
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe acidic nature of “NMR-invisible” tri-coordinated framework aluminum species in zeolites†
Shaohui
Xin
ad,
Qiang
Wang
 *a,
Jun
Xu
a,
Yueying
Chu
a,
Pengfei
Wang
b,
Ningdong
Feng
a,
Guodong
Qi
a,
Julien
Trébosc
c,
Olivier
Lafon
ce,
Weibin
Fan
b and
Feng
Deng
*a
*a,
Jun
Xu
a,
Yueying
Chu
a,
Pengfei
Wang
b,
Ningdong
Feng
a,
Guodong
Qi
a,
Julien
Trébosc
c,
Olivier
Lafon
ce,
Weibin
Fan
b and
Feng
Deng
*a
aNational Centre for Magnetic Resonance in Wuhan, State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, CAS Key Laboratory of Magnetic Resonance in Biological Systems, Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences, Wuhan 430071, China. E-mail: qiangwang@wipm.ac.cn; dengf@wipm.ac.cn
bState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, P.O. Box 165, Taiyuan, Shanxi 030001, P. R. China
cUniv. Lille, CNRS, ENSCL, UMR 8181, Unité de Catalyse et de Chimie du Solide, 59000 Lille, France
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
eInstitut Universitaire de France, 75231 Paris, France
First published on 12th September 2019
Abstract
The unambiguous characterization of different acid sites in zeolites is of great importance for understanding their catalytic performance and the rational design of highly efficient zeolite catalysts. In addition to various well-characterized extra-framework Al species, a tri-coordinated framework aluminum species can also serve as a Lewis acid site in zeolites, which is “NMR-invisible” owing to its extremely distorted local environment. Here we provide a feasible and reliable approach to elucidate the acidic nature of the tri-coordinated framework Al in dehydrated H-ZSM-5 zeolites via sensitivity-enhanced two-dimensional (2D) multiple nuclear correlation NMR experiments coupled with trimethylphosphine oxide (TMPO) probe molecules. Two types of tri-coordinated framework Al sites have been unambiguously identified, which amount to 11.6% of the total Brønsted and Lewis acid sites. Furthermore, it was found that synergistic effects arising from the close spatial proximity between the tri-coordinated framework Al site and the Brønsted acid site lead to the generation of superacidity (with an acid strength stronger than 100% H2SO4) in the zeolite.
Introduction
Heterogeneous catalysts with acid–base properties, such as zeolites have been widely used in the chemical and petrochemical industry for catalytic cracking,1 isomerization,2 alkylation,3 and disproportionation.4 In aluminosilicates-type zeolites, the Brønsted acid site (BAS) and Lewis acid site (LAS) are responsible for the active sites in numerous catalytic reactions. Therefore, understanding the structures and properties of the acidic sites in zeolites is essential to exploring the reaction mechanism and to the optimization of their catalytic performance.5–7 It is well-known that the BAS is associated with the tetrahedral framework aluminum (FAL) in the form of a bridging hydroxyl (Si–OH–Al) in zeolites. Furthermore, the LAS is generally recognized as an extra-framework or framework aluminum species, formed upon calcination or steaming of zeolites.8,9 The properties of BAS associated with the tetrahedral framework Al and LAS associated with various extra-framework Al (EFAL) species have been extensively studied, from their structural characteristics10–14 to their functions in catalytic reactions.15–18 In particular, the LAS in zeolites has been proved to play an important role in the catalytic transformations of hydrocarbons such as the hydrogen transfer process,7,19,20 and also generates the Brønsted/Lewis acid synergy owing to the vicinity of the corresponding Al species in the local architecture of dealuminated zeolites.11,21–23 Although the tri-coordinated FAL is commonly considered as a LAS in zeolites, only a few characterizations24–26 have been attempted to confirm the existence of the “moisture sensitive” tri-coordinated Al species in the zeolite framework. Up to now, no experimental approach has been reported to probe the intrinsic acidic properties of this important framework Al species in the zeolite.Solid-state NMR has emerged as an important technique for the characterization of heterogeneous catalysts at the atomic scale.22,27–31 The coordination state of both FAL and EFAL species in zeolites can be determined on the basis of one-dimensional (1D) 27Al magic-angle spinning (MAS) and 2D 27Al multiple-quantum magic-angle spinning (MQMAS) NMR spectroscopy. 2D 1H–1H and 27Al–27Al double-quantum single-quantum (DQ–SQ) homonuclear correlation MAS NMR methods can provide in-depth structural information on the spatial proximity of different acid sites in dealuminated zeolites, especially under high magnetic fields (>18.8 T).11,22,32 A recent report has shown that the perturbed aluminum species was attributed to terminal Al–OH in the hydrated zeolites, which was considered to be derived from hydrous tri-coordination of the Al species.33 However, most of these measurements were performed under hydration conditions. In the case of non-hydrated zeolites, it is difficult to distinguish FAL species owing to the resolution/sensitivity issues from the quadrupolar nature of the 27Al isotope. The stretch of the acidic proton in the BAS induces a larger distortion of the corresponding tetrahedral FAL in the H-form zeolites, leading to quadrupolar coupling constants (CQ) up to 14–18 MHz.34–36 Generally, mild calcination and dehydration treatments of zeolites would lead to partial breaking (or hydrolysis) of the framework ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Al–O– bond and generation of a tri-coordinated FAL and framework silanol group as illustrated in Scheme 1.37–39 Although the neighboring framework Si–OH would interact with the tri-coordinated FAL to restore the tetrahedral FAL after cooling down of the sample to room temperature, partial Si–OH derived from the breaking of the aluminum oxygen bond that bears the proton may be further dehydroxylated by a neighboring defect site (Si–OH group), thus causing the irreversible formation of a tri-coordinated coordinated aluminum.26 However, reducing the number of neighboring oxygen groups bound to FAL yields a dramatic distortion of its local environment, and the extreme asymmetry of its surrounding electric field results in a considerable line broadening of its 27Al NMR resonance (usually with CQ > 30 MHz).33,40 Therefore, the direct observation of tri-coordinated FAL species in a dehydrated sample is generally impossible using conventional 27Al MAS NMR owing to its huge quadrupolar broadening and relatively low concentration, and tri-coordinated FAL was suggested to be the “NMR-invisible” species in zeolites.26,33 After rehydration, the tri-coordinated FAL could be easily transformed into a symmetric tetra-coordinated FAL,33,38 which is usually indistinguishable from the FAL of BAS. Further hydrolysis of the framework Al–O bond leads to removal of the Al from the zeolite framework, forming EFAL species such as Al(OH)3, Al(OH)2+, AlOH2+, Al3+, AlO+ and so forth, which have been well characterized using solid-state NMR spectroscopy.11,22,32,36,41,42
Al–O– bond and generation of a tri-coordinated FAL and framework silanol group as illustrated in Scheme 1.37–39 Although the neighboring framework Si–OH would interact with the tri-coordinated FAL to restore the tetrahedral FAL after cooling down of the sample to room temperature, partial Si–OH derived from the breaking of the aluminum oxygen bond that bears the proton may be further dehydroxylated by a neighboring defect site (Si–OH group), thus causing the irreversible formation of a tri-coordinated coordinated aluminum.26 However, reducing the number of neighboring oxygen groups bound to FAL yields a dramatic distortion of its local environment, and the extreme asymmetry of its surrounding electric field results in a considerable line broadening of its 27Al NMR resonance (usually with CQ > 30 MHz).33,40 Therefore, the direct observation of tri-coordinated FAL species in a dehydrated sample is generally impossible using conventional 27Al MAS NMR owing to its huge quadrupolar broadening and relatively low concentration, and tri-coordinated FAL was suggested to be the “NMR-invisible” species in zeolites.26,33 After rehydration, the tri-coordinated FAL could be easily transformed into a symmetric tetra-coordinated FAL,33,38 which is usually indistinguishable from the FAL of BAS. Further hydrolysis of the framework Al–O bond leads to removal of the Al from the zeolite framework, forming EFAL species such as Al(OH)3, Al(OH)2+, AlOH2+, Al3+, AlO+ and so forth, which have been well characterized using solid-state NMR spectroscopy.11,22,32,36,41,42
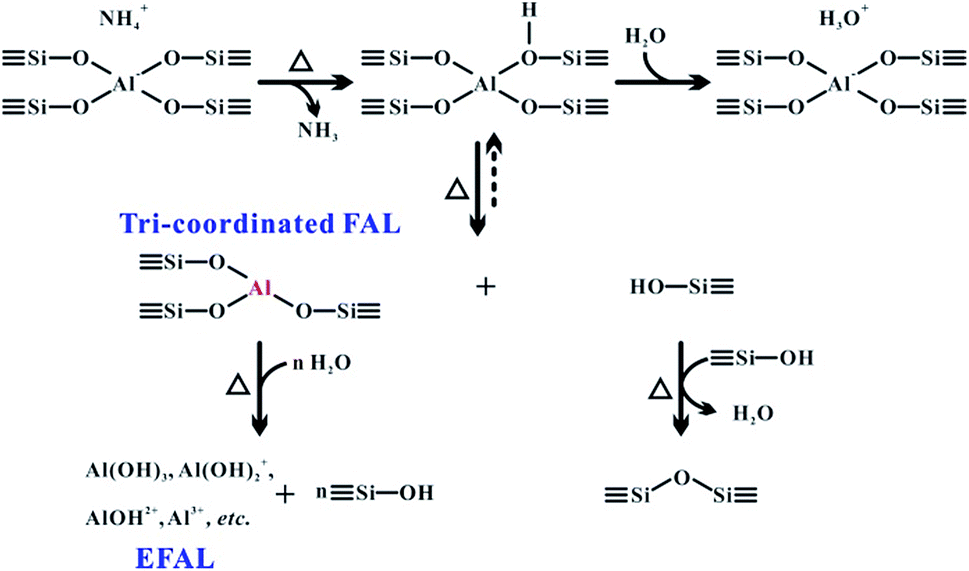 | ||
| Scheme 1 Formation route for the tri-coordinated FAL and EFAL species in zeolites. | ||
The adsorption of basic probe molecules such as 2-13C-acetone and trimethylphosphine oxide (TMPO) is a feasible approach to investigate the acidity of zeolites.22,43–45 The great majority of studies utilized the chemical shift of certain elements (e.g.13C or 31P) in probe molecules to determine the acidic features of zeolites, which were usually combined with density functional theory (DFT) calculations to reveal the relationship between chemical shifts and the corresponding acidic properties (e.g. type, distribution, and strength) on zeolites.22,45 However, it has previously been a great challenge to provide precise and localized information on the structure of the interface between the “guest” probe molecules and the specific acid sites of the “host” zeolite via analytical or spectroscopic techniques.
