
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlleviation of symptoms of Alzheimer's disease by diminishing Aβ neurotoxicity and neuroinflammation†
Tao
Yang‡
 a,
Zhenzhu
Zhu‡
a,
Enmao
Yin
a,
Yanqing
Wang
b,
Changli
Zhang
c,
Hao
Yuan
d,
Hongmei
Zhang
a,
Suxing
Jin
a,
Zijian
Guo
d and
Xiaoyong
Wang
*a
a,
Zhenzhu
Zhu‡
a,
Enmao
Yin
a,
Yanqing
Wang
b,
Changli
Zhang
c,
Hao
Yuan
d,
Hongmei
Zhang
a,
Suxing
Jin
a,
Zijian
Guo
d and
Xiaoyong
Wang
*a
aState Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing 210023, P. R. China. E-mail: boxwxy@nju.edu.cn
bSchool of Chemistry and Chemical Engineering, Yancheng Teachers University, Yancheng 224002, P. R. China
cDepartment of Chemistry, Nanjing Xiaozhuang College, Nanjing, 210017, P. R. China
dState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China
First published on 19th September 2019
Abstract
Alzheimer's disease (AD) is one of the most prevailing neurodegenerative illnesses in the elderly. Accumulation of amyloid-β peptide (Aβ) and inflammation play critical roles in the pathogenesis and development of AD. Multi-target drugs may interdict the progress of AD through a synergistic mechanism. A neuromodulator, 2-((1H-benzo[d]imidazole-2-yl)methoxy)benzoic acid (BIBA), consisting of an Aβ-targeting group and a derivative of anti-inflammatory aspirin was designed as a potential anti-AD agent. BIBA exhibits a remarkable inhibitory effect on the self- and metal-induced Aβ aggregations and shows outstanding anti-inflammatory activity simultaneously. The neurotoxicity of Aβ aggregates is attenuated, and the production of pro-inflammatory cytokines (PICs), such as IL-6, IL-1β and TNF-α, in microglia stimulated by lipopolysaccharide (LPS) or Aβ is reduced. Owing to the synergy between the inhibition of Aβ oligomerization and downregulation of PICs, BIBA markedly prolongs the lifespan and relieves the Aβ-induced paralysis of Aβ-transgenic Caenorhabditis elegans, thus showing the potential to ameliorate the symptoms of AD through inhibiting Aβ neurotoxicity and deactivating microglia. These findings demonstrate that both Aβ aggregation and neuroinflammation are therapeutic targets for anti-AD drugs, and dual-functional agents that integrate anti-Aβ and anti-inflammatory capabilities have great advantages over the traditional single-target agents for AD treatment.
Introduction
Alzheimer's disease (AD) is a fatal and age-related neurodegenerative disorder characterized by memory loss and progressive cognitive impairment.1,2 Currently, approved pharmacotherapies only provide short-term or mild relief from AD symptoms, but do not cure or prevent the process of AD.3 Although the exact pathogenesis of AD is elusive, amyloid cascade hypothesis is the major etiologic basis for AD over the past few decades. It is believed that AD is caused by the accumulation of amyloid-β peptide (Aβ), which initiates a sequence of pathogenic events, such as the formation of neurofibrillary tangles (NFTs) and the onset of synaptic and neuronal dysfunction or death.4 In AD pathogenesis, Aβ species, mainly Aβ40 and more toxic Aβ42,5 aberrantly aggregate from non-toxic monomers into toxic oligomers, fibrils, and eventually senile plaques. Plenty of evidence suggests that Aβ aggregates, especially soluble oligomers, could impair synaptic and cognitive functions.6 Thus, Aβ oligomers and fibrils are regarded as the main therapeutic targets for AD.7 Since Aβ aggregation and its neurotoxicity could be aggravated by metal ions such as Zn2+ and Cu2+ in the AD brain,8,9 metal chelators that can regulate the distribution of metal ions are potential therapeutic agents for AD.10,11 Extensive studies have focused on the reduction of Aβ or its aggregation by targeting Aβ production or clearance,12–15 and some compounds have effectively prevented the de novo aggregation of Aβ; however, they cannot eliminate the existing Aβ oligomers and plaques in the brain.16 Consequently, such strategies hardly improve the cognition of patients, and none of them have gone through clinical trials so far.17,18On the other hand, activated microglia in the central nervous system and increased inflammatory mediators around Aβ plaques in the brain of AD patients have been related to the pathogenesis of AD. Aβ and NFTs can indirectly activate immune response or microglia and lead to the release of various pro-inflammatory cytokines (PICs) and neurotoxic mediators.19 Microglia have two phenotypes of activation: pro-inflammatory M1 and immunosuppressive M2.20 M1 is associated with releasing PICs, such as interleukin-1β (IL-1β), interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), superoxide, nitric oxide (NO), reactive oxygen species (ROS), and expressing inducible nitric oxide synthase (iNOS). These PICs and mediators not only attenuate the phagocytosis of Aβ,21 but also affect the tau pathological metamorphosis, increasing tau phosphorylation and accelerating tangle formation,22 thereby inducing neurotoxicity. In addition, PICs and activated microglia could promote Aβ production, exacerbating the formation of Aβ fibrils and deposition.23,24 To make things worse, toxic Aβ oligomers and fibrils could be recognized by some receptors expressed by microglia,25,26 triggering the microglial activation through nuclear factor-kappa B (NF-κB) or mitogen-activated protein kinases (MAPKs).27 Aging and toxic conditions in the AD brain also favour the chronic activation of microglia and reduce their phagocytic capacity and prolong neuroinflammation.28 As a result, a vicious cycle between neuroinflammation and Aβ aggregation is formed, ultimately leading to the death of neurons and onset of AD.29,30 Thus, interfering with the vicious cycle may produce therapeutic effects on AD.
Anti-inflammatory drugs were expected to reduce the risk of AD, but clinical trials for these drugs have failed.31–34 Considering the pathogenic effect of Aβ aggregation and neuroinflammation in AD, we suppose that defence against both Aβ oligomers or plaques and PICs may produce a dual impact on the AD progression and protect the neurons in the brain. Herein we report a novel neuromodulator (BIBA, Fig. 1), which is a hybrid of a derivative of aspirin, a non-steroidal anti-inflammatory drug, and 2-(phenoxymethyl)-1H-benzoimidazole, an analogue of Aβ-targeting thioflavin T (ThT), for inhibiting the Aβ aggregation and neuroinflammation. As anticipated, BIBA showed remarkable inhibitory effects on self- and metal-induced Aβ aggregations, and exhibited obvious anti-inflammatory activity by suppressing intracellular lipopolysaccharide (LPS)- or Aβ-induced production of PICs in microglia. Moreover, it delayed the Aβ-induced paralysis and increased the lifespan of transgenic Caenorhabditis elegans (C. elegans). A preliminary test suggests that BIBA could rescue the memory deficits of the amyloid precursor protein/presenilin protein 1 (APP/PS1) mice through inhibiting Aβ oligomerization and overactivation of microglia. To the best of our knowledge, BIBA is the first anti-AD agent capable of regulating Aβ aggregation and neuroinflammation simultaneously.
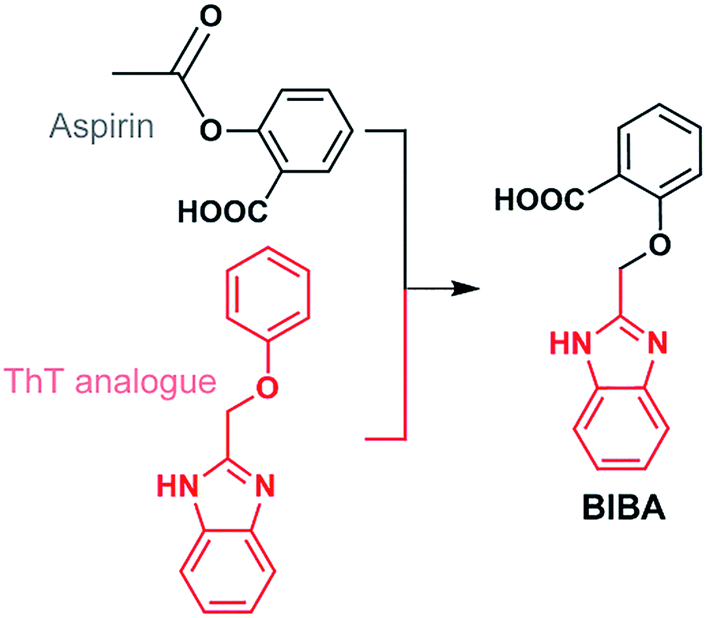 | ||
| Fig. 1 Construction of BIBA. | ||
Results and discussion
Design and synthesis of BIBA
BIBA was designed by linking an Aβ-targeting moiety with a derivative of aspirin. The 2-(phenoxymethyl)-1H-benzoimidazole group was selected due to its high lipophilicity and affinity for Aβ aggregates;35 the derivative of aspirin was chosen to suppress the generation of PICs by inhibiting cyclooxygenases (COXs).36 In addition, a low dose of aspirin could decrease Aβ plaques by upregulating the transcription factor EB,37 and benefit the inhibition of protein aggregation and corresponding toxicity through acetyl-donating activity.38 The synthesis and characterization of BIBA are shown in Scheme S1 and Fig. S1.† BIBA is stable over 24 h in buffer (pH 7.4), suggesting that it is suitable for biochemical experiments (Fig. S2†). The lipophilicity parameter (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) of BIBA determined by the shake-flask method is −0.04 ± 0.01, indicating that BIBA is more lipophilic than the aspirin derivative (logP = −1.40 ± 0.07). According to Lipinski's “rule of five”,39 BIBA could potentially cross the BBB (see Table S1†). To verify the BBB-penetrating ability, we analyzed the brains of C57BL/6J mice after intravenous injection of BIBA by HPLC. BIBA reached its maximum in the brain at around 10 min and attenuated gradually at 20 min after injection (Fig. S3†). The results confirm that BIBA can cross the BBB.
