZVPP//PBE0-D3(BJ)/def2-SVP level of theory (for details, see ESI†). The results for trisazobenzophane 2 are shown in Fig. 5. Interestingly, the (E,Z,Z) and the (E,E,Z) isomer were found to be almost equal in enthalpy. An explanation for this observation can be found in the different influence of ring strain in the corresponding isomers. The (E,E,Z) conformation featured the highest strain energy (54 kcal mol−1) of all isomers, whereas the (E,Z,Z) form exhibited the lowest (39 kcal mol−1). This causes a rise of the enthalpy of the (E,E,Z) and a decrease of the (E,Z,Z) isomer. Hence, the enthalpic gap between both isomers becomes narrower. Furthermore, the significant influence of ring strain on the isomer stabilities becomes obvious when taking a look at the transition state enthalpies. The highest barrier was found for the (E,Z,Z) → (E,E,Z) isomerization, followed by the (Z,Z,Z) → (E,Z,Z) and the lowest was found for the final (E,Z,Z) → (E,E,E) isomerization. The computations provided that in the (E,Z,Z) → (E,E,Z) step about 14 kcal mol−1 strain energy has to be built up, whereas the other two isomerizations occur almost strain-free (Table S6†).
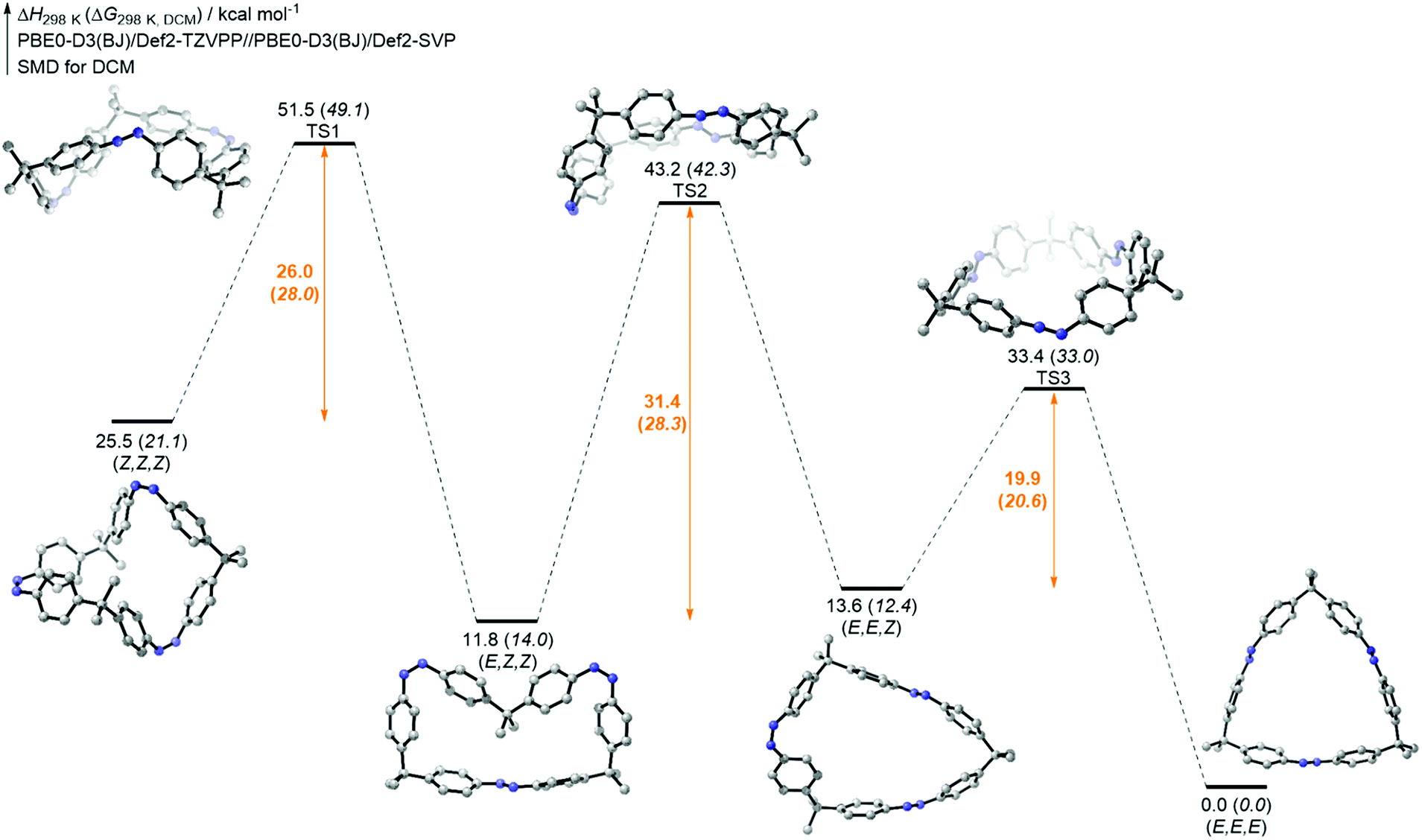 | ||
| Fig. 5 Enthalpy and Gibbs free energy (in parentheses) pathway of the all-Z-2 → all-E-2 thermal isomerization at the PBE0-D3(BJ)/def2-TZVPP//PBE0-D3(BJ)/def2-SVP level of theory. Solvation was considered implicitly using the SMD model for dichloromethane.34 | ||
Hence, the total ring strain of the system is built up in only one isomerization step, which is in agreement with the NMR experiments, that revealed the (E,Z,Z) → (E,E,Z) to be the rate determining step. However, the computed activation enthalpies are rather high for the first two steps and quite low for the last step. Nevertheless, the values are reasonable within the expected DFT error range33 and thus the qualitative trend of the thermal isomerization rates could be reproduced.