In this contribution, we provide a unique insight into the acidic nature of tri-coordinated FAL species on a H-ZSM-5 zeolite using solid-state NMR spectroscopy, and present a clear adsorption picture of the TMPO probe molecules on different framework Al species in a dehydrated H-ZSM-5 zeolite. In particular, the sensitivity-enhanced 2D 31P–27Al heteronuclear correlation (HETCOR) MAS NMR technique46 is employed to successfully discriminate the interactions between distinct adsorbed TMPO molecules and Al species in the zeolite framework. Thus, it is straightforward to ascertain the presence of LAS originating from the tri-coordinated FAL. Two tri-coordinated FAL species are unambiguously identified using the advanced solid-state NMR technique. Further quantitative NMR analysis reveals that a considerable amount (ca. 11.6%) of this type of LAS exists in the framework of the dehydrated H-ZSM-5 zeolite even with moderate thermal treatments. Moreover, we demonstrate that the presence of the tri-coordinated FAL is responsible for the formation of superacidity (characterized by a down-field 31P chemical shift of the adsorbed TMPO up to 85–88 ppm) owing to the Brønsted/Lewis acid synergy in the zeolite framework which was further confirmed using 2D 27Al–27Al DQ–SQ homonuclear correlation spectroscopy and DFT calculations.
Results and discussion
Structural characterization of the dehydrated zeolites
The 27Al MAS NMR spectra of the hydrated parent NH4-ZSM-5 acquired at two different magnetic fields (18.8 and 11.4 T) shown in Fig. 1a and d both displayed a typical narrow line around 54 ppm, respectively, corresponding to relatively symmetrical tetrahedral Al sites in the zeolite framework. Usually a significantly reduced quadrupolar broadening of the 27Al NMR line shape can be obtained at a high field (B0 = 18.8 T) and fast MAS speed (νR = 40 kHz) compared to that at a medium field (B0 = 11.7 T) and moderate MAS speed (νR = 10 kHz). After calcination and dehydration, the 27Al spectra of the dehydrated H-ZSM-5 zeolite exhibited a very broad resonance (Fig. 1b and e) with a full width of ca. 20 kHz even at 18.8 T, indicative of a dominant contribution from the large second-order quadrupolar broadening. Previous studies have demonstrated that this broad signal was mainly attributed to the more-distorted tetrahedral FAL species with a CQ ≈ 16 MHz from the Brønsted acidic site on the dehydrated zeolites.35,36 In fact, dealumination is inevitable in most sample preparation procedures, while the mild vacuum or flow temperature-programmed methods used here could make that framework dealumination rarely occurs, which would largely reduce the interference of EFAL species in the study of tri-coordinated FAL.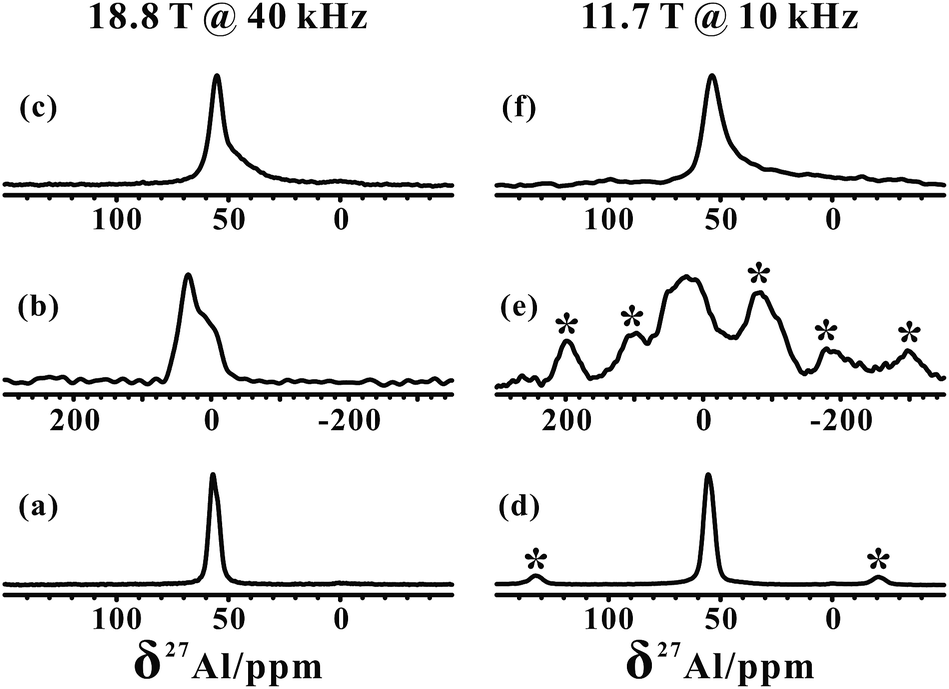 | ||
| Fig. 1 1D 27Al MAS NMR spectra of parent NH4-ZSM-5 (a) and (d); calcined and dehydrated H-ZSM-5 (b) and (e); and TMPO-adsorbed H-ZSM-5 with P/Al = 0.42 (c) and (f); obtained at B0 = 18.8 T (a)–(c) and B0 = 11.7 T (d)–(f). The asterisks denote spinning sidebands. | ||
The XRD patterns (Fig. S1†) of the ZSM-5 zeolites indicated no apparent change in the crystallinity after these treatments. To estimate the amount of EFAL in our calcined sample, the parent and calcined forms of these zeolites were treated using 1 M aqueous NaNO3 at 353 K three times. As the Na+ cations can balance the charge of the framework AlO4− tetrahedra, the Na/Al ratio is a reliable parameter to assess the variation of the fraction of the tetrahedral framework Al.47 The Na/Al ratio measured using inductively coupled plasma (ICP) was 1.01 and 0.98 respectively for the parent NH4-ZSM-5 and dehydrated H-ZSM-5 zeolites treated with Na+ exchange, suggesting that most of the Al species were still located in the zeolite framework. This is also supported by a slight loss (1%) of framework Si–O–Al moieties in the dehydrated zeolite via 1D 29Si MAS and 2D 29Si{1H} HETCOR NMR analysis (Fig. S2 and S3†). The quantitative 29Si MAS spectra shown in Fig. S3† indicates an increase of the Si–OH groups in the dehydrated H-ZSM-5 zeolite compared with those in the parent NH4-form spectra. In addition, further 1H MAS NMR analysis (Fig. S4†) showed that the number of Si–OH groups decreased when raising the dehydration temperature from 573 to 673 K, suggesting that the dehydroxylation of Si–OH groups may occur as depicted in Scheme 1.