P) of BIBA determined by the shake-flask method is −0.04 ± 0.01, indicating that BIBA is more lipophilic than the aspirin derivative (logP = −1.40 ± 0.07). According to Lipinski's “rule of five”,39 BIBA could potentially cross the BBB (see Table S1†). To verify the BBB-penetrating ability, we analyzed the brains of C57BL/6J mice after intravenous injection of BIBA by HPLC. BIBA reached its maximum in the brain at around 10 min and attenuated gradually at 20 min after injection (Fig. S3†). The results confirm that BIBA can cross the BBB.
Prevention of Aβ aggregation
Aβ aggregations, especially those induced by Zn2+ or Cu2+, play important roles in neurodegenerative diseases, influencing both protein structures and oxidative stress.40 Therefore, the chelating behaviour of BIBA towards Zn2+ or Cu2+ was initially investigated by fluorescence titration in Tris–HCl buffer. BIBA gave sensitive fluorescence responses to both Zn2+ and Cu2+ in a 2:1 binding ratio (Fig. S4†), indicating that it can interact with Zn2+ or Cu2+ and potentially affect the metal-induced Aβ aggregations. The inhibitory effect of BIBA on Aβ aggregation was examined by circular dichroism (CD) spectroscopy, which is extensively used to monitor the secondary structural conversion of proteins. As shown in Fig. 2A, the CD spectrum of Aβ displays two negative peaks around 198 and 217 nm, which are characteristics of random coils and β-sheet conformation, respectively. When Aβ was incubated with Zn2+ or Cu2+, the negative peak at 198 nm disappeared and a new positive peak emerged around 194 nm, along with the increase of the negative peak around 217 nm. These changes indicate that the conformation of Aβ is transformed from random coils to β-sheets, thus signifying the formation of Aβ fibrils. The results are consistent with reported observations.41,42 Upon addition of BIBA, the negative band centred at 217 nm diminished, indicating that the level of Aβ fibrils was decreased. The morphological changes of Aβ in the absence and presence of metal ions and/or BIBA were directly visualized by transmission electron microscopy (TEM). As shown in Fig. 2B, some fibrils were observed in the solution of Aβ, and large amounts of mature aggregates were seen in the presence of Zn2+ and Cu2+, indicating that soluble β-sheet-rich fibrils and insoluble Aβ aggregates are formed.42 Massive Aβ aggregates were changed into short fibrils or sporadic granule-like species after incubation with BIBA, showing that the Aβ aggregations with or without metal ions were suppressed. The Western blot also showed that BIBA could inhibit the formation of insoluble Aβ fibrils and increase the number of monomers (Fig. 2C). The effect of BIBA on Aβ42 aggregation was also analyzed by using CD spectra, TEM and Western blot (Fig. S5†); the results are consistent with those for Aβ40. The CD spectra of Aβ42 were somewhat different from those of Aβ40 possibly because Aβ42 aggregated into fibrils more quickly than Aβ40, which was consistent with previous reports.43–45
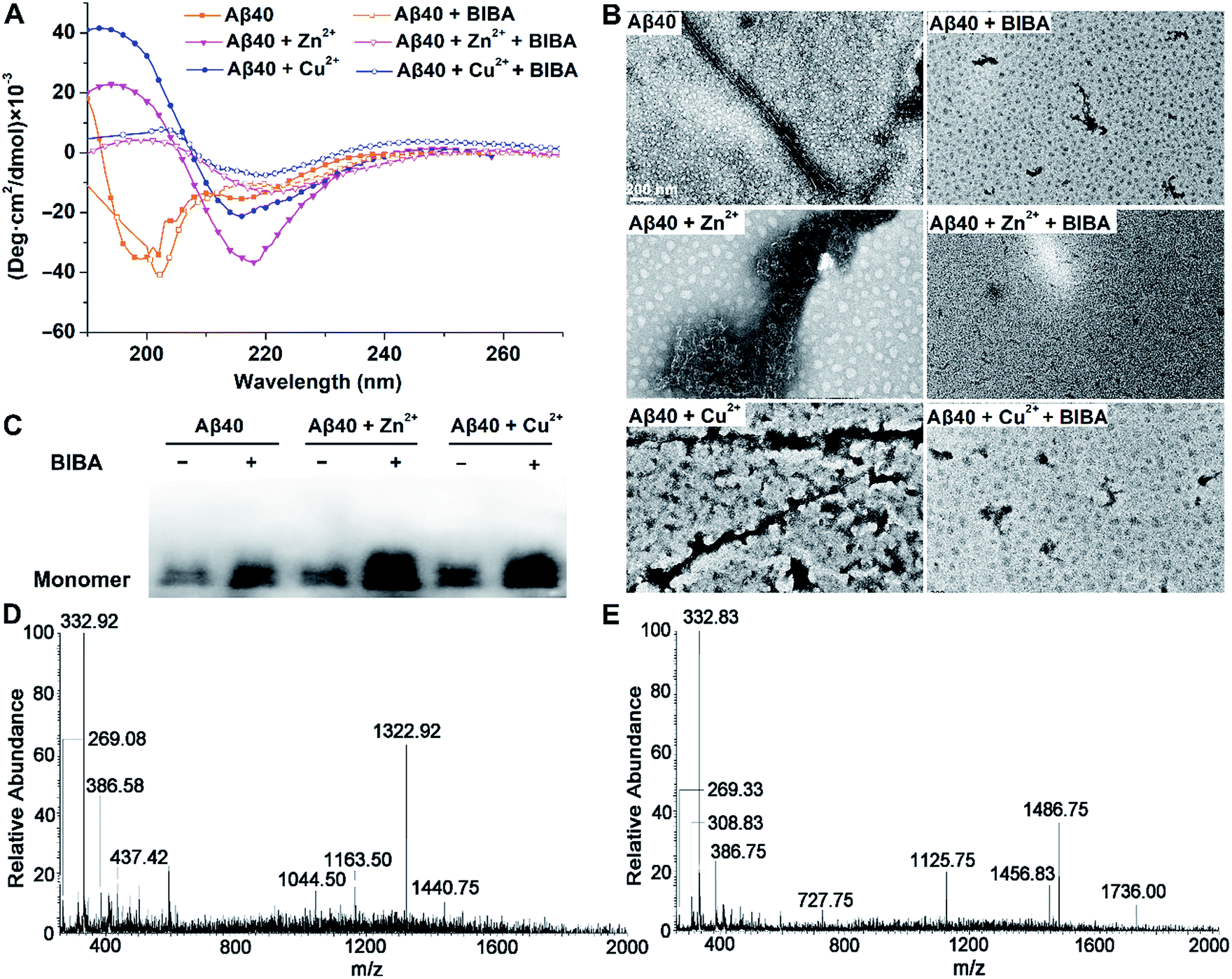 | ||
| Fig. 2 Prevention of self- or metal-induced Aβ40 aggregation by BIBA. (A) CD spectra, (B) TEM, and (C) Western blot of Aβ40 without or with metal ions and BIBA, and ESI-MS spectra of Aβ40 with (D) Zn2+ or (E) Cu2+ in the presence of BIBA ([Aβ] = [Zn2+/Cu2+] = 10 μM and [BIBA] = 20 μM). | ||
The interactions of BIBA with the Zn2+- or Cu2+-induced Aβ40 aggregates were further investigated by ESI-MS. As shown in Fig. 2D and E, no peaks assignable to Aβ40 species or BIBA–Aβ40 adducts are observed after the co-incubation of Aβ40, metal ions and BIBA (see Tables S2 and S3†), suggesting that no covalent interactions occurred between BIBA and Aβ40. Noticeably, several peaks corresponding to BIBA–metal ions and BIBA–metal–Aβ40 adducts were observed. All these results indicate that BIBA exhibits high potential to dissociate the Aβ aggregates without or with metal ions, and the chelation is involved in the inhibition of metal-induced Aβ aggregation.