The computations for the thermal isomerization of tetraazobenzophane 3 showed that the enthalpies of the isomers decrease with every consecutive Z → E switching step on the minimum energy pathway (see ESI, Fig. S11†). In the case of the (E,Z,Z,Z) isomer, there are two different possibilities of isomerization, namely the (E,Z,Z,Z) → (E,E,Z,Z) or the (E,Z,Z,Z) → (E,Z,E,Z) pathway. The latter was found to be preferred thermodynamically of 6 kcal mol−1 and also kinetically of 3.7 kcal mol−1. Moreover, the (E,E,Z,Z) isomer is slightly higher in enthalpy than the (E,Z,Z,Z) form (ca. 1 kcal mol−1). Additionally, the highest ring strain energy of all isomers was found in the (E,E,Z,Z) isomer, and the lowest in the all-Z isomer. In regard to the transition states, lower activation enthalpies and free energies compared to the trisazobenzene system 2 were computed. This is consistent with the NMR experiments, where full thermal isomerization of 3 was reached faster than in the case of compound 2. Although the computations predicted the (E,Z,Z,Z) isomer to be the most stable, the (E,Z,E,Z) → (E,Z,Z,Z) isomerization was identified experimentally to be the rate determining step. Nevertheless, the latter process was computed to require the second highest activation enthalpy. All in all, the computed values agree with the experiment regarding the error of the applied method.
After having gained insight into the photo- and thermal isomerization behavior of macrocycle 2, experiments towards the selective generation of the all-Z, (E,Z,Z) and all-E isomers were conducted. High performance liquid chromatography (HPLC) was applied as method of choice,22 since it allows direct and precise analysis of the relative isomer concentrations in solution. Furthermore, only small sample amounts, both in solution volume and concentration, are necessary.
A first analysis of the different PSS showed consistent results to the previous NMR experiments (see ESI, Table S2†). With this simple and relatively fast method in hand, conditions to selectively switch between three states were optimized. As a result, it was found that the all-Z, the all-E and the (E,Z,Z) isomers could be generated selectively in amounts of 70–80% by irradiation at 365 nm, irradiation at 448 nm, followed by heating at 80 °C, and irradiation at 365 nm followed by heating at 80 °C, respectively (Fig. 6). Furthermore, every state could be generated from any other state. Additionally, cycling of the states (21 states) showed reversibility and repeatability of the selective state generation. This represents the first triple state molecular switch solely composed from identical azobenzene moieties.
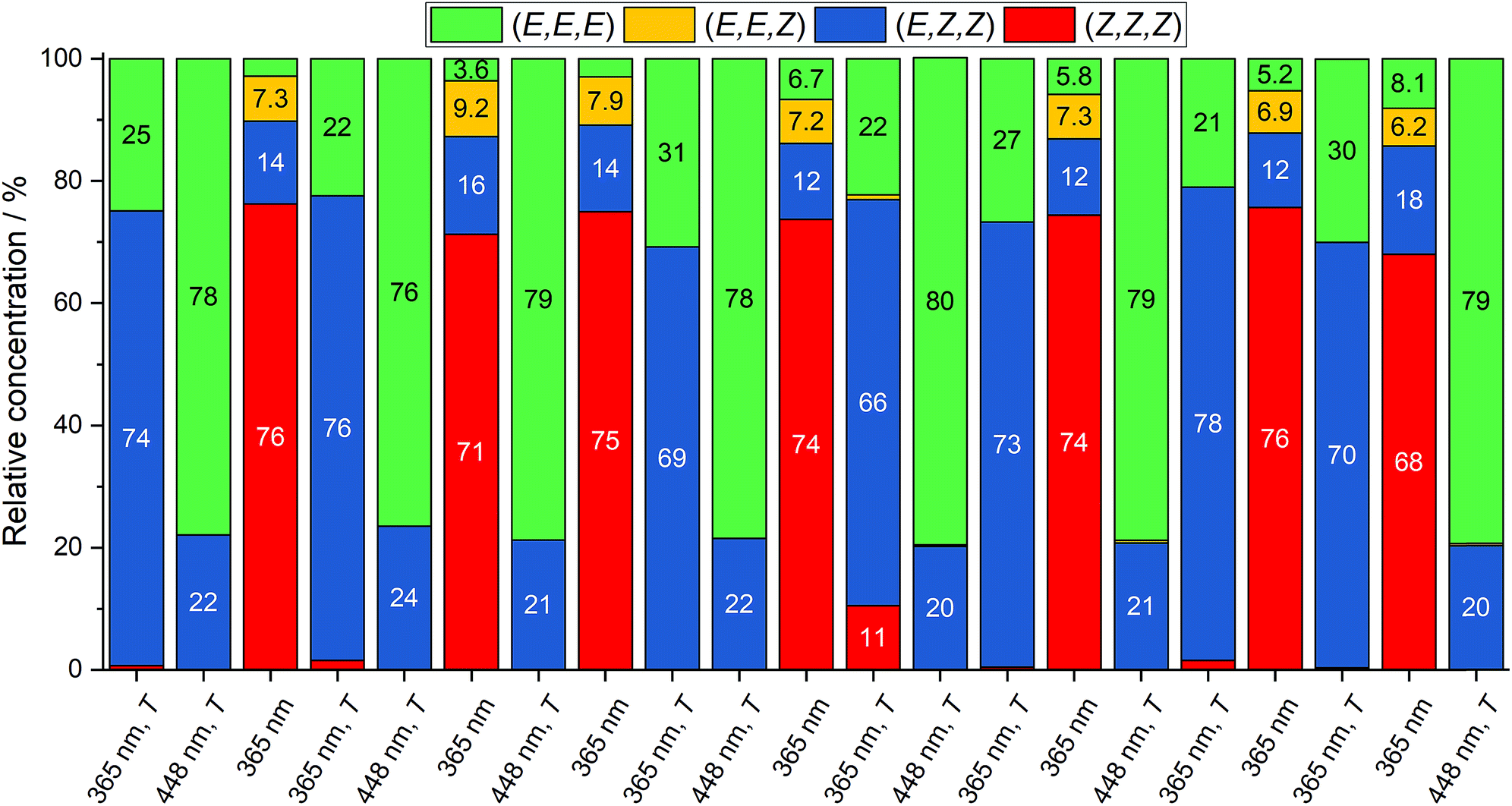 | ||
| Fig. 6 Switching cycles of macrocycle 2 in TCE-d2. All states [(E,E,E), (E,Z,Z) and (Z,Z,Z)] can be reversibly generated from all starting states. The same sample was irradiated at the given wavelength for 10 min, or irradiated for 10 min and heated for 10 min afterwards, respectively. | ||
Conclusions
In summary, a series of macrocyclic oligoazobenzenes 1–3 was synthesized, characterized and investigated towards photophysical and thermal isomerization behavior. It was discovered that macrocyclic ring strain has a major influence on the isomerization behavior. In the case of trisazobenzophane 2, remarkably high thermal stability was found for the (E,Z,Z) isomer, rationalized by a high (E,Z,Z) → (E,E,Z) activation barrier due to high ring strain that has to be overcome in this step. Moreover, it was possible to utilize this property to selectively generate three different isomerization states, namely the (E,E,E), (E,Z,Z) and the (Z,Z,Z) states. These states can easily be addressed via irradiation or a combination of irradiation and heating for short times. Hence, azobenzophane 2, to the best of our knowledge, represents the first symmetric oligoazobenzene that can be selectively isomerized to three different states with >70% relative isomer concentration. Thus, this compound represents a promising candidate for applications in material science, e.g. in data storage or optically controllable materials.The highly strained bisazobenzophane 1 could not, as expected from previous studies,19,20 isomerize photochemically or thermally within the timescale of the applied experiments. Furthermore, ring strain reversed the isomer stability from all-E to all-Z as the thermodynamically favored conformation. The isomerization behavior of the largest tetraazo macrocycle 3 was still significantly influenced by ring strain. Again, it was possible to identify three different PSS. However, the identified photoisomers showed lower thermal stabilities compared to those of trisazo 2. The thermal (E,Z,E,Z) → (E,Z,Z,Z) isomerization was identified as the rate determining step in the overall isomerization process, where most of the strain is built up. All in all, this study clearly revealed that that macrocyclic strain is a crucial tool in the design of multi-state photoswitches and opens new possibilities in the design of multi-state light-responsive materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged. Dr Heike Hausmann and Stefan Bernhardt are acknowledged for NMR and HPLC support, respectively. Furthermore, we thank Dr Urs Gellrich for advice concerning computations and Lea E. Schäfer for help with the graphical abstract.References
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC
.
- G. S. Hartley, Nature, 1937, 140, 281 CrossRef CAS
-
(a) A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC
-
(a) T. Ikeda and O. Tsutsumi, Science, 1995, 268, 1873–1875 CrossRef CAS PubMed
-
(a) Y. Jiang, J. Huang, W. Feng, X. Zhao, T. Wang, C. Li and W. Luo, Sol. Energy Mater. Sol. Cells, 2019, 193, 198–205 CrossRef CAS
-
(a) L. Schweighauser, M. A. Strauss, S. Bellotto and H. A. Wegner, Angew. Chem., Int. Ed., 2015, 54, 13436–13439 CrossRef CAS PubMed
- F. Cisnetti, R. Ballardini, A. Credi, M. T. Gandolfi, S. Masiero, F. Negri, S. Pieraccini and G. P. Spada, Chemistry, 2004, 10, 2011–2021 CrossRef CAS PubMed
- C. Slavov, C. Yang, L. Schweighauser, C. Boumrifak, A. Dreuw, H. A. Wegner and J. Wachtveitl, Phys. Chem. Chem. Phys., 2016, 18, 14795–14804 RSC
- D. Bléger, J. Dokić, M. V. Peters, L. Grubert, P. Saalfrank and S. Hecht, J. Phys. Chem. B, 2011, 115, 9930–9940 CrossRef PubMed
-
(a) F. Zhao, L. Grubert, S. Hecht and D. Bléger, Chem. Commun., 2017, 53, 3323–3326 RSC
- A. Fihey, A. Perrier, W. R. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44, 3719–3759 RSC
- R. Reuter and H. A. Wegner, Chem. Commun., 2011, 47, 12267–12276 RSC
- L. Schweighauser and H. A. Wegner, Chem. Commun., 2013, 49, 4397–4399 RSC
- L. Schweighauser, D. Häussinger, M. Neuburger and H. A. Wegner, Org. Biomol. Chem., 2014, 12, 3371–3379 RSC
-
(a) D. Gräf, H. Nitsch, D. Ufermann, G. Sawitzki, H. Patzelt and H. Rau, Angew. Chem., Int. Ed., 1982, 21, 373–374 CrossRef
- D. Röttger and H. Rau, J. Photochem. Photobiol. A, 1996, 101, 205–214 CrossRef
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, C. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595 CrossRef CAS PubMed
- M. Hammerich, C. Schütt, C. Stähler, P. Lentes, F. Röhricht, R. Höppner and R. Herges, J. Am. Chem. Soc., 2016, 138, 13111–13114 CrossRef CAS PubMed
- A. H. Heindl, L. Schweighauser, C. Logemann and H. A. Wegner, Synthesis, 2017, 49, 2632–2639 CrossRef CAS
- C. Slavov, C. Yang, A. H. Heindl, T. Stauch, H. A. Wegner, A. Dreuw and J. Wachtveitl, J. Phys. Chem. Lett., 2018, 9, 4776–4781 CrossRef CAS PubMed
-
(a) Y. Norikane, K. Kitamoto and N. Tamaoki, Org. Lett., 2002, 4, 3907–3910 CrossRef CAS PubMed
- N. Tamaoki, K. Koseki and T. Yamaoka, Tetrahedron Lett., 1990, 31, 3309–3312 CrossRef CAS
- N. Tamaoki, K. Ogata, K. Koseki and T. Yamaoka, Tetrahedron, 1990, 46, 5931–5942 CrossRef CAS
- R. Reuter and H. A. Wegner, Org. Lett., 2011, 13, 5908–5911 CrossRef CAS PubMed
- C. Mills, J. Chem. Soc., Trans., 1895, 67, 925–933 RSC
- S. Bellotto, R. Reuter, C. Heinis and H. A. Wegner, J. Org. Chem., 2011, 76, 9826–9834 CrossRef CAS PubMed
- C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 6174–6177 CrossRef CAS PubMed
-
(a) H. Hilpert, L. Hoesch and A. S. Dreiding, Helv. Chim. Acta, 1985, 68, 325–333 CrossRef CAS
- R. Salvio, L. Mandolini and C. Savelli, J. Org. Chem., 2013, 78, 7259–7263 CrossRef CAS PubMed
- B. Priewisch and K. Rück-Braun, J. Org. Chem., 2005, 70, 2350–2352 CrossRef CAS PubMed
- S. E. Wheeler, K. N. Houk, P. v. R. Schleyer and W. D. Allen, J. Am. Chem. Soc., 2009, 131, 2547–2560 CrossRef CAS PubMed
-
(a) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data, NMR spectra and computational data are provided (PDF). X-ray crystal structure data for 1–3. See DOI: 10.1039/c9sc02347j |
| This journal is © The Royal Society of Chemistry 2019 |