Framework aluminum studied using 2D 27Al 3QMAS
In our measurements, TMPO molecules were adsorbed onto the H-ZSM-5 zeolite at a concentration much lower than that of the framework acid sites, meaning the interference between the TMPO molecules themselves can be largely avoided. Interestingly, in addition to the narrow 27Al signal at ca. 54 ppm, a new broad shoulder feature (at ca. 45 ppm) was visible in the 1D 27Al MAS spectra of the dehydrated zeolite loaded with TMPO (P/Al = 0.42, Fig. 1c and f). As the tri-coordinated FAL with Lewis acidity (if present) can directly interact with the oxygen atom of TMPO, which would significantly decrease the asymmetry of the local environment of the Lewis acidic FAL, yielding its “NMR-detectable” 27Al resonance on non-hydrated zeolites. Then, a 2D 27Al 3QMAS experiment at 18.8 T was employed to discriminate between specific aluminum sites in the dehydrated and TMPO-loaded sample (Fig. 2a). Three signals, denoted as Ala, Alb and Alc, were well-resolved with isotropic chemical shifts of 54.4, 51.6 and 58.8 ppm, respectively. For the dehydrated zeolite with a lower TMPO concentration (P/Al = 0.18), an obvious decline of the relative proportion of Alc and Alb signals (Fig. 2b, S10†) was evident. The NMR parameters deduced from the 2D spectrum are listed in Table 1. The Ala signal with PQ = 2.3 MHz could be attributed to the regular tetrahedral FAL species from Brønsted acidic sites analogous to that of the NH4-form or hydrated zeolites. The local structure of the tetrahedral FAL in dehydrated H-ZSM-5 became more symmetrical stemming from a shrinkage of the Al–O(H) bond length in BAS after the TMPO adsorptions. According to their isotropic chemical shifts, Alb and Alc should both be associated to tetrahedral FAL species as well, while exhibiting a larger inhomogeneous second-order quadrupolar broadening (PQ > 7 MHz). However, their assignments were ambiguous, as they could be from either the FAL in another type of BAS, or the tri-coordinated FAL of LAS directly bound to a TMPO molecule, yielding a new distorted tetrahedral FAL state. In particular, the Alb and Alc resonances contributed to a considerable fraction in the quantitative 27Al NMR spectra of the dehydrated zeolites with different TMPO loadings (Fig. 1c). Therefore, further experiments are required to identify the host–guest correlations/interactions between the adsorbed TMPO probe molecules and the framework of zeolites.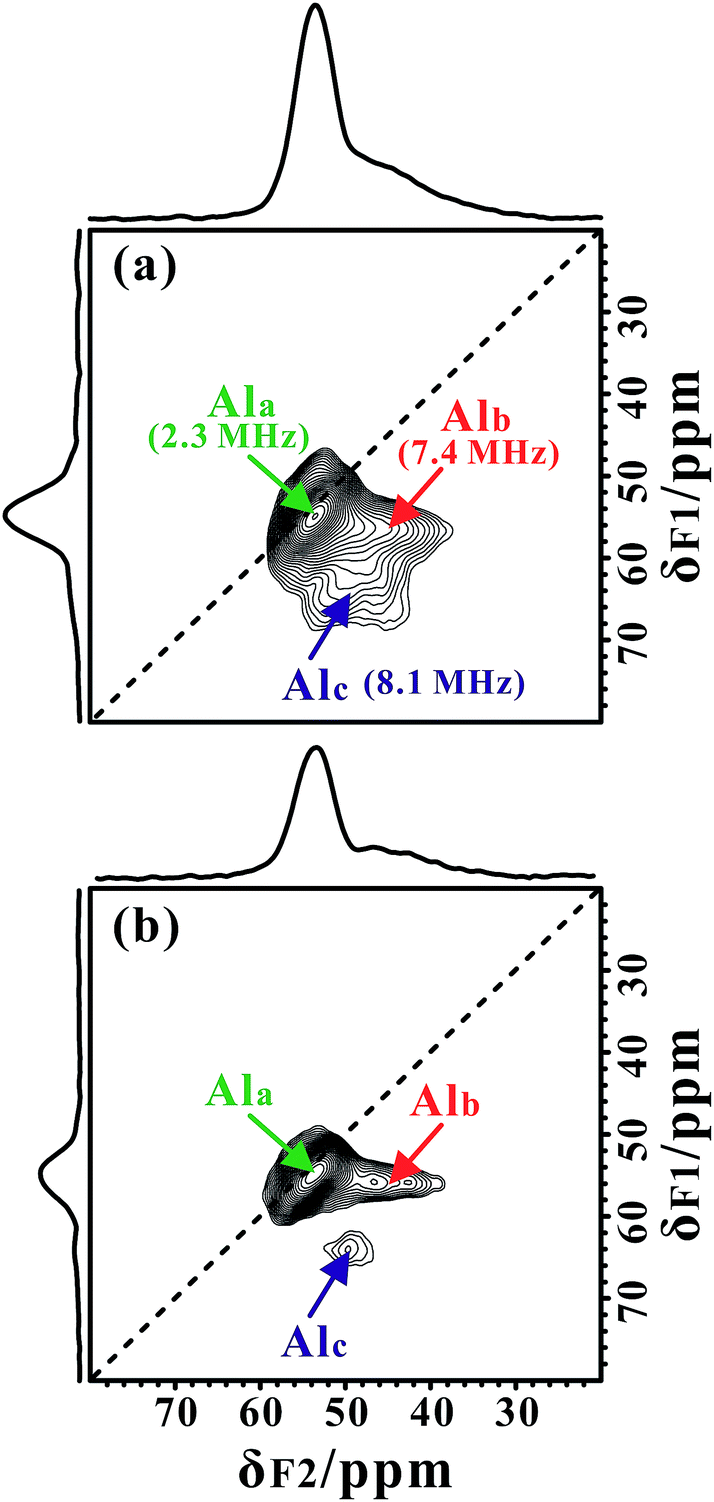 | ||
| Fig. 2 Sheared 2D 27Al 3QMAS NMR spectra of dehydrated H-ZSM-5 zeolites with different TMPO loadings, (a) P/Al = 0.42 and (b) P/Al = 0.18, obtained at B0 = 18.8 T. Skyline projections are drawn along the F1 and F2 dimensions. The determined quadrupolar parameter PQ are indicated in the brackets for different Al species. | ||
| Site | δ F2 /ppm | δ F1 /ppm | δ CS /ppm | P Q /MHz | C Q /MHz |
|---|---|---|---|---|---|
| a δ F2 and δF1 represent the center of gravity of each particular species measured along the direct dimension F2 and isotropic dimension F1 on the sheared spectra, respectively. b Isotropic chemical shift δCS and quadrupole interaction product PQ of each aluminum species were deduced from δF2 and δF1. c The quadrupolar coupling constant (CQ) was extracted from the slice of the 3QMAS spectra by fitting the corresponding second-order quadrupolar line shape (η = 0.5) with the DMFIT program. | |||||
| Ala | 53.6 | 54.8 | 54.4 | 2.3 | 2.2 |
| Alb | 44.1 | 56.0 | 51.6 | 7.4 | 7.1 |
| Alc | 49.7 | 64.1 | 58.8 | 8.1 | 7.8 |
Acid properties analyzed using 2D 31P{1H} HETCOR
For the acidity characterization using TMPO probe molecules, dichloromethane (CH2Cl2) is usually used as the solvent in order to disperse the TMPO molecules into zeolite well.45,48 Although the evacuation at 323 K seems to be insufficient to dispose of all of the solvent, in the 2D 13C{1H} and 31P{1H} HETCOR spectra of the TMPO/H-ZSM-5 zeolite (Fig. S5†) acquired with a relatively short cross polarization (CP) contact time (4 ms or 1 ms) there is no correlation between the CH2Cl2 and TMPO molecules, indicating that the residual solvent and adsorbed TMPO molecules are well separated. Furthermore, the 27Al MAS NMR spectrum of the H-ZSM-5 zeolite with only CH2Cl2 adsorbed (Fig. S6†) still exhibits a very broad resonance (Δδ ≈ 90 ppm), indicating that the residual CH2Cl2 hardly interacts with the acidic sites on the zeolite, and thus has almost no influence on the acidity characterization using TMPO probe molecules. Note that, after removing the residual solvent on the TMPO/H-ZSM-5 zeolite at a higher temperature (363 K, Fig. S7†), the 27Al MAS NMR spectrum of TMPO/H-ZSM-5 was the same as that treated at 323 K (Fig. S8†), which clearly indicated that the influence of the solvent (CH2Cl2) residues on the framework Al species can be negligible.The 1D 31P MAS NMR spectra of the H-ZSM-5 zeolites with different TMPO loadings (P/Al = 0.18 and 0.42) are shown in Fig. 3a and c, respectively. Up to six characteristic peaks at 88, 85, 76, 69, 65 and 51 ppm were identified from the 31P MAS NMR spectra by using Gaussian deconvolutions (also see Table S1†). Indeed, quantitative analyses of 27Al MAS NMR are difficult owing to either different quadrupolar broadenings of distinct FAL species or only partial acid sites interacting with the TMPO probe molecules, however, here the 31P (I = 1/2) MAS NMR spectra can provide quantitative information on the framework BAS and LAS that react with the TMPO molecules. The 2D 31P{1H} CP-HETCOR spectrum of the TMPO/H-ZSM-5 zeolite (P/Al = 0.18) acquired with a CP contact time of 8 ms in Fig. 3e exhibited two broad distributions of the correlation peaks in “sloped” ovals (dotted line) centered at (76, 12.2) ppm and (88, 6.4) ppm, respectively, which could be assigned to distinct protonated TMPOH+ ionic pair complexes. These correlations were also observable when a short CP contact time (1 ms) was used (see Fig. S5†). The interaction of the adsorbed TMPO molecules with BAS in the H-ZSM-5 zeolite will result in a downfield shift of the 1H resonance of the acidic proton, and the electron cloud density surrounding the 31P nucleus in the TMPO molecule decreases with the increasing acidic strength of BAS, leading to movement of the 31P resonance towards a higher chemical shift (downfield).22,44,45 Furthermore, it was also found that the 31P chemical shift of the adsorbed TMPO molecules linearly increases with the increase of the Brønsted acid strength, and a 31P chemical shift of ca. 86 ppm was demonstrated for the threshold of superacidity (with acid strength stronger than 100% H2SO4).45 Therefore, the 31P peak at δ31P = 88 ppm reflected the strongest acid site with a strength over the threshold of superacidity in the zeolite. In addition, the correlation peaks between all 31P signals and methyl groups (δ1H = 1.8 ppm) of the adsorbed TMPO molecules can be observed at all times. As the 1H–1H spin diffusion will promote the detection of long range interactions in the case of a long CP contact time (8 ms), weak correlations between the protons of the residual CH2Cl2 solvent (δ1H ≈ 5.4 ppm) and the 31P of TMPO molecules were also detectable in the 2D spectrum.