Attenuation of Aβ neurotoxicity
The transition of Aβ from nontoxic monomers to toxic oligomers and plaques was believed to be a crucial reason for the neurotoxicity in AD.46 In addition, Zn2+ and Cu2+ are able to stabilize the soluble Aβ42 oligomers and hence aggravate the neuron death.47,48 The effect of BIBA on the Aβ-induced neurotoxicity in the absence and presence of Zn2+ or Cu2+ was tested in the mouse neuroblastoma Neuro2a (N2a) cell line, which was usually used to evaluate the drug toxicity in the study of AD.49 As shown in Fig. 3A, the viability of N2a cells treated with Aβ42 alone or in the presence of metal ions was less than 70%, which is consistent with a literature report;50 the viability increased remarkably when the cells were co-incubated with Aβ42 and BIBA in the presence and absence of metal ions. The results suggest that BIBA can effectively attenuate the cytotoxicity of Aβ42 aggregates with or without metal ions. BIBA per se was almost non-toxic toward N2a cells even at 50 μM (Fig. S6†).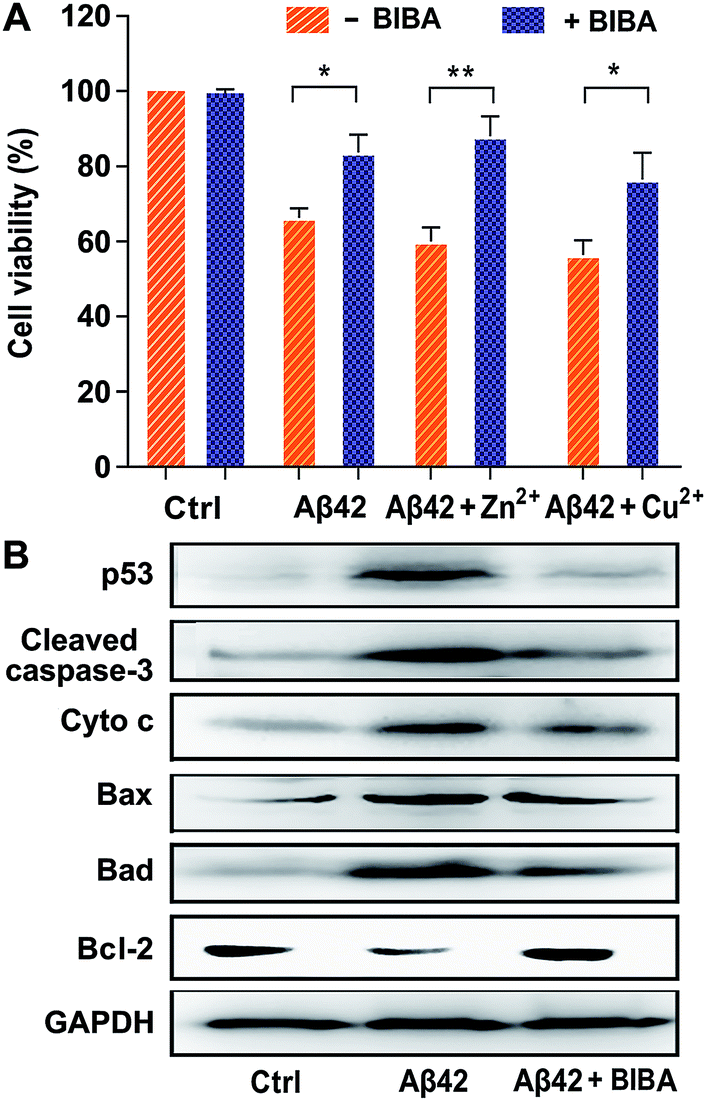 | ||
| Fig. 3 (A) Viability of N2a cells in the presence of Aβ42 without or with metal ions and BIBA after incubation for 24 h determined by the MTT assay, and (B) expression of apoptosis-related proteins in N2a cells stimulated with Aβ42 aggregates in the presence and absence of BIBA for 24 h detected by the Western blot (n = 3; **P < 0.001 and *P < 0.01; [Aβ] = [Zn2+/Cu2+] = 10 μM, and [BIBA] = 20 μM). | ||
It is known that neuronal apoptosis induced by neurotoxic Aβ aggregates plays a central role in triggering AD. The prevention of such apoptosis may represent an effective way for the treatment of AD. To explore the capability of BIBA to block the signalling pathway to Aβ-induced neuronal cell death, the expression of apoptotic proteins in N2a cells stimulated by Aβ42 in the presence of BIBA was measured by the Western blot. Among them, p53 is related to the induction of neuronal apoptosis in AD and believed to be one of the targets of nuclear Aβ;51 cleaved caspase-3 plays a critical role in the apoptotic signalling pathway;52 cytochrome c (Cyto c) is released from the mitochondria into the cytoplasm to initiate apoptosis; Bax and Bad are pro-apoptotic proteins in the Bcl-2 protein family; and Bcl-2 is an anti-apoptotic protein that inhibits the activation of the apoptotic pathway. As shown in Fig. 3B, after treatment with Aβ42, the expression of p53, cleaved caspase-3, Cyto c, Bax, and Bad was dramatically increased, but that of Bcl-2 was decreased; however, after addition of BIBA, the expression of pro-apoptotic proteins was down-regulated obviously, while that of Bcl-2 was up-regulated. The results suggest that BIBA can prevent the apoptosis of neural cells induced by Aβ.
Effect on paralysis and lifespan of transgenic C. elegans
Transgenic C. elegans strains expressing the human Aβ gene in muscles or neurons are widely used as in vivo models of AD for examining the accumulation and toxicity of Aβ and screening potential drugs, because the worms are simple in culturing and fast in generation, and have 302 neurons in hermaphrodites, a well-characterized nerve system, and a high level of homology with the human genome.53–57 The expression of the Aβ gene in muscles of C. elegans is associated with severe age-progressive paralysis,58 which is an apparent symptom and also a measurable phenotype regarded as a consequence of Aβ toxicity in these AD models. Hence, the effect of BIBA on the paralysis was investigated. In transgenic C. elegans strain CL4176, the aggregation of Aβ1–42 in the muscle tissue is temperature-inducible,59 which results in the time-dependent aggregation of Aβ and paralytic phenotype.60 This kind of worms produces low levels of Aβ and moves as rollers when grown at 15 °C (Fig. 4A top), and generates a large amount of Aβ and becomes straight and loses the moving ability when the temperature was upshifted to 25 °C (Fig. 4A bottom). Meanwhile, transgenic C. elegans strain CL802 that does not express the Aβ gene in muscle tissue61 and aspirin-treated CL4176 were used as the controls. As expected, BIBA and aspirin decreased the percent of paralysis from 58.7% to 6.3% and 17.4%, respectively, at 34 h after the temperature upshift (Fig. 4B). In addition, 92.6% of the untreated worms became paralyzed after 36 h, while only 27.2% of the BIBA-treated worms were paralyzed. By contrast, the mobility of the CL802 strain did not alter. These results indicate that BIBA can more effectively protect transgenic C. elegans against the Aβ-induced paralysis than aspirin.