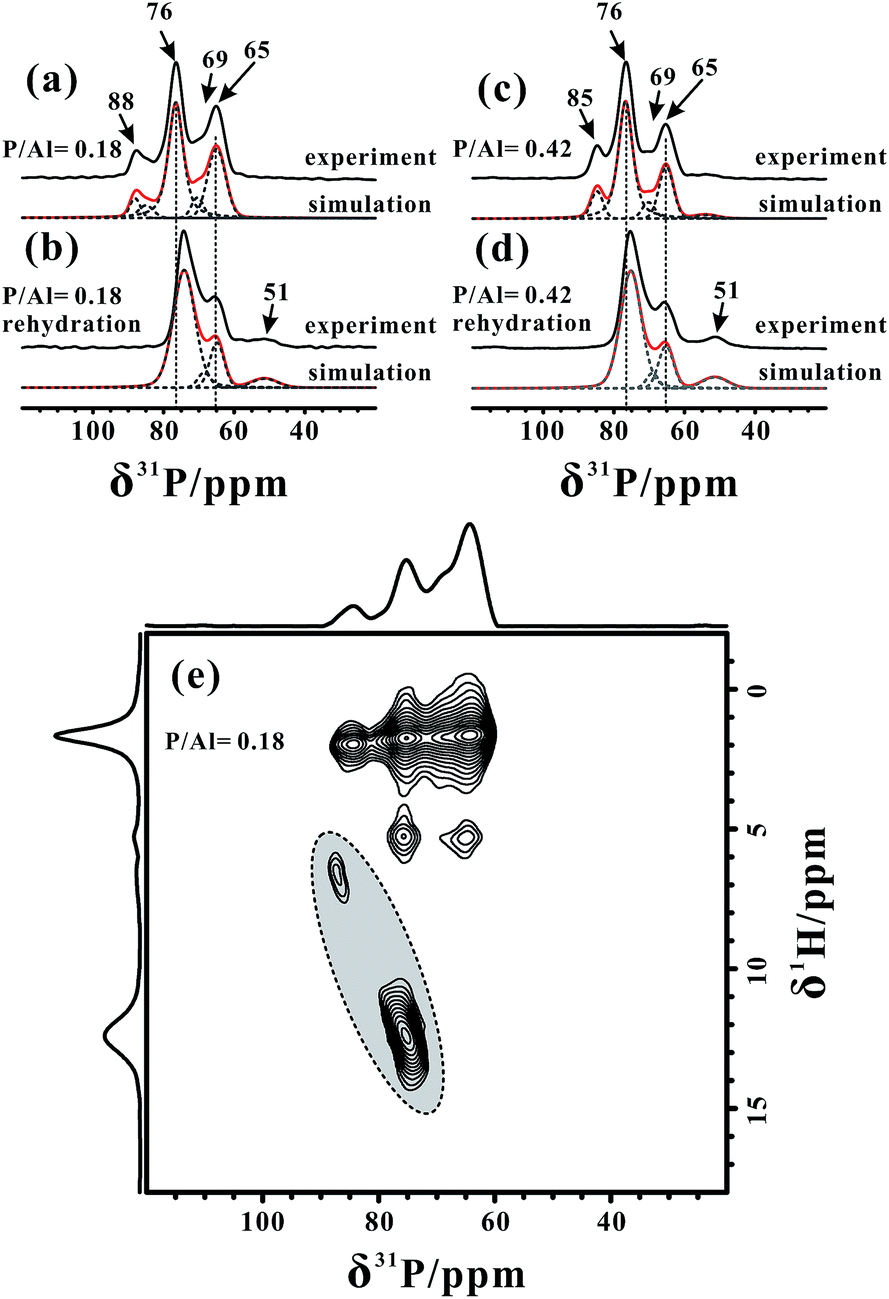 | ||
| Fig. 3 1D one-pulse 31P MAS spectra of dehydrated H-ZSM-5 zeolites with different TMPO loadings (a) and (c), and subsequent rehydration (b) and (d). 2D 31P{1H} CP HETCOR MAS NMR spectrum (e) of the dehydrated H-ZSM-5 zeolite with a low TMPO loading (P/Al = 0.18). | ||
However, as illustrated in the 2D 31P{1H} HETCOR spectrum of Fig. 3e, even though the 1H–1H spin diffusion also contributed to the 1H–31P correlations when the CP contact time was set to 8 ms, no correlation can be observed for the 31P signals around 65 ppm with the 1H signals of BAS, which suggested that the 31P signals do not originate from the protonated TMPOH+ complexes. It is generally recognized that H2O could competitively react with the Lewis acid sites, such as the tri-coordinated Al species, to form weak BAS.48 After exposing our samples to humidity for 2 h, the corresponding 31P MAS NMR spectra in Fig. 3b and d exhibited an apparent decline in the relative intensity of the signals at 65 and 69 ppm (also see Table S1†), and the signal at 88 ppm (or 85 ppm for the sample with P/Al = 0.42) disappeared completely. In addition, the peak at 76 ppm was slightly shifted upfield with a broader distribution, accompanying a notable increase of the peak at 51 ppm from the TMPO adsorbed on the weak acid sites. Therefore, we can reasonably consider that the 31P signals at 65 and 88 ppm (or 85 ppm) are both associated with Lewis acid sites directly or indirectly. Although the peaks at ca. 88 ppm have proved to be from TMPO molecules adsorbed on Brønsted acid sites, it seems that BAS together with the neighboring LAS should contribute to the formation of the superacidity (e.g. δ31P = 88 ppm), indicative of the presence of a synergy effect. Accordingly, after humidity exposure of the dehydrated zeolite, the LAS was preferentially hydrated, thus the influence on its neighboring BAS (the superacid feature) vanished, which in turn caused the BAS to revert back to its original acidity. The two 31P signals at 65 and 69 ppm were probably from the TMPO molecule adsorbed on the Lewis acidic FAL species. Concerning the abundance of Alb and Alc in Fig. 2 (also in Fig. S10†), we proposed that the aforementioned 31P and 27Al species belong to the interacting phosphorus–aluminum pairs in the Lewis acid complexes, (CH3)3PO⋯Al. In addition, we also acquired the 2D 31P{1H} HETCOR spectrum (Fig. S9†) of the TMPO/H-ZSM-5 zeolite with the complete removal of solvent at 363 K, which exhibited similar 1H–31P correlations between the TMPO molecules and the framework acid sites in the zeolite compared with that treated at 323 K, and further excluded the influence of the residual solvent.
Tri-coordinated framework Al characterized using 31P{27Al} HMQC and 31P{27Al} S-RESPDOR
To verify this assumption, the Lewis complex (CH3)3PO⋯Al was identified directly using our recently developed 31P–27Al population-transfer dipolar-mediated HMQC (PT-D-HMQC) experiment which can establish the direct correlation/connectivity between TPMO and various Al species. As shown in Fig. S5,† the 1D 31P{27Al} PT-D-HMQC MAS NMR spectra via31P → 27Al → 31P filtering on TMPO-loaded H-ZSM-5 zeolite (P/Al = 0.42) revealed that the two main 31P resonances around 76 and 65 ppm were correlated with the 27Al species in the zeolite. When using a short recoupling time (τmix = 1.33 ms), the peaks at ca. 65 ppm became more intense compared to that at ca. 76 ppm (Fig. S11†). This indicated the stronger 31P–27Al dipolar interaction of the corresponding spin pairs, further suggesting the presence of the Lewis complex [(CH3)3PO⋯Al].
Notably, the 2D 31P{27Al} PT-D-HMQC spectrum acquired at 11.7 T for the TMPO-adsorbed H-ZSM-5 zeolite in Fig. 4b clearly illustrated two correlation groups. The correlation peaks at around (76, 54) ppm were associated with TMPO adsorbed on the bridging hydroxyl (Si–OH–Al) proton (i.e. the Brønsted acid sites). Importantly, other correlations indicated that the 31P resonance peaks at 65 and 69 ppm are both correlated with broad 27Al resonances from distorted four-coordinated aluminum, reflecting the presence of two types of Lewis acid sites. We also conducted 2D 27Al{31P} D-HMQC experiments at 18.8 T. Owing to the short transverse relaxation times (T2) of the 27Al resonances, we could only observe 27Al–31P correlations from the Lewis complex [(CH3)3PO⋯Al] in Fig. 4c, in which the 31P signals at 65 and 69 ppm were correlated with Alb and Alc, respectively (Fig. 4d and e). Meanwhile, a reduction of the TMPO concentration led to a decrease in the proportion of the 31P signal at 69 ppm relative to that at 65 ppm (Table S1†), this was consistent with the relative proportion variation of Alc and Alb (Fig. 2). Therefore, we can confirm that Alc and Alb are both from tri-coordinated FAL species bound to the O atom of the TMPO molecules, which were discriminated well from their different Lewis acid strengths.
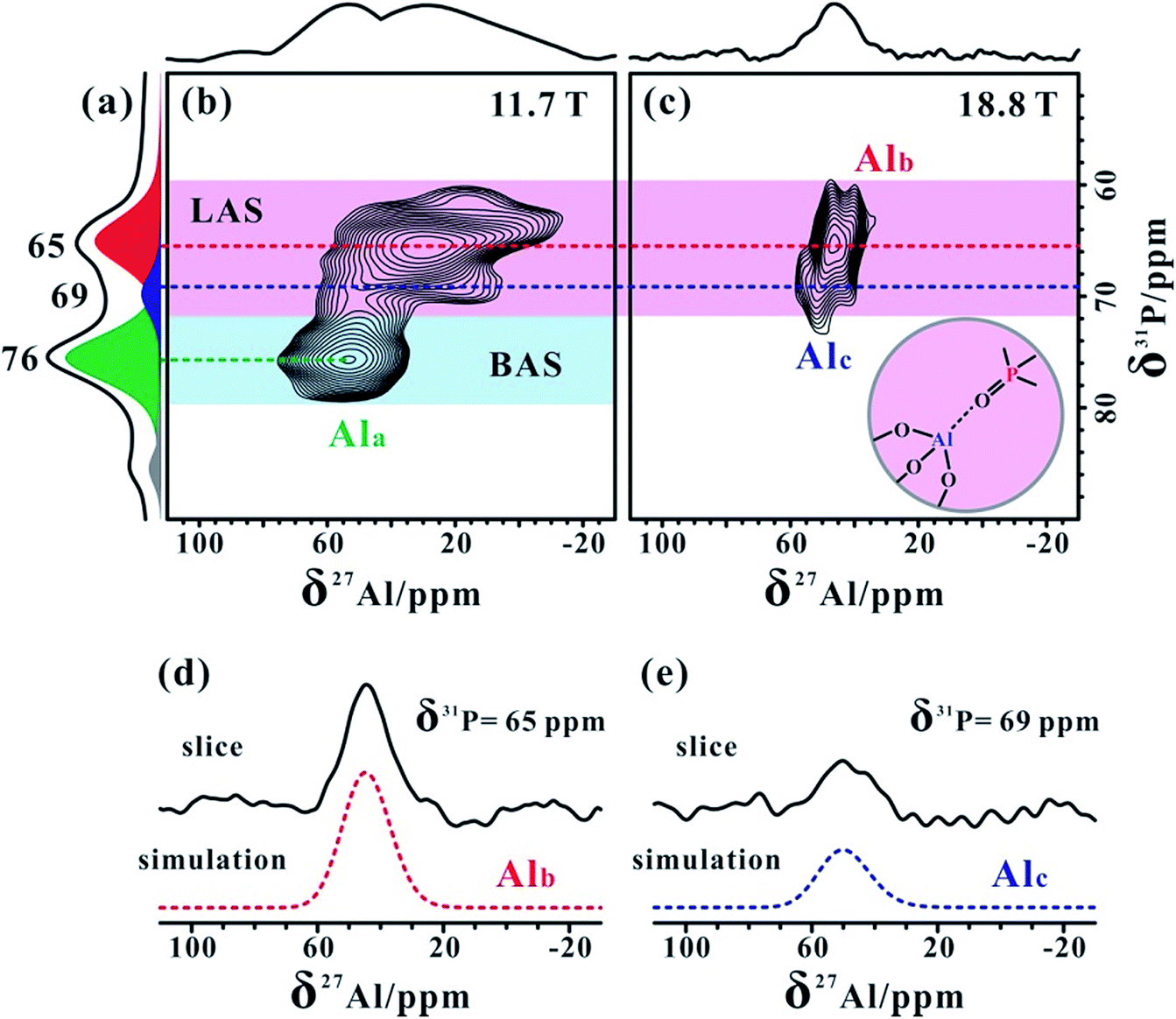 | ||
| Fig. 4 1D one-pulse 31P MAS (a) and 2D 31P{27Al} PT-D-HMQC MAS (b) NMR spectra recorded at 11.7 T, and 2D 27Al{31P} D-HMQC MAS NMR spectrum (c) recorded at 18.8 T of the dehydrated H-ZSM-5 zeolite (Si/Al = 25) with adsorbed TMPO (P/Al = 0.42). Selected slices with simulations are shown in (d) and (e). | ||
Owing to its low content and Brønsted acid feature, we did not observe the 31P–27Al correlations from the 31P signal at 88 ppm (or 85 ppm) in Fig. 4. Fortunately, the symmetry-based resonance-echo saturation-pulse double-resonance (S-RESPDOR) method49 can be employed here to estimate the 31P–27Al heteronuclear dipolar interactions. The results are usually analyzed by plotting the signal fraction ΔS/S0 = (S0 − S′)/S0 (S′ and S0 represent the signal intensity with and without dipolar dephasing, respectively) as a function of the recoupling time τ. The 31P difference spectrum (ΔS = S0 − S′, shown in Fig. S12†) obtained using 1D 31P{27Al} S-RESPDOR experiments still suggested spatial interactions/proximities between all of the 31P species of the adsorbed TMPO molecules (including those giving rise to the resonance at 88 ppm) and 27Al species in the framework of H-ZSM-5. Furthermore, the reduced concentration of TMPO molecules (P/Al = 0.18) allowed us to estimate the distances between 31P atoms and 27Al atoms, as we could approximately consider the Lewis complex [(CH3)3PO⋯Al] with 31P signals at 65 ppm or the Brønsted acid complex [(CH3)3PO⋯H–OAl] with 31P signals at 76 ppm as “isolated” phosphor–aluminum spin pairs in this case. Therefore, the P–Al distance of the former was determined to be 2.9 ± 0.2 Å by fitting the dephasing curve (Fig. 5a), which was rationalized in terms of the structural model of the TMPO molecule adsorbed on the framework tri-coordinated aluminum species (Fig. 5c). The later complex had a reasonable P–Al distance of 4.1 ± 0.4 Å (Fig. 5b), in which the 31P atom was about four bonds away from the Al site (Fig. 5d). We also considered the dipolar coupling networks of multiple-spin systems (shown in Fig. S13†). The representative three spin systems are depicted in Fig. S13b–d.† In the normalized dipolar dephasing curve with respect to the first extremum (or maximum) in each spin system, it was found that the dephasing curves of the observed 31P signals in multi-spin systems have a similar upward trend to that of the isolated 31P–27Al spin pair. This result suggests that the upward trend of the dipolar dephasing curve is dominated by the short-range (strongest) 31P–27Al dipolar interaction. The long-range couplings have little impact on the measurement of the short distance, which mainly affects the oscillation of the dephasing curves after reaching its first extremum. Moreover, the 31P–27Al distances extracted using the dipolar dephasing curves were in good agreement with the DFT calculations, in which the theoretically predicted 31P–27Al distances of the Lewis acid complex [(CH3)3PO⋯Al] and Brønsted acid complex [(CH3)3PO⋯H–OAl] were 3.1 Å and 4.1 Å (Fig. 5c and d), respectively.