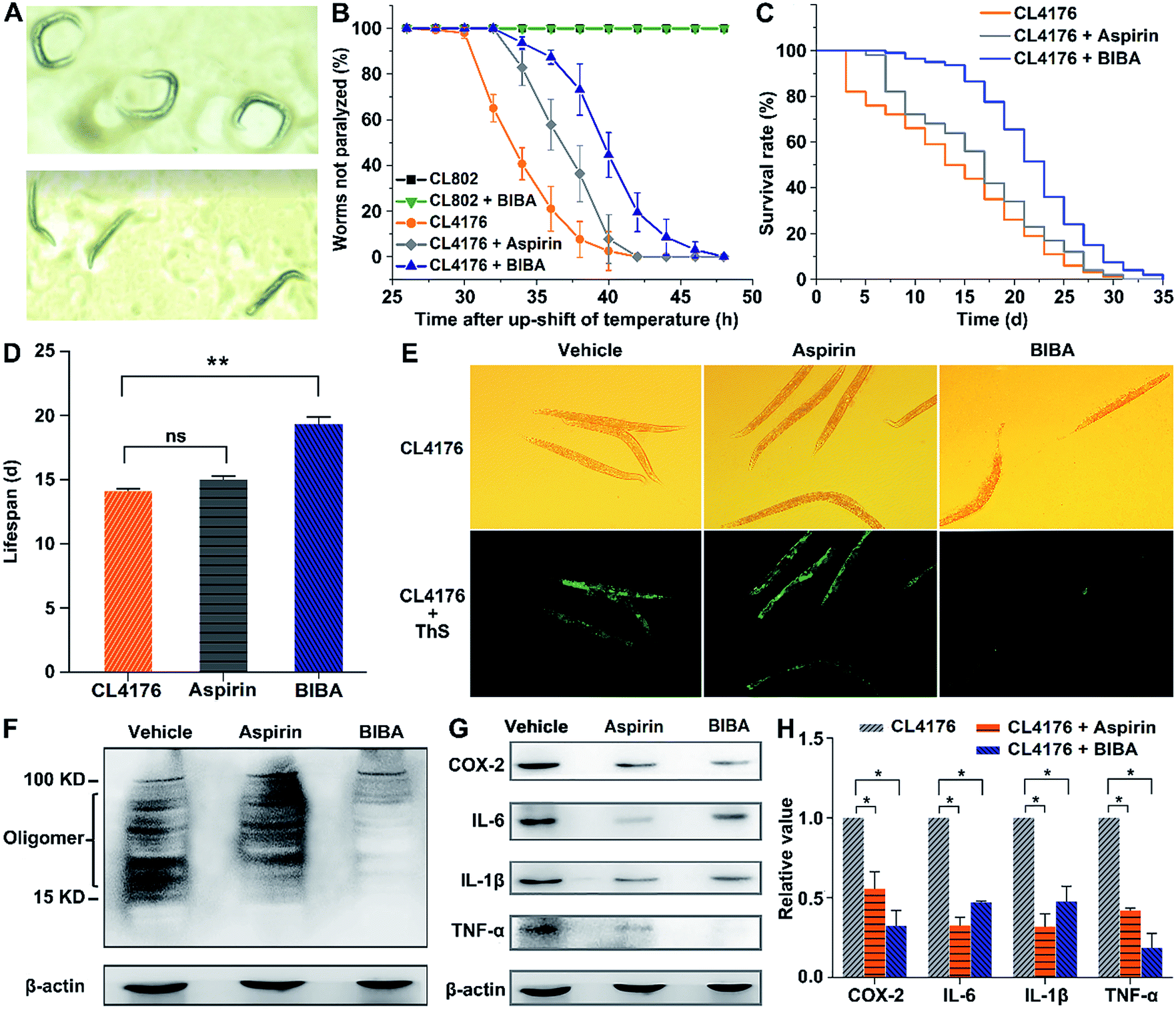 | ||
| Fig. 4 (A) Different motion modes of CL4176, (B) effects of drugs on the paralysis of CL802 or CL4176 worms at 25 °C, (C) survival curves of CL4176 worms after treatment with drugs at 15 °C, (D) mean lifespan of CL4176 worms after drug treatment calculated by using GraphPad Prism 7, (E) photos of CL4176 worms after drug treatment at 25 °C for 48 h and then stained with ThS, (F) Western blot of Aβ species and (G) inflammatory mediators in CL4176 worms treated with drugs at 25 °C for 48 h, and (H) quantitative analysis of inflammatory cytokines by using ImageJ and GraphPad Prism 7. All results are presented as mean ± SEM (n = 2); *p < 0.0001 ([aspirin] = [BIBA] = 20 μM). | ||
The effect of BIBA on the lifespan of CL4176 worms was further examined by analyzing the overall nematode survival. As shown in Fig. 4C and D, BIBA caused a significant increase in the lifespan of CL4176 worms as compared with the control, suggesting that it can mitigate the Aβ-related pathological threat to the worms. The same dose of aspirin was less effective for increasing the lifespan, which was consistent with the paralysis assay. We speculate that the advantages may arise from the synergistic effect of two functional moieties in BIBA. The administration of BIBA to wild-type (N2) worms at the L2 larval stage did not modify their survival (Fig. S7†), indicating that its toxicity is negligible.
The Aβ deposits in transgenic CL4176 were first examined by thioflavin S (ThS, a dye for Aβ plaques) staining, which could reflect the inhibitory effect of BIBA on Aβ aggregation through green fluorescence. Fig. 4E shows that the Aβ deposits were greatly reduced in worms fed with BIBA, while they did not respond obviously to aspirin. Furthermore, the Aβ species from the CL4176 worms fed with or without BIBA were analyzed by the Western blot. As shown in Fig. 4F, the Aβ-immunoreactive (6E10) bands of the tissues indicate that BIBA evidently reduced the amount of toxic Aβ oligomeric species. The expression of COX-2, TNF-α, IL-6 and IL-1β in CL4176 worms was also measured by the Western blot after the worms were fed with BIBA, aspirin or vehicle. As shown in Fig. 4G and H, the expression of PICs decreased markedly as compared with the control group. Apparently, the extension of lifespan and delay of paralysis in CL4176 by aspirin result from its anti-inflammatory ability rather than inhibitory ability to Aβ oligomerization, while the protective effect of BIBA against Aβ-induced toxicity in the worms arises from its inhibition of Aβ oligomerization and deposition as well as its anti-inflammatory ability. Aspirin alone exhibits a much weaker effect than BIBA due to the lack of anti-Aβ aggregation ability.
Inhibition of PICs
The immune system could be overactivated indirectly and lead to the release of various neurotoxic mediators and PICs, which may contribute to neuronal damage or degeneration in the brain of AD patients. The pro-inflammatory M1 phenotype of microglia is predominant at the site of neuroinflammation and related to the release of PICs, which may cause tissue damage and cell death. During chronic activation of microglia, sustained exposure of neurons to PICs, such as IL-6, IL-1β, and TNF-α, can cause neuronal dysfunction and cell death.62 To verify the anti-inflammatory activity of BIBA, the expression of PICs and neurotoxic mediators in BV-2 microglia was investigated by the Western blot after incubation with LPS or Aβ42 and BIBA, with LPS- or Aβ42-treated BV-2 cells as controls. As shown in Fig. 5A and B, the expression of PICs and mediators increased markedly when the cells were stimulated by LPS or Aβ42 as compared with the control group; however, BIBA significantly inhibited their expression under the same conditions.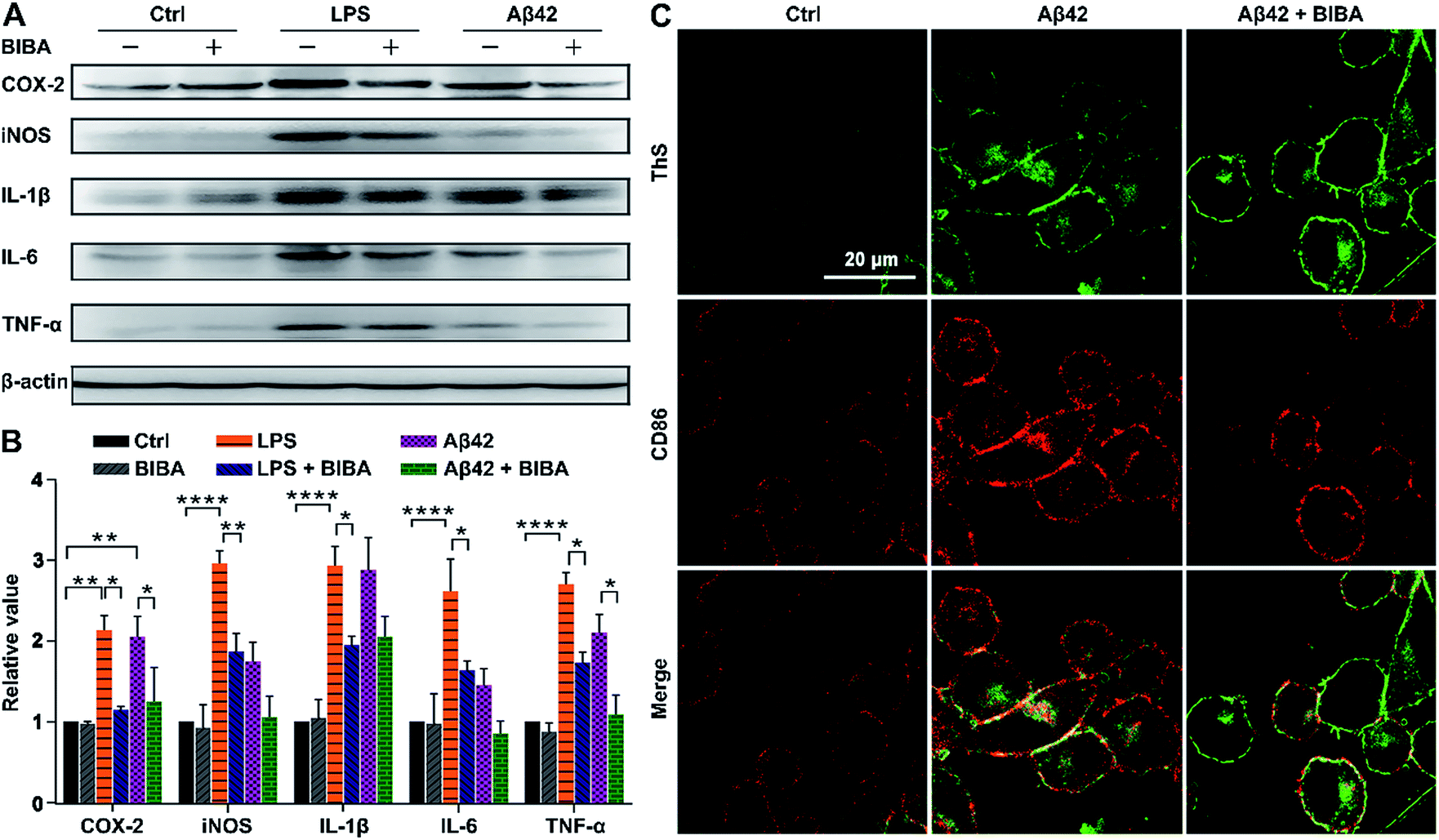 | ||