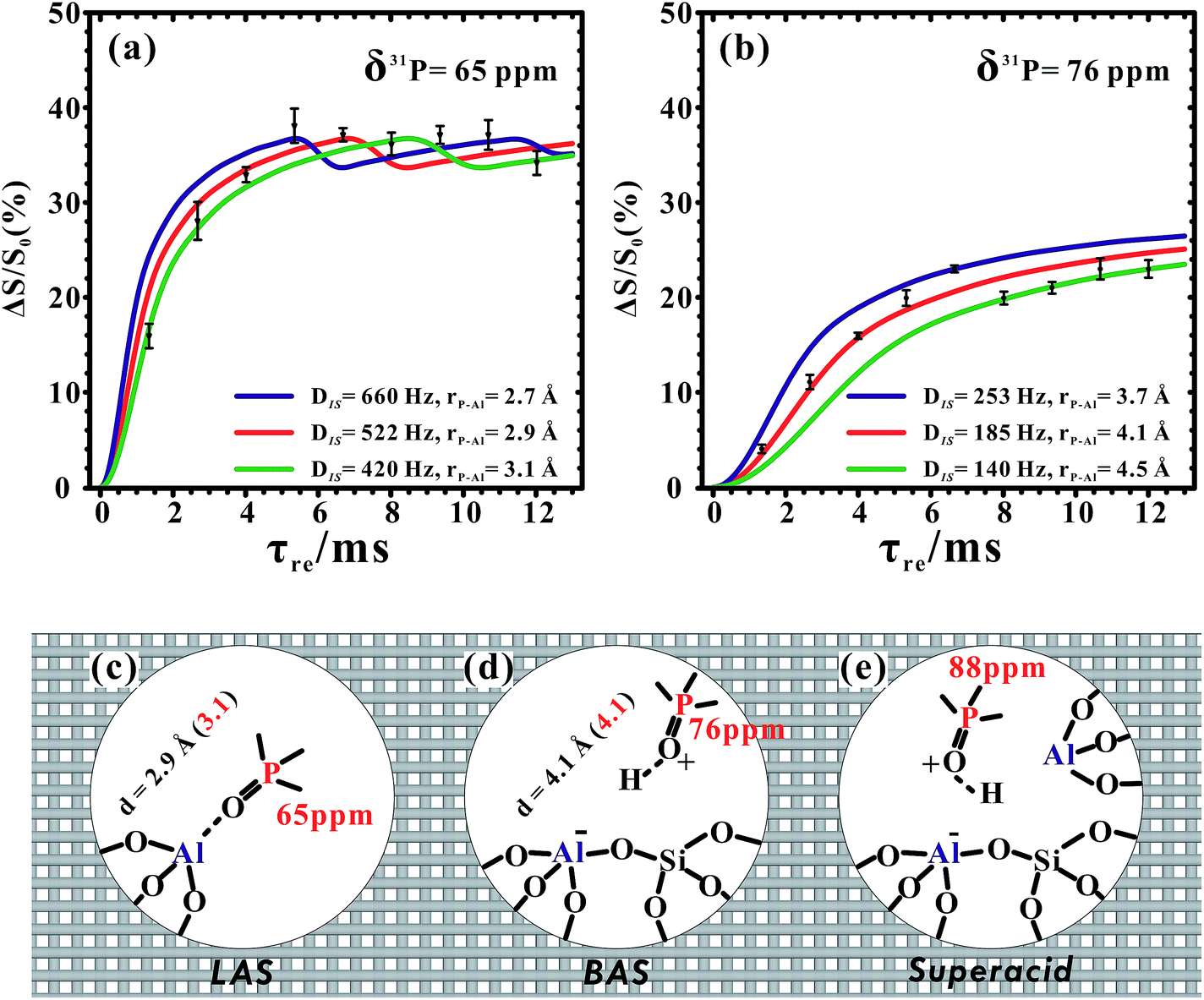 | ||
| Fig. 5 31P{27Al} S-RESPDOR built-up curves of TMPO adsorbed on (a) LAS (tri-coordinated FAL) and (b) BAS of dehydrated H-ZSM-5 zeolite, fitted using an analytical formula. DIS and rP–Al represent the dipolar interaction constant and internuclear distance of the 31P–27Al spin pair, respectively. Three adsorption structure models of: (c) TMPO adsorbed on tri-coordinated FAL, forming a distorted tetrahedral FAL; (d) TMPO adsorbed on a BAS; and (e) TMPO adsorbed on a BAS with a tri-coordinated FAL in close proximity. The red numbers in parentheses are the distances obtained using DFT calculations. | ||
Although it is difficult to calculate the accurate amount of the tri-coordinated FAL in our sample, an estimation of the lower limit of the concentration via the quantitative analysis of the 31P MAS NMR spectrum (Fig. 3) is possible using the following equation:
| Ω(FALIII) = χ(P/Al) × Ω(31PLAS) |
In which Ω(FALIII) is the fraction of the tri-coordinated FAL species which interacted with the TMPO in the total Al content, Ω(31PLAS) = 27.0% is the proportion of the 31P signals at 65 and 69 ppm in the quantitative 31P MAS NMR spectrum (see Table S1†), and χ(P/Al) = 0.42 is the atomic ratio of the P to Al of the sample obtained using ICP. Therefore, the calculated Ω(FALIII) = 11.6% suggested that a considerable amount of LAS formed and was located in the framework of the dehydrated H-ZSM-5 zeolite, this was in agreement with the relative content of the Lewis acid sites (13.3%) determined using our Fourier transform infrared (FT-IR) analysis of the pyridine adsorption measurements (Table 2 and Fig. S14†).
Brønsted/Lewis synergy effect studied using 2D 27Al DQ–SQ NMR and DFT calculations
We have successfully obtained a high-resolution 27Al–27Al DQ–SQ homonuclear correlation spectra of the hydrated zeolites (HY, MOR, ZSM-5, etc.) using our developed BR212 recoupling scheme at a high magnetic field,32,41 in which only Al species from the framework BAS and extra-framework LAS were considered. In the case of non-hydrated zeolite catalysts, the broadening and overlap of the 27Al quadrupolar patterns usually results in considerable barriers to the implementation of the 2D 27Al DQ–SQ experiment. Fortunately, the improved resolution of the 27Al MAS NMR spectra of the dehydrated H-ZSM-5 zeolite after TMPO adsorptions offers an opportunity to investigate the intrinsic structure of different framework Al species under the dehydrated state, the acidic natures of which have been clearly characterized. Fig. 6a displays the 27Al–27Al DQ–SQ MAS NMR spectrum of the TMPO-loaded H-ZSM-5 with P/Al = 0.42 at 18.8 T. Two auto-correlation peaks can be observed, in which the intense blue part indicates that the tetra-coordinated framework Al species (Ala) is in close proximity to another (Ala), and the weak orange part is ascribed to the spatial proximity of a low content of Alb–Alb or Alc–Alc pairs. Note that, it is still hard to discriminate the Alb and Alc in the 27Al DQ–SQ MAS NMR spectrum. In addition, the appearance of an intense cross-peak pair between Ala and Alb (red part in Fig. 6a) provided direct evidence of the spatial proximity between the framework Brønsted and Lewis acid sites, in which Ala and Alb had been confirmed from the Brønsted acid complex [(CH3)3PO⋯H–OAl] and the Lewis acid complex [(CH3)3PO⋯Al] in the TMPO-loaded H-ZSM-5, respectively. Therefore, it implied that the superacidity characterized by the 31P NMR signals at 85–88 ppm (Fig. 3) on the specific BAS could be derived from the influence of the neighboring tri-coordinated FAL (LAS), generating a Brønsted/Lewis synergy effect. The assignment was also confirmed by the absence of the 31P signals at 85–88 ppm on a quantitatively TMPO-adsorbed H-ZSM-5 zeolite with a low Si/Al of 140 (see Fig. S15†), which has a significantly reduced probability of a Al–Al pair being in close proximity in the zeolite framework owing to the much lower Al content.
 | ||
| Fig. 6 2D 27Al–27Al DQ–SQ MAS NMR spectrum (a) of dehydrated and TMPO-loaded H-ZSM-5 zeolite (P/Al = 0.42), recorded at 18.8 T with a spinning speed of 20 kHz. Local theoretically optimized structures of TMPO adsorbed at the isolated Brønsted site (b) and the Brønsted/Lewis acid synergetic site (c) with a tri-coordinated FAL in close proximity. The predicted 31P chemical shifts of the TMPO (blue numbers) and 27Al chemical shifts of the corresponding acid sites (black numbers) are labeled in ppm. | ||
In order to gain further insights into the Brønsted/Lewis synergy effect, we also performed DFT calculations (see Fig. S16†). As shown in the local theoretically optimized structures (Fig. 6b and c), for TMPO adsorbed on the BAS with a neighboring tri-coordinated FAL site, a 31P chemical shift of 83 ppm was theoretically predicted, while for TMPO adsorbed on the isolated BAS, a 31P chemical shift of 76 ppm was achieved, this was consistent with our experimental observations. The calculated 31P and 27Al isotropic chemical shifts for different adsorption models are listed in Table S3.† Except for the unreacted “NMR-invisible” framework LAS (tri-coordinated FAL), the chemical shift parameters extracted from the DFT calculations were consistent with our experimental results. These results indicate that the synergistic effect between the framework BAS and the framework LAS (tri-coordinated FAL) owing to their spatial proximity (Fig. 5e) leads to an enhancement of the Brønsted acidity and thus generates the superacidity in the H-ZSM-5 zeolite.
Conclusions
Tri-coordinated framework aluminum species can also serve as Lewis acid sites in principle, however, one of the fundamental challenges is to observe and distinguish these tri-coordinated framework aluminum species in zeolites. Their relatively low concentration and largely distorted local environment make them “NMR-invisible” in zeolites, thus experimental studies on their acidic properties are rarely reported. Benefiting from current state-of-the-art multi-nuclear and multi-dimensional solid-state NMR spectroscopy, unique insights into the structure and properties of tri-coordinated FAL species acting as Lewis acid sites in H-ZSM-5 have been achieved in conjunction with TMPO probe molecule adsorptions. As the unsaturated tri-coordinated FAL species can act with the basic TMPO probe molecules to form distorted tetrahedral FAL sites, two tri-coordinated FAL species with different Lewis acidities were discriminated using sensitivity-enhanced 2D 31P–27Al D-HMQC MAS NMR experiments. In particular, the adsorption models of the TMPO probe molecules on distinct acidic sites in H-ZSM-5 were clearly illustrated, and the strong 31P–27Al dipolar interaction from the Lewis complex [(CH3)3PO⋯Al] was confirmed using measurements of the distance between the corresponding 31P and 27Al atoms. Furthermore, the remarkably improved resolution and sensitivity of the 27Al MAS NMR spectrum of the TMPO-adsorbed H-ZSM-5 zeolite allowed us to obtain a well-resolved 2D 27Al DQ–SQ homonuclear correlation spectrum, and thus to understand the structure of the different framework acid sites in the non-hydrated zeolite. Consequently, the superacidity of BAS (with acid strength stronger than 100% H2SO4) induced by the synergistic effect between the framework BAS and framework LAS (tri-coordinated FAL) was evidenced by their close spatial proximity in dehydrated H-ZSM-5 zeolites and the observation of a 31P chemical shift of up to 85–88 ppm for the adsorbed TMPO. Notably, apart from the extra-framework Al as a LAS in the zeolite, the existence of a considerable amount of tri-coordinated framework Al serving as LAS in the zeolite framework suggests that further studies are needed to understand its catalytic function, which may be essential to rationally design highly efficient zeolite catalysts. The detailed characterization of the tri-coordinated framework Al in H-ZSM-5 in this work provides a useful strategy to explore the acidic nature of this type of “moisture-sensitive” species in zeolite catalysts, and may shed light on the experimental exploration of its structure–function relationship in zeolite catalysis.