| Fig. 5 Inhibition effect of BIBA on PICs. (A) Western blot and (B) quantitative analysis of PICs and mediators in BV-2 cells stimulated by LPS or Aβ42 in the presence or absence of BIBA at 37 °C for 12 h, and (C) immunofluorescence images showing the expression of CD86 in Aβ-exposed BV2 microglia in the presence or absence of BIBA. The results are presented as mean ± SEM (n = 2); ****P < 0.0001, ***P < 0.001, **P < 0.01 and *P < 0.1 ([LPS] = 1 μg mL−1, [Aβ42] = 10 μM, and [BIBA] = 20 μM). | ||
Cluster of differentiation 86 (CD86) is closely correlated with microglia overactivation and is regarded as a marker of the M1 phenotype of microglia.63 To clarify the phenotype of BV-2 cells in the presence of BIBA, we detected the level of CD86 using immunohistochemistry. As shown in Fig. 5C, the expression of CD86 (red) was obviously increased in the Aβ-exposed microglia, and decreased after treatment with BIBA, suggesting that BIBA can protect BV-2 cells from overactivation induced by Aβ42. This deactivation ability towards microglia is more obvious than that of adiponectin, an adipokine derived from adipose tissue.64
Reduction of Aβ oligomers and overactivation of microglia in vivo
APP/PS1 double-TG model mice produce elevated levels of human Aβ by expressing mutant human APP and PS1, forming amyloid plaques as early as 4 months old and losing cognition when 6–8 months old.65 To verify the inhibitory effect of BIBA on the formation of Aβ oligomers and plaques in vivo, the brains of the BIBA-treated APP/PS1 mice were examined by immunohistochemical analysis and the Western blot. As shown in Fig. 6, green fluorescence spots of Aβ plaques are observed in the brain of untreated and aspirin-treated 9-month old mice after staining with ThS, which could visualize the dense-core Aβ plaques,66 while the formation of plaques was dramatically inhibited in the BIBA-treated APP/PS1 mice. The formation of Aβ oligomers was also examined by the Western blot. BIBA greatly decreased the amount of oligomeric species in soluble fractions; however, the effect of aspirin seems to be weaker than the reported observations possibly due to the lower dosage (Fig. S8†).34 Aβ monomers did not appear because in APP/PS1 mice Aβ exists mainly in the form of neurotoxic oligomers. These results are consistent with those observed for C. elegans, thus confirming that BIBA can effectively reduce the Aβ plaques and oligomers formed in the brain of AD mice.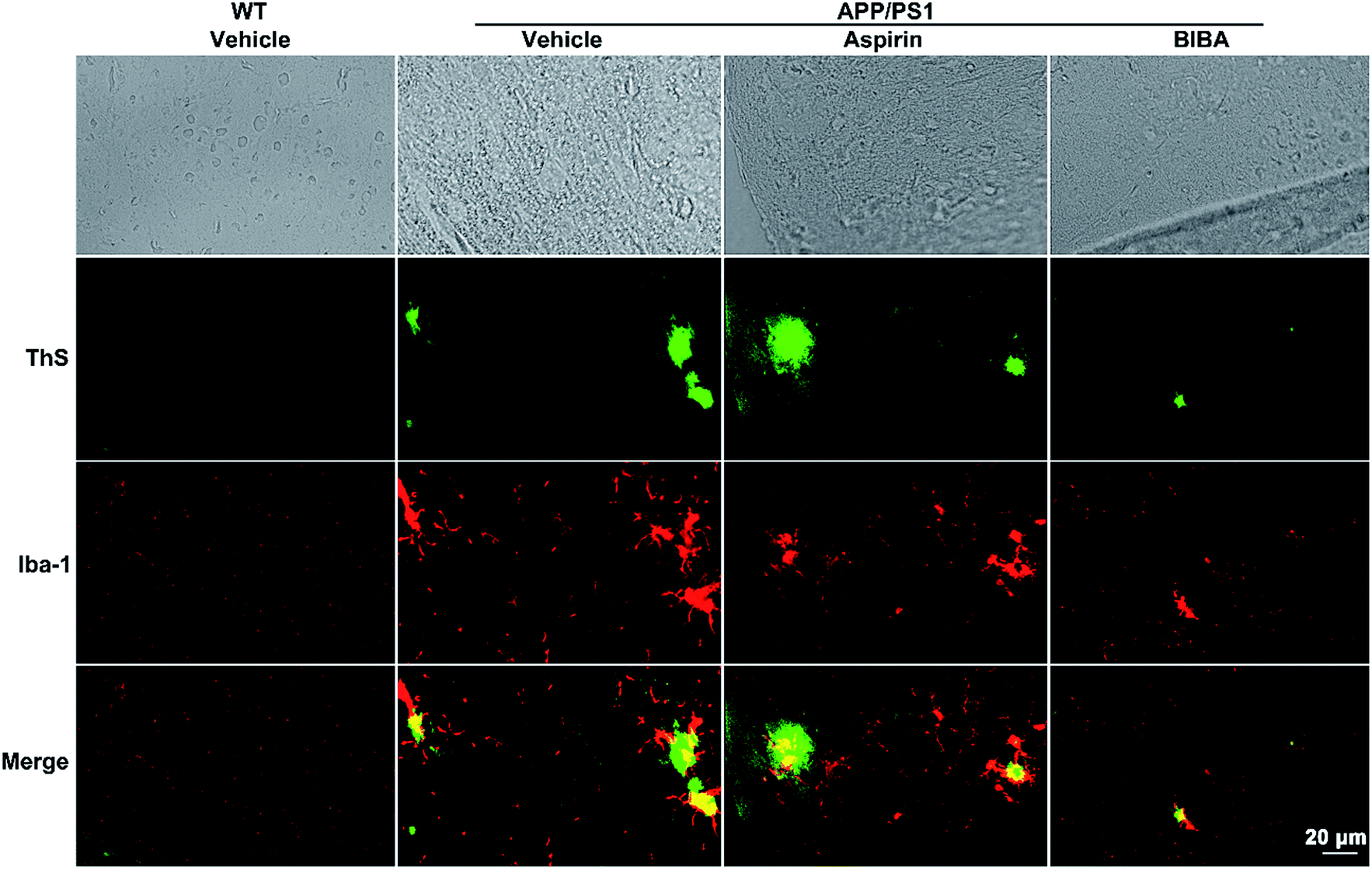 | ||
| Fig. 6 Reduction of Aβ expression and microglia overactivation. Histochemical analyses of Aβ deposition (green) and Iba-1 (red) in WT and APP/PS1 mice brain after treatment with vehicle, aspirin (1.4 mg kg−1) and BIBA (2 mg kg−1), respectively, every 3 days for 3 months. | ||
Abnormal accumulation of Aβ induces overactivation of microglia in the brain, which is closely associated with neuron injury in AD.67 Calcium-binding protein Iba-1 is specifically expressed in brain microglia and up-regulated in activated microglia.68 Therefore, we detected the Iba-1-positive cells and the expression of Iba-1 in the brain of APP/PS1 mice by immunofluorescence staining. As shown in Fig. 6 and S9,† strong activation of microglia (red spots) was found around the Aβ plaques (green spots) in the brain of APP/PS1 mice; by contrast, BIBA markedly reduced the expression of Iba-1. Aspirin did not show significant effects on the activation of microglia. These results show that the aberrant elevation of microglia activation in the APP/PS1 mice was alleviated and the surrounding Aβ species was reduced after the administration of BIBA.