Experimental section
Sample preparation
The parent NH4-ZSM-5 with Si/Al = 25 and Si/Al = 140 was purchased from Zeolyst International. The sample was calcined at 773 K under a dry air atmosphere for 10 h to remove the template and the H-form of ZSM-5 was obtained. Prior to the adsorption of TMPO, the sample was dehydrated on a vacuum line. The temperature was gradually increased at a rate of 1 °C min−1 and the sample was kept at a final temperature of 673 K at a pressure below 10−3 Pa overnight. The detailed procedures involved in introducing the TMPO probe molecule onto the sample can be found elsewhere.45 In brief, a known amount of TMPO dissolved in anhydrous CH2Cl2 was first added into a vessel containing the dehydrated H-ZSM-5 sample in a N2 glovebox, followed by removal of the CH2Cl2 solvent by evacuation on a vacuum line at 323 or 363 K. To ensure a uniform adsorption of the TMPO molecules in the pores/channels of the zeolites, the sealed vessel was further subjected to a thermal treatment at 453 K for 2 h. Finally, the sample was transferred into a ZrO2 MAS rotor in the N2 glovebox prior to the solid-state NMR experiments.ICP experiment
Atomic emission spectroscopy (AES-ICP) was used to determine the relative amounts of silicon, aluminum and phosphorus in the TMPO-loaded H-ZSM-5 samples. The Na and Al contents were measured using AES-ICP for a 10 mg sample dissolved in 50 mL HF solution.FT-IR of pyridine adsorption
The FT-IR analysis of the pyridine adsorption measurements were conducted on a Bruker Tensor 27 spectrometer. About 20 mg of the sample was pressed into a self-supported wafer with a diameter of 13 mm. Prior to the measurement, the catalysts were evacuated to 10−2 Pa at 673 K for 2 h. After cooling down to room temperature, pyridine vapor was introduced into the sample cell at room temperature for 30 min to allow it to reach equilibrium, the residual pyridine was removed by vacuum. The FT-IR spectra of the pyridine-adsorbed samples were measured at 423, 523, and 623 after evacuation for 30 min, respectively.Solid-state NMR experiments
For all 2D HETCOR experiments, the isotope within the braces indicates that it is indirectly detected in the F1 dimension. 1D 1H MAS NMR and 31P MAS NMR experiments were performed on an 9.4 T Bruker AVANCE-III spectrometer using 4 mm rotors at a spinning frequency of 10 kHz (operating at a Larmor frequency of 399.3 MHz and 161.7 MHz, respectively). The 1H single-pulse acquisition was employed with a pulse width of 3.9 μs (ca. π/2 pulse), a recycle delay of 3 s, and 32 scans. The 31P single-pulse acquisition was employed with a pulse width of 3 μs (ca. π/2 pulse), a recycle delay of 30 s, and 1024 scans, in which the RF amplitude of 1H TPPM decoupling was set to ca. 50 kHz.The 29Si MAS NMR experiments were performed on an 11.7 T Bruker AVANCE-III spectrometer using 7 mm rotors at a spinning frequency of 6 kHz (operating at a Larmor frequency of 99.4 MHz). The 29Si single-pulse acquisitions were employed with a pulse width of 8.4 μs (ca. π/2 pulse), a recycle delay of 30 s, and 320 scans, in which the RF amplitude of 1H TPPM decoupling was set to ca. 38 kHz.
The single-pulse 27Al MAS NMR data were collected on an 11.7 T Bruker AVANCE-III spectrometer with a 4 mm double-resonance probe at a spinning rate of 10 kHz, a pulse width of 0.4 μs (ca. π/6 pulse), a recycle delay of 1 s, and 2048 scans. To improve the resolution, the single-pulse 27Al MAS NMR data were also collected on an 18.8 T Bruker AVANCE-III spectrometer (corresponding to a 27Al Larmor frequency of 208.6 MHz) with an 1.9 mm double-resonance probe at a spinning rate of 40 kHz, a pulse width of 0.3 μs (ca. π/9 pulse), a recycle delay of 1 s, and 4096 scans.
The 2D 27Al triple-quantum (3Q) MAS z-filtering NMR experiments with fast amplitude modulation (FAM) enhancement50 were also carried out at 18.8 T. The pulse duration was set to 3.6 μs for the first strong pulse with νRF ≈ 125 kHz. A fast amplitude-modulated (FAM) pulse train consisting of four loops of two short pulses (νRF ≈ 125 kHz) and two delays with the same duration of 0.7 μs was used to convert p = ±3 to ±1 coherences. The “soft” π/2 pulses were set to 10.4 μs.
The 1D 31P{27Al} S-RESPDOR and 2D 31P{27Al} PT-D-HMQC MAS NMR experiments were performed on an 11.7 T Bruker AVANCE-III spectrometer using commercial 7 mm rotors with an o-ring cap at a spinning frequency of 6 kHz, tuned and matched to the 1H, 31P and 27Al Larmor frequencies (500.6 MHz, 202.6 MHz and 130.5 MHz, respectively). 1H–31P CP with a contact time of 8 ms was employed to prepare the initial 31P signal.
For the 31P{27Al} S-RESPDOR experiments, a saturation pulse on the 27Al channel with an amplitude of ca. 25 kHz and a length of 166.67 μs (TR) was irradiated at ca. 30 ppm to transfer the 31P–27Al interactions. For the 2D 31P{27Al} PT-D-HMQC experiments, repetitive sideband-selective (SS) WURST-80 adiabatic pulses with a length of 166.67 μs were employed on the Al channel during SR4 recoupling to accelerate the coherence transfers between 31P and 27Al. The values for the peak RF amplitude and offset of WURST-80 were optimized with (νmax1, νoffset) = (7.5, 175) kHz on the 27Al channel. For the 2D 27Al{31P} D-HMQC and 27Al DQ–SQ MAS NMR experiments, the central transition (CT) enhancement of the initial magnetizations (27Al) was obtained by using a SS-WURST-80 irradiation with a length of 1 ms. More details on the NMR parameters of the 2D or double-resonance experiments can be found in Table S2.†
All simulations of the 1D MAS NMR spectra or slices were performed with the DMFIT software.51 The chemical shifts for 1H, 31P, 29Si and 27Al were referenced to adamantane, 85% H3PO4, kaolinite and 1 M aqueous Al(NO3)3, respectively.
DFT calculations
All of the structures of the ZSM-5 zeolite were represented by a 64 T model (containing the complete double 10-membered ring (MR) intersection pores formed by 10-MR straight and 10-MR sinusoidal pore channels), which were extracted from their crystallographic structural data (http://www.iza-structure.org/databases/). The terminal Si–H was fixed at a bond length of 1.47 Å, oriented along the direction of the corresponding Si–O bond. The geometries of TMPO adsorbed at the isolated Brønsted acid site and the Brønsted/Lewis synergetic site of ZSM-5 were optimized over the 64 T cluster model by the combined theoretical ONIOM method. In the calculations, the TMPO and the high layer atoms of the zeolite framework were allowed to fully relax with the dispersion-corrected ωB97XD functional52 with a 6-31G (d, p) basis set, whereas the remaining atoms were fixed at their crystallographic positions using the AM1 method. Based on the optimized structures, the 31P and 27Al NMR chemical shifts were then calculated at the ωB97XD/TZVP level using the GIAO method.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants, 91745111, 21573278, 21733013, 21622311 and 21673283), the Bureau of International Cooperation (112942KYSB20180009) and the Youth Innovation Promotion Association, The Chinese Academy of Sciences. J. Trébosc and O. Lafon thank the Chevreul Institute (FR 2638) supported by the Ministère de l’Enseignement Supérieur, de la Recherche et de l’innovation, the Région Nord-Pas de Calais and the Fonds Européen de Développement des Régions. Financial support from the TGIR-RMN-THC Fr3050 CNRS for conducting the research is gratefully acknowledged. We gratefully thank Prof. Christophe Copéret for the helpful discussions.References
- J. Biswas and I. E. Maxwell, Appl. Catal., 1990, 63, 197–258 CrossRef CAS
.
- M. Moliner, Y. Roman-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed
- A. Corma, V. Martínez-Soria and E. Schnoeveld, J. Catal., 2000, 192, 163–173 CrossRef CAS
- J. Wahlen, S. Dehertogh, D. Devos, V. Nardello, S. Bogaert, J. Aubry, P. Alsters and P. Jacobs, J. Catal., 2005, 233, 422–433 CrossRef CAS
- A. Vjunov, J. L. Fulton, T. Huthwelker, S. Pin, D. H. Mei, G. K. Schenter, N. Govind, D. M. Camaioni, J. Z. Hu and J. A. Lercher, J. Am. Chem. Soc., 2014, 136, 8296–8306 CrossRef CAS PubMed
- T. Y. Liang, J. L. Chen, Z. F. Qin, J. F. Li, P. F. Wang, S. Wang, G. F. Wang, M. Dong, W. B. Fan and J. G. Wang, ACS Catal., 2016, 6, 7311–7325 CrossRef CAS
- K. Z. Chen, M. Abdolrhamani, E. Sheets, J. Freeman, G. Ward and J. L. White, J. Am. Chem. Soc., 2017, 139, 18698–18704 CrossRef CAS PubMed
- H. M. Kao, C. P. Grey, K. Pitchumani, P. H. Lakshminarasimhan and V. Ramamurthy, J. Phys. Chem. A, 1998, 102, 5627–5638 CrossRef CAS
- S. M. Maier, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2011, 115, 8005–8013 CrossRef CAS
- W. O. Haag, R. M. Lago and P. B. Weisz, Nature, 1984, 309, 589–591 CrossRef CAS
- S. H. Li, A. M. Zheng, Y. C. Su, H. L. Zhang, L. Chen, J. Yang, C. H. Ye and F. Deng, J. Am. Chem. Soc., 2007, 129, 11161–11171 CrossRef CAS PubMed
- D. E. Perea, I. Arslan, J. Liu, Z. Ristanovic, L. Kovarik, B. W. Arey, J. A. Lercher, S. R. Bare and B. M. Weckhuysen, Nat. Commun., 2015, 6, 7589 CrossRef PubMed
- H. Koller, T. Uesbeck, M. R. Hansen and M. Hunger, J. Phys. Chem. C, 2017, 121, 25930–25940 CrossRef CAS
- C. Schroeder, M. R. Hansen and H. Koller, Angew. Chem., Int. Ed., 2018, 57, 14281–14285 CrossRef CAS PubMed
- M. Milina, S. Mitchell, P. Crivelli, D. Cooke and J. Perez-Ramirez, Nat. Commun., 2014, 5, 3922 CrossRef
- Z. Wang, L. Wang, Y. Jiang, M. Hunger and J. Huang, ACS Catal., 2014, 4, 1144–1147 CrossRef CAS
- S. Prodinger, M. A. Derewinski, A. Vjunov, S. D. Burton, I. Arslan and J. A. Lercher, J. Am. Chem. Soc., 2016, 138, 4408–4415 CrossRef CAS PubMed
- I. Yarulina, K. De Wispelaere, S. Bailleul, J. Goetze, M. Radersma, E. Abou-Hamad, I. Vollmer, M. Goesten, B. Mezari, E. J. M. Hensen, J. S. Martinez-Espin, M. Morten, S. Mitchell, J. Perez-Ramirez, U. Olsbye, B. M. Weckhuysen, V. Van Speybroeck, F. Kapteijn and J. Gascon, Nat. Chem., 2018, 10, 804–812 CrossRef CAS PubMed
- R. Weingarten, G. A. Tompsett, W. C. Conner and G. W. Huber, J. Catal., 2011, 279, 174–182 CrossRef CAS
- M. Orazov and M. E. Davis, Chem. Sci., 2016, 7, 2264–2274 RSC
- P. Batamack, C. Doremieuxmorin, R. Vincent and J. Fraissard, Microporous Mater., 1994, 2, 515–524 CrossRef CAS
- A. Zheng, S. Li, S. B. Liu and F. Deng, Acc. Chem. Res., 2016, 49, 655–663 CrossRef CAS PubMed
- C. Wang, Y. Y. Chu, J. Xu, Q. Wang, G. D. Qi, P. Gao, X. Zhou and F. Deng, Angew. Chem., Int. Ed., 2018, 57, 10197–10201 CrossRef CAS PubMed
- A. Zecchina, S. Bordiga, G. Spoto, D. Scarano, G. Petrini, G. Leofanti, M. Padovan and C. O. Arean, J. Chem. Soc., Faraday Trans., 1992, 88, 2959–2969 RSC
- F. Collignon, P. A. Jacobs, P. Grobet and G. Poncelet, J. Phys. Chem. B, 2001, 105, 6812–6816 CrossRef CAS
- J. A. van Bokhoven, A. M. J. van der Eerden and D. C. Koningsberger, J. Am. Chem. Soc., 2003, 125, 7435–7442 CrossRef CAS PubMed
- W. P. Zhang, S. T. Xu, X. W. Han and X. H. Bao, Chem. Soc. Rev., 2012, 41, 192–210 RSC
- S. E. Ashbrook and S. Sneddon, J. Am. Chem. Soc., 2014, 136, 15440–15456 CrossRef CAS PubMed
- Z. C. Wang, Y. J. Jiang, O. Lafon, J. Trebosc, K. D. Kim, C. Stampfl, A. Baiker, J. P. Amoureux and J. Huang, Nat. Commun., 2016, 7, 13820 CrossRef CAS PubMed
- C. Coperet, W. C. Liao, C. P. Gordon and T. C. Ong, J. Am. Chem. Soc., 2017, 139, 10588–10596 CrossRef CAS PubMed
- D. Xiao, S. T. Xu, X. W. Han, X. E. Bao, Z. M. Liu and F. Blanc, Chem. Sci., 2017, 8, 8309–8314 RSC
- Z. W. Yu, A. M. Zheng, Q. Wang, L. Chen, J. Xu, J. P. Amoureux and F. Deng, Angew. Chem., Int. Ed., 2010, 49, 8657–8661 CrossRef CAS PubMed
- J. Brus, L. Kobera, W. Schoefberger, M. Urbanova, P. Klein, P. Sazama, E. Tabor, S. Sklenak, A. V. Fishchuk and J. Dedecek, Angew. Chem., Int. Ed., 2015, 54, 541–545 CAS
- C. P. Grey and A. J. Vega, J. Am. Chem. Soc., 1995, 117, 8232–8242 CrossRef CAS
- A. P. M. Kentgens, D. Iuga, M. Kalwei and H. Koller, J. Am. Chem. Soc., 2001, 123, 2925–2926 CrossRef CAS PubMed
- J. Jiao, J. Kanellopoulos, W. Wang, S. S. Ray, H. Foerster, D. Freude and M. Hunger, Phys. Chem. Chem. Phys., 2005, 7, 3221–3226 RSC
- P. A. Jacobs and H. K. Beyer, J. Phys. Chem., 1979, 83, 1174–1177 CrossRef CAS
- Q. L. Wang, G. Giannetto, M. Torrealba, G. Perot, C. Kappenstein and M. Guisnet, J. Catal., 1991, 130, 459–470 CrossRef CAS
- G. L. Woolery, G. H. Kuehl, H. C. Timken, A. W. Chester and J. C. Vartuli, Zeolites, 1997, 19, 288–296 CrossRef CAS
- E. Lam, A. Comas-Vives and C. Copéret, J. Phys. Chem. C, 2017, 121, 19946–19957 CrossRef CAS
- Z. W. Yu, S. H. Li, Q. Wang, A. M. Zheng, X. Jun, L. Chen and F. Deng, J. Phys. Chem. C, 2011, 115, 22320–22327 CrossRef CAS
- X. F. Yi, K. Y. Liu, W. Chen, J. J. Li, S. T. Xu, C. B. Li, Y. Xiao, H. C. Liu, X. W. Guo, S. B. Liu and A. M. Zheng, J. Am. Chem. Soc., 2018, 140, 10764–10774 CrossRef CAS PubMed
- J. F. Haw, J. B. Nicholas, T. Xu, L. W. Beck and D. B. Ferguson, Acc. Chem. Res., 1996, 29, 259–267 CrossRef CAS
- E. F. Rakiewicz, A. W. Peters, F. Wormsbecher, K. J. Sutovich and K. T. Mueller, J. Phys. Chem. B, 1998, 102, 2890–2896 CrossRef CAS
- A. Zheng, S. B. Liu and F. Deng, Chem. Rev., 2017, 117, 12475–12531 CrossRef CAS PubMed
- Q. Wang, Y. X. Li, J. Trebosc, O. Lafon, J. Xu, B. W. Hu, N. D. Feng, Q. Chen, J. P. Amoureux and F. Deng, J. Chem. Phys., 2015, 142 Search PubMed
- A. Janda and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 19193–19207 CrossRef CAS PubMed
- Q. Zhao, W. H. Chen, S. J. Huang, Y. C. Wu, H. K. Lee and S. B. Liu, J. Phys. Chem. B, 2002, 106, 4462–4469 CrossRef CAS
- L. Chen, X. Y. Lu, Q. A. Wang, O. Lafon, J. Trebosc, F. Deng and J. P. Amoureux, J. Magn. Reson., 2010, 206, 269–273 CrossRef CAS PubMed
- P. K. Madhu, A. Goldbourt, L. Frydman and S. Vega, Chem. Phys. Lett., 1999, 307, 41–47 CrossRef CAS
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J. O. Durand, B. Bujoli, Z. H. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS
- J. D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02634g |
| This journal is © The Royal Society of Chemistry 2019 |