In view of the above properties, we assessed the protective effect of BIBA on the spatial judgement and hippocampal memory of APP/PS1 mice (6-month-old, male) by Morris water maze (MWM) tests after drug administration for 3 months.69 On the 4th day during the acquisition phase, the BIBA-treated APP/PS1 mice seem to perform better than the aspirin-treated and the control group, as reflected by a reduction in escape latency time (Fig. S10B†), with more annulus crossing and longer time spending in the target quadrant in the probe trial (Fig. S10C and D†). No obvious toxicity was observed on BIBA at a dose of 10 mg kg−1 based on the mortality, changes in body weight and hair loss (Fig. S11†). These preliminary results suggest that BIBA could ameliorate the Aβ-related learning and memory deficits in the APP/PS1 mice.
Mode of interaction between BIBA and Aβ
Given that BIBA can inhibit the self- or metal-induced Aβ aggregation and prevent Aβ-induced cell death, we hypothesized that BIBA may interact with Aβ monomers or oligomers noncovalently and interfere with the formation of fibrils. In order to further understand the possible molecular mechanism for the inhibiton of Aβ aggregation by BIBA, we adopted the crystallographic data of Aβ40 and optimized the geometry of BIBA to perform the blind docking calculations using Autodock software.42,70 As shown in Fig. 7A, there are seven amino acid residues in Aβ40, which are Glu-22, Phe-19, Val-18, Lys-16, Gln-15, His-6, and Arg-5, taking part in the interaction with BIBA. Among them, Val-18 and Phe-19 are hydrophobic, and the rest are hydrophilic. Two hydrogen bonds involving Gln-15 (2.6871 Å) and Lys-16 (2.4672 Å) are associated with BIBA, implying that hydrogen bonding as well as hydrophobic and van der Waals forces contributed to the interaction. In Aβ40 fibrils, BIBA is located in the central hydrophobic residues and remains in contact with Ile-31 and -32 through hydrogen bondings (Fig. 7C). The interaction energies between BIBA and related amino acids of Aβ40 monomers or fibrils are listed in Table S4.† These results illustrate that BIBA may interfere in the Aβ self-aggregation through weak interactions with the amino acid residues in Aβ40. Fig. 7B and D depict the lowest energy conformation of BIBA with monomeric and fibrillar Aβ40, respectively. The weak interaction between BIBA and Aβ40 was further studied by 1H NMR. The proton signals assignable to the imidazole of His residues only slightly shifted in the presence of BIBA (Fig. S12†), which suggests that BIBA may affect the chemical environment of protons in Aβ40 through H-bonding interactions.71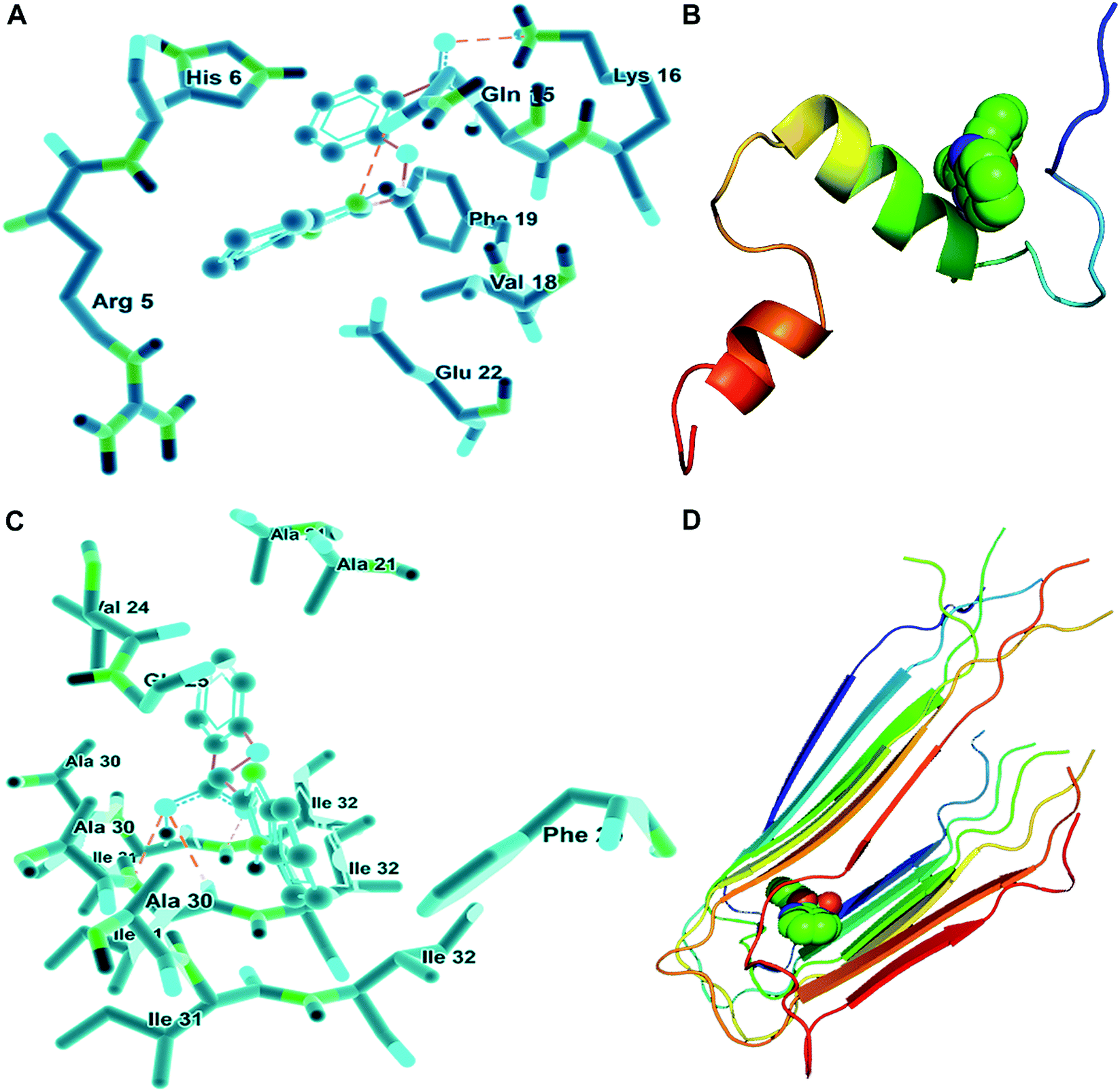 | ||
| Fig. 7 Calculated surroundings of BIBA–Aβ40 monomers (A and B) and fibrils (C and D). | ||
Conclusions
Alzheimer's disease is characterized by several hallmarks, including Aβ plaques, NFTs, overactivation of microglia, neuronal loss and cognitive impairment.72 Currently, many single-targeted drug candidates are designed to solubilize Aβ aggregates or dissociate NFTs, and some of them have entered different stages of pre-clinical or clinical development. However, most of them failed finally in clinical trials, suggesting that single-target drugs only achieve limited effects on the AD progression.73,74 AD is usually diagnosed at late stages when substantial Aβ aggregates and PICs are produced, and irreversible neuron death and cognitive impairments have occurred. Hence, drugs with Aβ aggregates or inflammation as the single target cannot break the vicious cycle between Aβ aggregation and neuroinflammation. By contrast, dual-target BIBA significantly decreased the level of Aβ fibrils and inhibited the self- or metal-induced Aβ aggregation; meanwhile, it suppressed the intracellular expression of pro-inflammatory mediators and cytokines, thereby protecting microglia from overactivation induced by Aβ42. In addition, BIBA could reduce the expression of apoptosis-related proteins and prevent the apoptosis of neural cells induced by Aβ aggregates. The synergistic effect between the inhibition of Aβ aggregation and suppression of neuroinflammation exerted by BIBA is depicted in Fig. 8.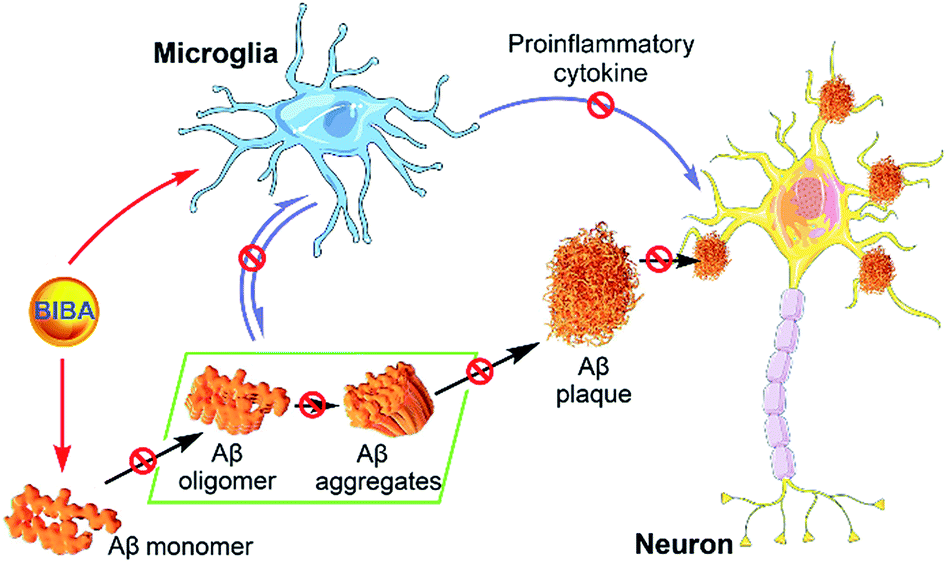 | ||
| Fig. 8 Schematic diagram showing the mechanism of BIBA-mediated protection of neurons in the AD brain. | ||
In conclusion, a difunctional neuromodulator (BIBA) was designed in this study. BIBA can (1) inhibit self- and metal-induced Aβ aggregations, (2) diminish neuroinflammation in microglia, (3) prolong the lifespan and attenuate Aβ-induced paralysis in Aβ-transgenic C. elegans, and (4) reduce Aβ plaques, oligomers and neuroinflammation in the brain of AD model mice. Mechanism studies suggest that BIBA holds back the metal-induced Aβ aggregation by chelating with metal ions, interferes with the Aβ self-aggregation by interacting with amino acid residues, and inhibits the neuroinflammation by suppressing PICs in microglia. Therefore, BIBA is a multi-target anti-AD agent that may alleviate the progress of AD through a synergistic mechanism. The design of BIBA represents a new conception for the treatment of AD and opens a promising avenue for the development of anti-AD agents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (Grants 21877059, 31570809, and 31700714) for financial support.Notes and references
- W. Q. Henry and M. L. Frank, N. Engl. J. Med., 2010, 362, 329–344 CrossRef PubMed.
- L. M. Ittner and J. Götz, Nat. Rev. Neurosci., 2011, 12, 67–72 CrossRef PubMed.
- A. Lleó, S. M. Greenberg and J. H. Growdon, Annu. Rev. Med., 2006, 57, 513–533 CrossRef PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- I. Kuperstein, K. Broersen, I. Benilova, J. Rozenski, W. Jonckheere, M. Debulpaep, A. Vandersteen, I. Segers-Nolten, K. V. D. Werf, V. Subramaniam, D. Braeken, G. Callewaert, C. Bartic, R. D'Hooge, I. C. Martins, F. Rousseau, J. Schymkowitz and B. D. Strooper, EMBO J., 2010, 29, 3408–3420 CrossRef CAS PubMed.
- J. P. Cleary, D. M. Walsh, J. J. Hofmeister, G. M. Shankar, M. A. Kuskowski, D. J. Selkoe and K. H. Ashe, Nat. Neurosci., 2005, 8, 79–84 CrossRef CAS PubMed.
- K. Blennow, Nat. Med., 2010, 16, 1218–1222 CrossRef CAS PubMed.
- M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS PubMed.
- X. H. Wang, X. Y. Wang and Z. J. Guo, Coord. Chem. Rev., 2018, 362, 72–84 CrossRef CAS.
- S. Bolognin, D. Drago, L. Messori and P. Zatta, Med. Res. Rev., 2009, 29, 547–570 CrossRef CAS PubMed.
- X. H. Wang, X. Y. Wang, C. L. Zhang, Y. Jiao and Z. J. Guo, Chem. Sci., 2012, 3, 1304–1312 RSC.
- J. McLaurin, M. E. Kierstead, M. E. Brown, C. A. Hawkes, M. H. L. Lambermon, A. L. Phinney, A. A. Darabie, J. E. Cousins, J. E. French, M. F. Lan, F. Chen, S. S. N. Wong, H. T. J. Mount, P. E. Fraser, D. Westaway and P. S. George-Hyslop, Nat. Med., 2006, 12, 801–808 CrossRef CAS PubMed.
- F. S. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, P. Chen, R. Kayed, C. G. Glabe, S. A. Frautsch and G. M. Cole, J. Biol. Chem., 2005, 280, 5892–5901 CrossRef CAS PubMed.
- S. Kumar, A. Henning-Knechtel, I. Chehade, M. Magzoub and A. D. Hamilton, J. Am. Chem. Soc., 2017, 139, 17098–17108 CrossRef CAS PubMed.
- K. Rezai-Zadeh, D. Shytle, N. Sun, T. Mori, H. Hou, D. Jeanniton, J. Ehrhart, K. Townsend, J. Zeng, D. Morgan, J. Hardy, T. Town and J. Tan, J. Neurosci., 2005, 25, 8807–8814 CrossRef CAS PubMed.
- D. J. Selkoe, Science, 2012, 337, 1488–1492 CrossRef CAS PubMed.
- F. Panza, G. Logroscino, B. P. Imbimbo and V. Solfrizzi, Curr. Opin. Psychiatr., 2014, 27, 128–137 CrossRef PubMed.
- M. Staderini, M. A. Martín and M. L. Bolognesi, Chem. Soc. Rev., 2015, 44, 1807–1819 RSC.
- L. I. Labzin, M. T. Heneka and E. Latz, Annu. Rev. Med., 2018, 69, 437–449 CrossRef CAS PubMed.
- C. Nicolás, L. Fernandez-Sanchez, L. Campello, V. Maneu, P. D. l. Villa, P. Lax and I. Pinilla, Prog. Retinal Eye Res., 2014, 43, 17–75 CrossRef PubMed.
- J. Koenigsknecht-Talboo and G. E. Landreth, J. Neurosci., 2005, 25, 8240–8249 CrossRef CAS PubMed.
- N. Zilka, Z. Kazmerova, S. Jadhav, P. Neradil, A. Madari, D. Obetkova, O. Bugos and M. Novak, J. Neuroinflammation, 2012, 9, 47–56 CAS.
- G. Azizi, N. Khannazer and A. Mirshafiey, Am. J. Alzheimer's Dis. Other Dement. , 2014, 29, 415–425 CrossRef PubMed.
- V. Calsolaro and P. Edison, Alzheimer's Dementia, 2016, 12, 719–732 CrossRef PubMed.
- C. R. Stewart, L. M. Stuart, K. Wilkinson, J. M. Gils, J. S. Deng, A. Halle, K. J. Rayner, L. Boyer, R. Zhong, W. A. Frazier, A. Lacy-Hulbert, J. E. Khoury, D. T. Golenbock and K. J. Moore, Nat. Immunol., 2010, 11, 155–161 CrossRef CAS PubMed.
- F. J. Sheedy, A. Grebe, K. J. Rayner, P. Kalantari, B. Ramkhelawon, S. B. Carpenter, C. E. Becker, H. N. Ediriweera, A. E. Mullick, D. T. Golenbock, L. M. Stuart, E. Latz, K. A. Fitzgerald and K. J. Moore, Nat. Immunol., 2013, 14, 812–820 CrossRef CAS PubMed.
- B. Kaminska, Biochim. Biophys. Acta, 2005, 1754, 253–262 CrossRef CAS PubMed.
- L. Zuroff, D. Daley, K. L. Black and M. Koronyo-Hamaoui, Cell. Mol. Life Sci., 2017, 74, 2167–2201 CrossRef CAS PubMed.
- Y. Tang and W. D. Le, Mol. Neurobiol., 2016, 53, 1181–1194 CrossRef CAS PubMed.
- S. Piirainen, A. Youssef, C. Song, A. V. Kalueff, G. E. Landreth, T. Malm and L. Tian, Neurosci. Biobehav. Rev., 2017, 77, 148–164 CrossRef CAS PubMed.
- J. C. Breitner and P. P. Zandi, N. Engl. J. Med., 2001, 345, 1567–1568 CrossRef CAS PubMed.
- P. S. Aisen, Lancet Neurol., 2002, 1, 279–284 CrossRef CAS PubMed.
- ADAPT Research Group, Neurology, 2007, 68, 1800–1808 CrossRef PubMed.
- S. Côté, P.-H. Carmichaela, R. Verreault, J. Lindsaya, J. Lefebvred and D. Laurin, Alzheimer's Dementia, 2012, 8, 219–226 CrossRef PubMed.
- A. A. Reinke and J. E. Gestwicki, Chem. Biol. Drug Des., 2011, 77, 399–411 CrossRef CAS PubMed.
- J. R. Vane and R. M. Botting, Thromb. Res., 2003, 110, 255–258 CrossRef CAS PubMed.
- S. Chandra, M. Jana and K. Pahan, J. Neurosci., 2018, 38, 6682–6699 CrossRef CAS PubMed.
- S. Ayyadevara, M. Balasubramaniam, S. Kakraba, R. Alla, J. L. Mehta and R. J. S. Reis, Antioxid. Redox Signaling, 2017, 27, 1383–1396 CrossRef CAS PubMed.
- H. van de Waterbeemd and E. Gifford, Nat. Rev. Drug Discovery, 2003, 2, 192–204 CrossRef CAS PubMed.
- H. Kozlowski, M. Luczkowski, M. Remelli and D. Valensinc, Coord. Chem. Rev., 2012, 256, 2129–2141 CrossRef CAS.
- L. J. Zhu, Y. Song, P.-N. Cheng and J. S. Moore, J. Am. Chem. Soc., 2015, 137, 8062–8068 CrossRef CAS PubMed.
- X. Ma, J. A. Hua, K. Wang, H. M. Zhang, C. L. Zhang, Y. F. He, Z. J. Guo and X. Y. Wang, Inorg. Chem., 2018, 57, 13533–13543 CrossRef CAS PubMed.
- Y.-S. Cheng, Z.-t. Chen, T.-Y. Liao, C. Lin, H. C.-H. Shen, Y.-H. Wang, C.-W. Chang, R.-S. Liu, R. P.-Y. Chen and P.-H. Tu, EMBO Mol. Med., 2017, 9, 703–715 CrossRef CAS PubMed.
- S. Kumar and A. D. Hamilton, J. Am. Chem. Soc., 2017, 139, 5744–5755 CrossRef CAS PubMed.
- G. Wen, W. J. Qin, D. Y. Chen, Y. Q. Wang, X. Yue, Z. Y. Liu, Y. N. Cao, J. Du, B. H. Zhou and X. Z. Bu, Chem. Commun., 2017, 53, 3886–3889 RSC.
- J. W. Um, H. B. Nygaard, J. K. Heiss, M. A. Kostylev, M. Stagi, A. Vortmeyer, T. Wisniewski, E. C. Gunther and S. M. Strittmatter, Nat. Neurosci., 2012, 15, 1227–1285 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 6625–6636 CrossRef CAS PubMed.
- D. J. Tew, S. P. Bottomley, D. P. Smith, G. D. Ciccotosto, J. Babon, M. G. Hinds, C. L. Masters, R. Cappai and K. J. Barnham, Biophys. J., 2008, 94, 2752–2766 CrossRef CAS PubMed.
- K. Bettayeb, J. C. Chang, W. J. Luo, S. Aryal, D. Varotsis, L. Randolph, W. J. Netzer, P. Greengard and M. Flajolet, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5412–5417 CrossRef CAS PubMed.
- S. Lee, X. Zheng, J. Krishnamoorthy, M. G. Savelieff, H. M. Park, J. R. Brender, J. H. Kim, J. S. Derrick, A. Kochi, H. J. Lee, C. Kim, A. Ramamoorthy, M. T. Bowers and M. H. Lim, J. Am. Chem. Soc., 2014, 136, 299–310 CrossRef CAS PubMed.
- S. M. Monte, Y. K. Sohn and J. R. Wands, J. Neurol. Sci., 1997, 152, 73–83 CrossRef PubMed.
- C. B. Lu, W. M. Fu, G. S. Salvesen and M. P. Mattson, NeuroMol. Med., 2002, 1, 69–79 CrossRef CAS.
- Z. Z. Zhu, T. Yang, L. Zhang, L. L. Liu, E. M. Yin, C. L. Zhang, Z. J. Guo, C. Xu and X. Y. Wang, Eur. J. Med. Chem., 2019, 168, 330–339 CrossRef CAS PubMed.
- Y. J. Guan, Z. Du, N. Gao, Y. Cao, X. H. Wang, P. Scott, H. L. Song, J. S. Ren and X. G. Qu, Sci. Adv., 2018, 4, eaao6718 CrossRef PubMed.
- S. Alavez, M. C. Vantipalli, D. J. S. Zucker, I. M. Klang and G. J. Lithgow, Nature, 2011, 472, 226–229 CrossRef CAS PubMed.
- G. McColl, B. R. Roberts, T. L. Pukala, V. B. Kenche, C. M. Roberts, C. D. Link, T. M. Ryan, C. L. Masters, K. J. Barnham, A. I. Bush and R. A. Cherny, Mol. Neurodegener., 2012, 7, 57–66 CrossRef CAS PubMed.
- T. Kaletta and M. O. Hengartner, Nat. Rev. Drug Discovery, 2006, 5, 387–398 CrossRef CAS PubMed.
- C. D. Link, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9368–9372 CrossRef CAS PubMed.
- G. McColl, B. R. Roberts, A. P. Gunn, K. A. Perez, D. J. Tew, C. L. Masters, K. J. Barnham, R. A. Cherny and A. I. Bush, J. Biol. Chem., 2009, 284, 22697–22702 CrossRef CAS PubMed.
- Y. J. Wu, Z. X. Wu, P. Butko, Y. Christen, M. P. Lambert, W. L. Klein, C. D. Link and Y. Luo, J. Neurosci., 2006, 26, 13102–13113 CrossRef CAS PubMed.
- D. L. Rebolledo, R. Aldunate, R. Kohn, I. Neira, A. N. Minniti and N. C. Inestrosa, J. Neurosci., 2011, 31, 10149–10158 CrossRef CAS PubMed.
- M. T. Heneka, M. P. Kummer and E. Latz, Nat. Rev. Immunol., 2014, 14, 463–477 CrossRef CAS PubMed.
- F. Zhang, R. J. Zhong, S. Li, Z. F. Fu, C. Cheng, H. B. Cai and W. D. Le, Front. Aging Neurosci., 2017, 9, 282 CrossRef PubMed.
- J. Y. Song, S.-M. Choi and B. C. Kim, Front. Cell. Neurosci., 2017, 11, 64 Search PubMed.
- D. F. Cao, H. L. Lu, T. L. Lewis and L. Li, J. Biol. Chem., 2007, 282, 36275–36282 CrossRef CAS PubMed.
- T. Yang, L. Yang, C. L. Zhang, Y. Q. Wang, X. Ma, K. Wang, J. Luo, C. Yao, X. Y. Wang and X. H. Wang, Inorg. Chem. Front., 2016, 3, 1572–1581 RSC.
- M. T. Heneka, M. J. Carson, J. E. Khoury, G. E. Landreth, F. Brosseron, D. L. Feinstein, A. H. Jacobs, T. Wyss-Coray, J. Vitorica, R. M. Ransohoff, K. Herrup, S. A. Frautschy, B. Finsen, G. C. Brown, A. Verkhratsky, K. Yamanaka, J. Koistinaho, E. Latz, A. Halle, G. C. Petzold, T. Town, D. Morgan, M. L. Shinohara, V. H. Perry, C. Holmes, N. G. Bazan, D. J. Brooks, S. Hunot, B. Joseph, N. Deigendesch, O. Garaschuk, E. Boddeke, C. A. Dinarello, J. C. Breitner, G. M. Cole, D. T. Golenbock and M. P. Kummer, Lancet Neurol., 2015, 14, 388–405 CrossRef CAS PubMed.
- D. G. Walker and L.-F. Lue, Alzheimer's Res. Ther., 2015, 7, 56 CrossRef PubMed.
- S.-H. Yang, D. K. Lee, J. Shin, S. Lee, S. Baek, J. Kim, H. Jung, J.-M. Hah and Y.-S. Kim, EMBO Mol. Med., 2017, 9, 61–77 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- J. E. Del Bene and M. J. T. Jordan, J. Am. Chem. Soc., 2000, 122, 4794–4797 CrossRef CAS.
- E. E. Spangenberg, R. J. Lee, A. R. Najafi, R. A. Rice, M. R. P. Elmore, M. Blurton-Jones, B. L. West and K. N. Green, Brain, 2016, 139, 1265–1281 CrossRef PubMed.
- S. Salloway, R. Sperling, N. C. Fox, K. Blennow, W. Klunk, M. Raskind, M. Sabbagh, L. S. Honig, A. P. Porsteinsson, S. Ferris, M. Reichert, N. Ketter, B. Nejadnik, V. Guenzler, M. Miloslavsky, D. Wang, Y. Lu, J. Lull, I. C. Tudor, E. Liu, M. Grundman, E. Yuen, R. Black and H. R. Brashear, N. Engl. J. Med., 2014, 370, 322–333 CrossRef CAS PubMed.
- R. S. Doody, R. G. Thomas, M. Farlow, T. Iwatsubo, B. Vellas, S. Joffe, K. Kieburtz, R. Raman, X. Y. Sun, P. S. Aisen, E. Siemers, H. Liu-Seifert and R. Mohs, N. Engl. J. Med., 2014, 370, 311–321 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03042e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |