
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual near infrared (NIR) fluorescent palladium(II) macrocyclic complexes containing M–C bonds with bioimaging capability†
Yuhang
Yao
a,
Chun-Liang
Hou
b,
Zi-Shu
Yang
a,
Guangliu
Ran
c,
Lei
Kang
d,
Cuicui
Li
d,
Wenkai
Zhang
 *c,
Jing
Zhang
*b and
Jun-Long
Zhang
*a
*c,
Jing
Zhang
*b and
Jun-Long
Zhang
*a
aBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: zhangjunlong@pku.edu.cn
bCenter of Materials Science and Optoelectronics Engineering, College of Materials Science and Opto-Electronic Technology, University of Chinese Academy of Sciences, Beijing 100049, P. R. China. E-mail: zhangj271@ucas.ac.cn
cCenter for Advanced Quantum Studies, Department of Physics and Applied Optics Beijing Area Major Laboratory, Beijing Normal University, Beijing 100875, P. R. China. E-mail: wkzhang@bnu.edu.cn
dDepartment of Nuclear Medicine, Peking University First Hospital, Beijing 100034, P. R. China
First published on 11th September 2019
Abstract
Near infrared (NIR) luminescent metal complexes are promising probes in bioimaging and biosensing, however they generally suffer from oxygen interference arising from heavy metal effects. We designed new tetradentate macrocyclic benzitripyrrin (C^N^N^N) ligands by combination of M–C bond formation and reducing the π-conjugation to achieve NIR fluorescent Pd complexes (700–1000 nm) with quantum yields up to 14%. To understand the origin of NIR fluorescence, detailed analyses by density functional theory/time-dependent density functional theory (DFT/TDDFT) calculations together with femtosecond and nanosecond transient absorption spectroscopies suggest that M–C bond formation indeed leads to destabilization of the d–d excited state and less effective quenching of emission; and importantly, small spin–orbital coupling (SOC) and the large singlet-triplet energy gap are the primary causes of the non-population of triplet states. Comparison of PdII and PtII analogues shows that the non-radiative channel of the out-plane vibration of the tripyrrin plane effectively quenches the fluorescence of the PtII complex but not the PdII congener. We also demonstrate the proof-of-concept applications of PdII complexes (Pd-1 and Pd-3) encapsulated in silica nanoparticles, in both in vitro and in vivo bioimaging experiments without oxygen interference. Moreover, pH-induced reversible switching of NIR fluorescence was achieved even intracellularly using the Pd complex (Pd-2), which shows the potential to further develop perspective stimuli-responsive NIR materials.
Introduction
Imaging in the near infrared (NIR) region (700–1700 nm), known as the biological transparency window, attracts increasing attention for its unique advantages such as low tissue auto-absorption, small photon scattering, and deep tissue penetration, which is highly desirable to improve disease detection and treatment in clinical applications.1 Exploring NIR molecular fluorophores with both high luminescence efficiency and biocompatibility, to circumvent the safety issues such as toxicity and metabolism arising from inorganic and carbon-based nanomaterials such as carbon nanotubes, quantum dots, and Ln doped nanoparticles, is essential to boost clinical translations.2 Toward this goal, many organic fluorophores such as cyanines, donor–acceptor–donor (D–A–D) dyes, and borondipyrromethanes have been extensively studied as promising candidates in NIR imaging.3 In sharp contrast to the tremendous progress made by organic fluorophores, metal coordination compounds that are equally important molecular fluorophores have been seldom employed as the candidates in NIR biological imaging.4 Metal complexes possess intriguing photophysical properties such as brightness, large Stokes shifts, long lifetimes and anti-photobleaching and have been extensively used in visible bioimaging.5 However, they generally suffer from severe photocytotoxicity because the population of triplet states arising from heavy metal effects initiates reactive oxygen species such as 1O2, O2˙−etc., which hamper the direct application in NIR bioimaging. As such, further effort is required to design NIR metal complexes on par with their organic counterparts in biological applications.Thanks to the fast growth of palladacycles,6 notable examples of luminescent PdII complexes containing Pd–C bonds have emerged, because introducing a strong σ-donating carbanion can destabilize the antibonding 4dx2−y2 orbital, allow metal to ligand charge transfer and thus enhance the luminescence.6,7 However, extending this strategy to PdII porphyrinoids, which are typical phosphorescent complexes as red/NIR photosensitizers or probes (Fig. 1a),6,7 to improve NIR luminescence has not been successful. Replacement of one of the pyrrolic nitrogen donors by a σ-donating carbanion afforded carbaporphyrinoids including N-confused porphyrins (Fig. 1b)8 previously developed by Lash, Latos-Grazynski, Furuta and others. Deprotonation of the inner C–H bonds indeed offers the opportunity to form Pd–C bonds in porphyrin macrocycles, yet their photophysical properties have been much less frequently reported.8 Another issue is the extent of π-conjugation. Che et al. pointed out the importance of the π-conjugation of ligands on the excited states and photophysical properties, for example, increasing π-conjugation leads to more allowed low-lying singlet transitions for the lowest triplet state to borrow intensity from and more likely displays phosphorescence than fluorescence.9 By taking advantage of the merits of each approach, we reason that incorporation of O-bridging to replace the methine linker reduces the extent of the π-conjugation (Fig. 1c), which might give rise to new ligands with electronic structures different from those of traditional N4 or CN3 porphyrinoids.10 Importantly, we envision that this design could impact upon the photophysical properties and achieve NIR fluorescent PdII complexes to minimize the oxygen interference.
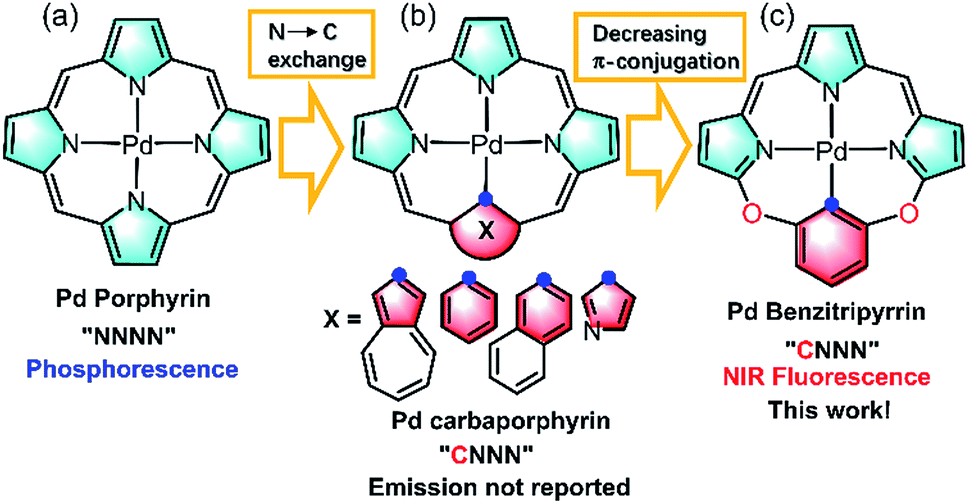 | ||
| Fig. 1 Schematic view of the ligand design: (a) porphyrinoids, (b) carbaporphyrinoids, and (c) benzitripyrrins. | ||
In this work, we synthesized a series of new macrocyclic tetradentate C^N^N^N ligands, namely benzitripyrrin, (11E,12Z,51E,52Z)-2,4-bis(perfluorophenyl)-12H,31H,52H-6,8-dioxa-1,3,5(2,5)-tripyrrola-7(1,3)-benzenacyclo-octaphane derivatives, (1–5) by connecting tripyrrin with 1,3-dihydroxybenzene (resorcinol) derivatives (Fig. 1c). Surprisingly, PdII complexes displayed strong NIR fluorescence (700–1000 nm) with quantum yields up to 14%, but not the phosphorescence often observed in PdII porphyrins,11 while the Pt(II) congener displays negligible NIR fluorescence. To the best of our knowledge, only a handful of Pd fluorescent complexes in the visible to deep red region have been previously reported.12 Rybtchinski et al. ascribed the lack of phosphorescence to the weak electronic interactions between the metal centers and ligand π-system.13 To gain insight into the luminescence mechanism, we performed fs/ns transient absorption (TA) spectroscopy and density functional theory/time-dependent density functional theory (DFT/TDDFT) calculations so that comparisons between PdII and PtII systems can be made. In view of the strong NIR fluorescence of the Pd complexes, proof-of-concept applications are performed in living cell imaging as well as in vivo imaging using a mice model by encapsulating PdII complexes including a pH sensitive compound into the nanochannels of mesoporous silica nanoparticles (MSN). Collectively, this work offers a new molecular scaffold to design NIR fluorescent PdII complexes with the capability of bioimaging without oxygen interference.
Results and discussion
Synthesis and characterization
As shown in Scheme 1a, we synthesized a series of new tripyrrin ligands (11E,12Z,51E,52Z)-2,4-bis(perfluorophenyl)-12H,31H,52H-6,8-dioxa-1,3,5(2,5)-tripyrrola-7(1,3)-benzenacyclooctaphane with different substituents on the bridging phenyl group (1–5, R1 = R2 = H, 1; R1 = NMe2, R2 = H, 2; R1 = CN, R2 = H, 3; R1 = H, R2 = OMe, 4; R1 = H, and R2 = CN, 5) in 3 steps.10 Synthesis of the PdII (Pd-1–3) or PtII (Pt-1) complexes was achieved in the yields of ca. 10–25%. 1–5, Pd-1–3 and Pt-1 have been characterized using HR ESI-MS, 1H, 13C and 19F NMR, FT-IR spectrometers and spectroscopes and the detailed synthetic procedures are listed in the ESI (Schemes S1–S11, Fig. S1–S45†).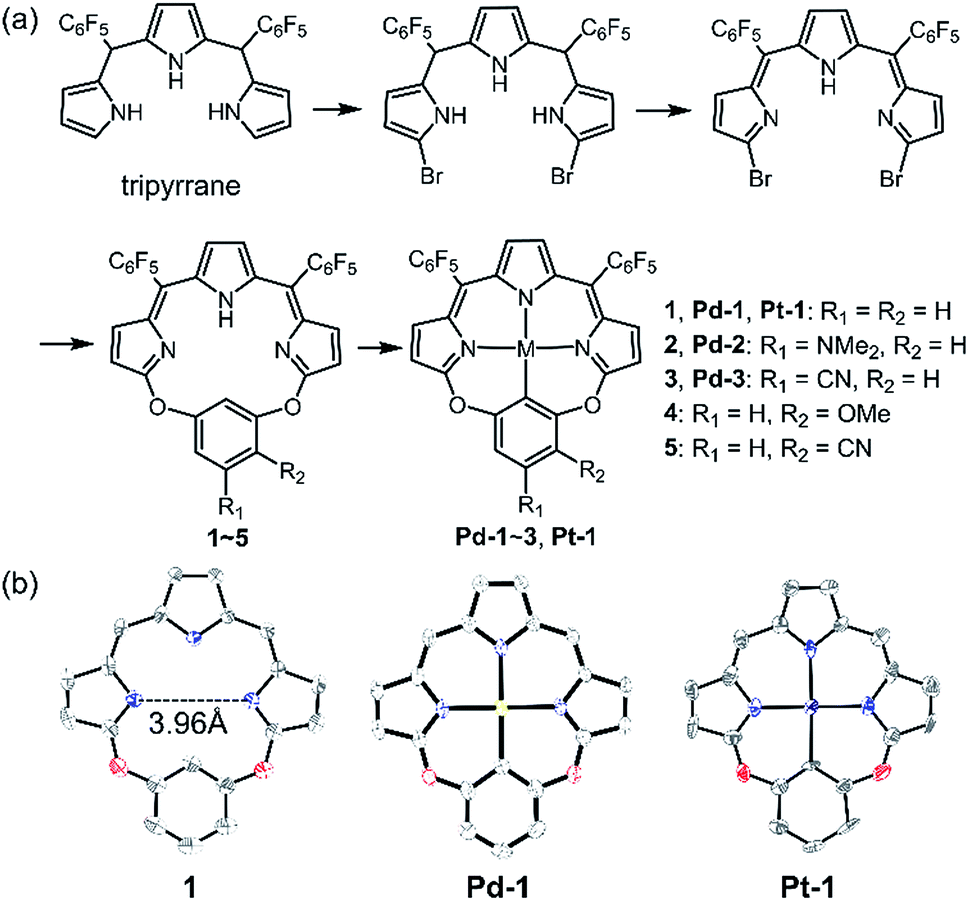 | ||
| Scheme 1 (a) Synthetic routes of 1–5, Pd-1–3 and Pt-1. (b) X-ray crystal structures of 1, Pd-1 and Pt-1. Hydrogen atoms, meso-substituents and solvent molecules are omitted for clarity. | ||
Single crystals of 1, Pd-1 and Pt-1 suitable for X-ray diffraction were obtained by slow diffusion of hexane to chloroform solution (CCDC: 1912139, 1912140, 1912141 in Table S1†). As shown in Scheme 1b and Fig. S46–S48,†1 displays a larger N3C cavity than carbaporphyrin with a distance of opposite N atoms of ∼3.96 Å. Pd-1 and Pt-1 show a planar-square structure with the average Pd–N/Pt–N bond distances of ∼2.01 Å. The short distances of 2.00 and 2.01 Å of the M–C bond are shown in Pd-1 and Pt-1, respectively, which are comparable to those (1.95–2.06 and 1.95–2.05 Å) observed in palladacycles or platinum complexes.7
Photophysical properties
Absorption and emission spectra of 1–5, Pd-1–3 and Pt-1 were recorded at 298 K in CH2Cl2 (Fig. 2 and S49–S77†) and the photophysical data are tabulated in Table S2.†1–5 show a sharp and intense band at around 300–400 nm and a broad, split band at 500–600 nm with extinction coefficients in the order of 104 dm3 mol−1 cm−1. The absorption could be assigned to the intraligand (IL) [π → π*] transitions.10 Interestingly, the aryl substitution has a slight effect on the absorption of 1–5, indicating that the vertical transition mainly involves HOMO → LUMO transition localized on the tripyrrin core.10 Upon excitation at 500 nm, 1, 3 and 5 display intense vibronic-structured emission with a maximum at 600 or 615 nm, while 2 and 4 show a weak, structureless emission around 550–800 nm. It is worthy to note that the electron-donating NMe2 quenches the fluorescence of 2, probably due to the photoinduced electron transfer (PET) process.14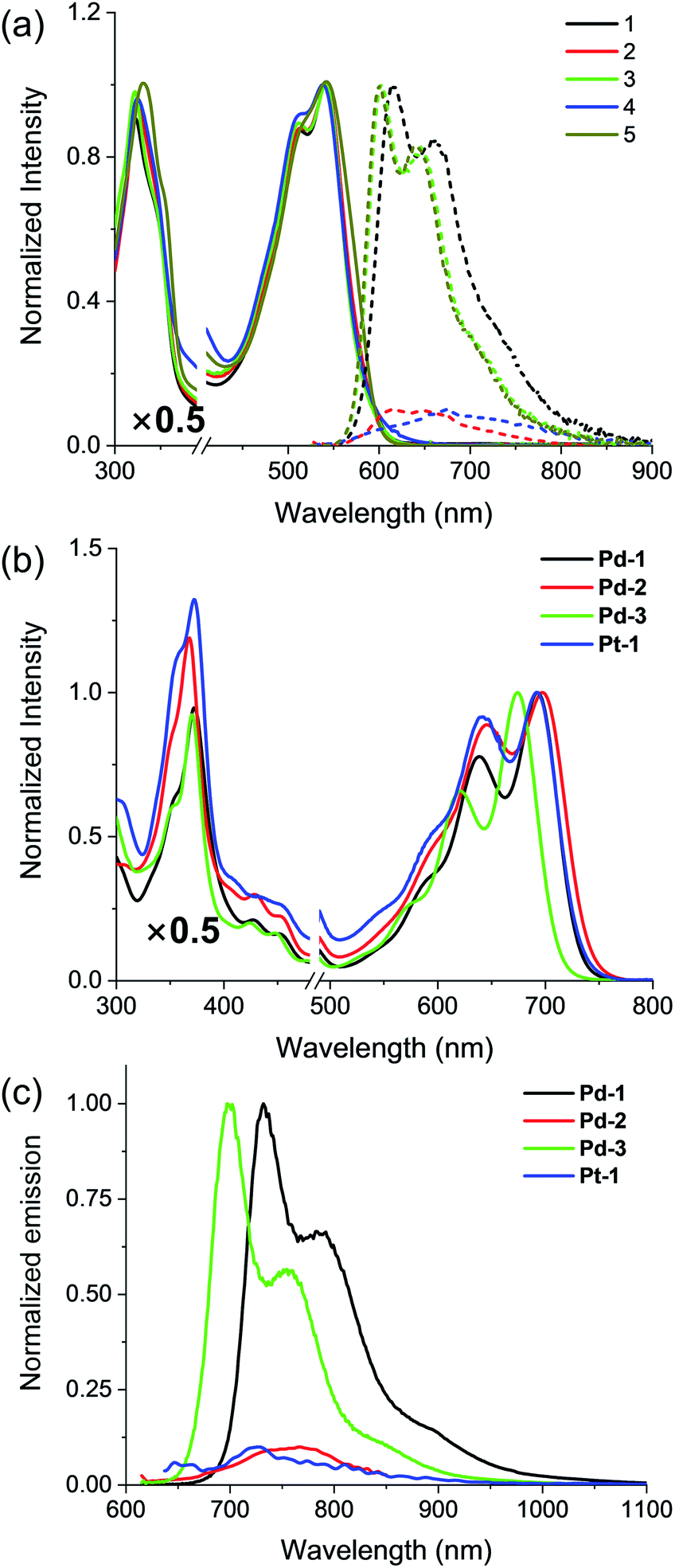 | ||
| Fig. 2 (a) Absorption (solid line) and emission (dashed line) spectra of the free base ligands 1–5 and (b) absorption and (c) emission spectra of Pd-1–3 and Pt-1 in CH2Cl2. | ||
Pd-1–3 and Pt-1 exhibit a sharp and intense absorbance in the range of 320–400 and 500–750 nm, with red-shifts of ca. 45 nm in the high energy region (320–400 nm) and ca. 150 nm in the low energy region (500–750 nm) compared to the corresponding ligands. With the electron-withdrawing CN, Pd-3 leads to a ca. 17 nm hypsochromic shift to Pd-1, while Pd-2 displays a bathochromic shift of ca. 8 nm. This indicates the more significant effect of the aryl substituents on the metal complexes than on the ligands. Upon excitation at 600 or 700 nm, all of the metal complexes emit at 700–1000 nm, red-shifts of ca. ∼130 nm compared to the corresponding ligands (Fig. 2c). Interestingly, exposure of such a solution to air does not quench the emission (Fig. S49–S52†). The emission lifetimes are in the nanosecond scale (<4 ns) even in 77 K glass (Fig. S65–S67†). Small Stokes shifts (<40 nm) are observed under either aerobic or anaerobic conditions. These photophysical data are suggestive of fluorescence for Pd-1–3 and Pt-1. Among them, Pd-1 and Pd-3 exhibit intense fluorescence maxima at 732 and 700 nm with a vibronic band at 792 and 758 nm (ΦF = 6.0 and 14.0%, τ = 1.06 and 2.40 ns), respectively, while Pd-2 displays a weak, less discernible vibrational emission centered at 767 nm with bi-exponential decay lifetimes of 0.26 (9%) and 2.05 ns (91%). Pt-1 also exhibits a weak fluorescence centered at 729 nm with a bi-exponential decay lifetime of 0.17 (20%) and 3.34 ns (80%). As shown in Fig. S77,† decreasing temperature from 300 to 77 K enhances the emission of Pt-1 (∼5.2 fold) more than that of Pd-1 (∼1.3 fold), indicating that emission of Pt-1 is more temperature- and vibration-dependent and will be discussed in context.
Electrochemical properties
The redox properties of 1, Pd-1 and Pt-1 were examined by cyclic voltammetry (CV) using tetra-n-butylammonium hexafluorophosphate (nBu4NPF6, 0.1 M) as the supporting electrolyte (Fig. 3, S78–S80†) and Table S3† summarizes the electrochemical data. Two quasi-reversible reduction waves at about −0.85 and −1.03 V vs. SCE and one irreversible oxidation wave at ca. 1.25 V vs. SCE are tentatively assigned to the cyclic tripyrrin ligand-centered redox potentials for 1 (Fig. S78†), similar to the previously reported cyclic tripyrrins (Ered = −0.92, −1.17 V and Eox = 1.35 V).10Pd-1 and Pt-1 display two quasi-reversible reduction waves (−0.75 and −1.18 V vs. SCE for Pd-1, −0.67 and −1.11 V vs. SCE for Pt-1, Fig. 3a) and one irreversible oxidation wave (1.45 V vs. SCE for Pd-1, 1.25 V vs. SCE for Pt-1, Fig. 3a). These results suggest that both reduction and oxidation are cyclic tripyrrin ligand-centered with mixing of the metal-centered contribution.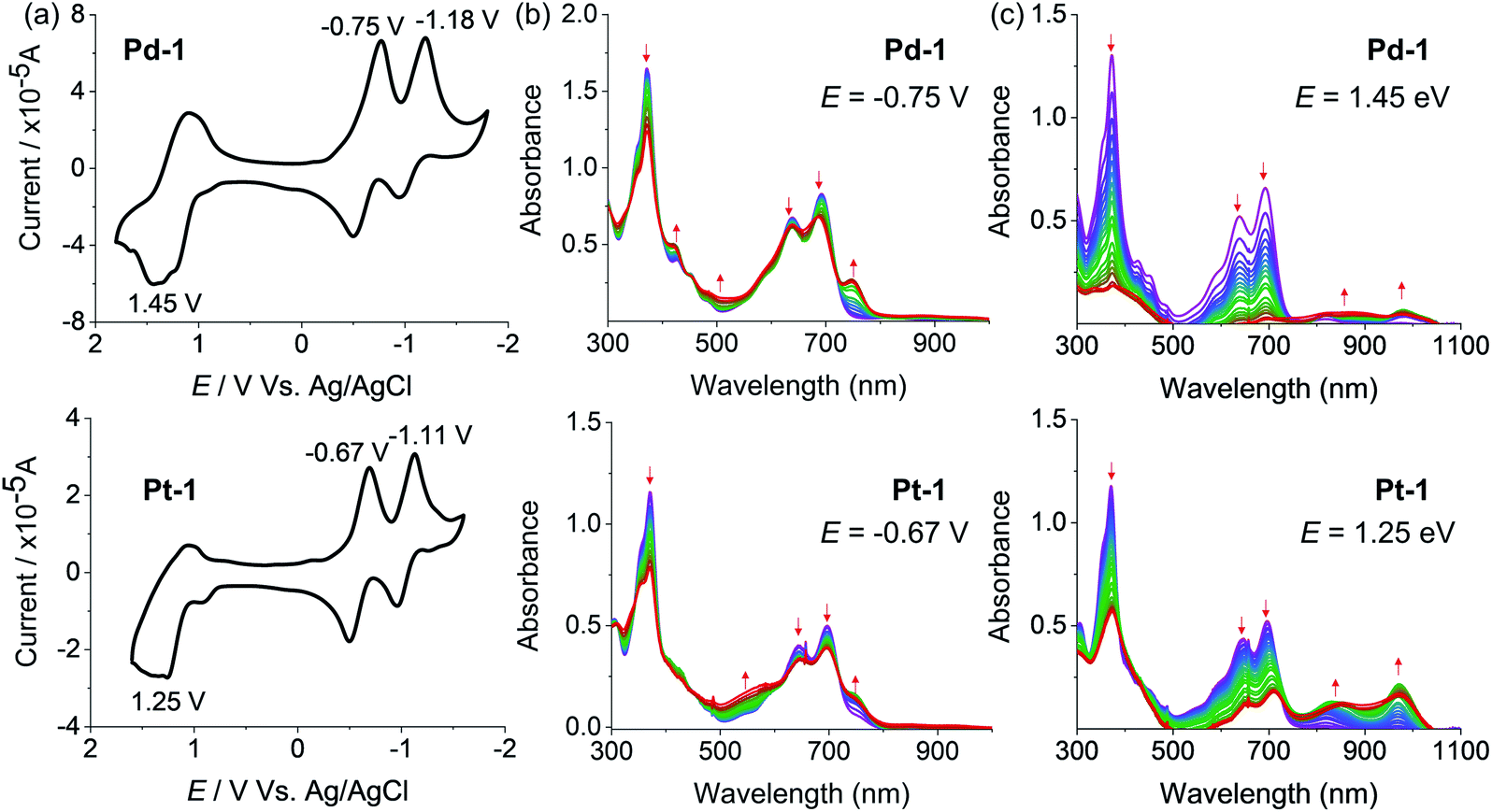 | ||
| Fig. 3 (a) Cyclic voltammograms and spectroelectrochemistry at (b) reduction potentials (−0.75 and −0.67 V, respectively) and (c) oxidation potentials (1.45 and 1.25 V, respectively) of Pd-1 and Pt-1 in CH2Cl2. | ||
The spectroelectrochemistry of 1, Pd-1 and Pt-1 were performed to monitor the spectral changes during the reduction and oxidation processes. As shown in Fig. S79,† one-electron reduction of 1 at −0.85 V causes a decreased absorption centered at 320 and 500 nm, and new peaks centered at 420, 600 and 750 nm appear with an isosbestic point at 366, 471 and 540 nm. One-electron reduction of Pd-1 at −0.75 V and Pt-1 at −0.67 V causes a slightly decreased absorption at 373, 640 and 692 nm with the appearance of a new peak at ∼750 nm (Fig. 3b). Such bands could be ascribed to the feature of the anionic radicals of the ligands. Oxidation of 1 at 1.25 V slightly decreases the absorption at 323, 508 and 540 nm, and new absorption centered at 380 and 600 nm appears with an isosbestic point at 350, 453 and 556 nm (Fig. S80†). When Pd-1 or Pt-1 was oxidized at ca. 1.45 or 1.25 V, respectively, the absorption intensity sharply decreases with a slight red-shift, and a new broad absorption peak at 750–1050 nm appears assigned to the cationic radicals (Fig. 3c).
Transient absorption spectroscopy
Insights into the nature of the excited states, exemplified by Pd-1 and Pt-1, were obtained using nanosecond (ns) and femtosecond (fs) transient absorption (TA) spectroscopes in degassed tetrahydrofuran (THF). The ns-TA spectra display negligible signals of ground-state bleaching (GSB) (Fig. S81–S83†) upon excitation at 355 nm, indicating that there is no population of the triplet excited state for Pd-1 and Pt-1. This is consistent with no phosphorescence observed in degassed solution or glass state in 77 K.Fig. 4 shows the contour plot of the time-resolved absorption difference spectra for Pd-1 and Pt-1 upon excitation at 700 nm. We observed a GSB band maximum at 700 nm and the excited-state absorption (ESA) band centered in the range of 750–850 nm and 875–925 nm for Pd-1. Pd-1 displays single exponential decay lifetime of 1.28 ns at 770 nm and bi-exponential decay lifetimes (86.34 ps and 1.32 ns) as monitored at 910 nm (Fig. 4c). This is assigned to the structural relaxation dynamics (∼100 ps) and intrinsic S1 state lifetime (Table S2†). The ESA bands at 910 nm for Pd-1 and ∼950 nm for Pt-1 are identical to the absorption under one electron oxidation (Fig. 3c), suggesting that the structures at the singlet state may be similar to the cation radicals of Pd-1 and Pt-1.
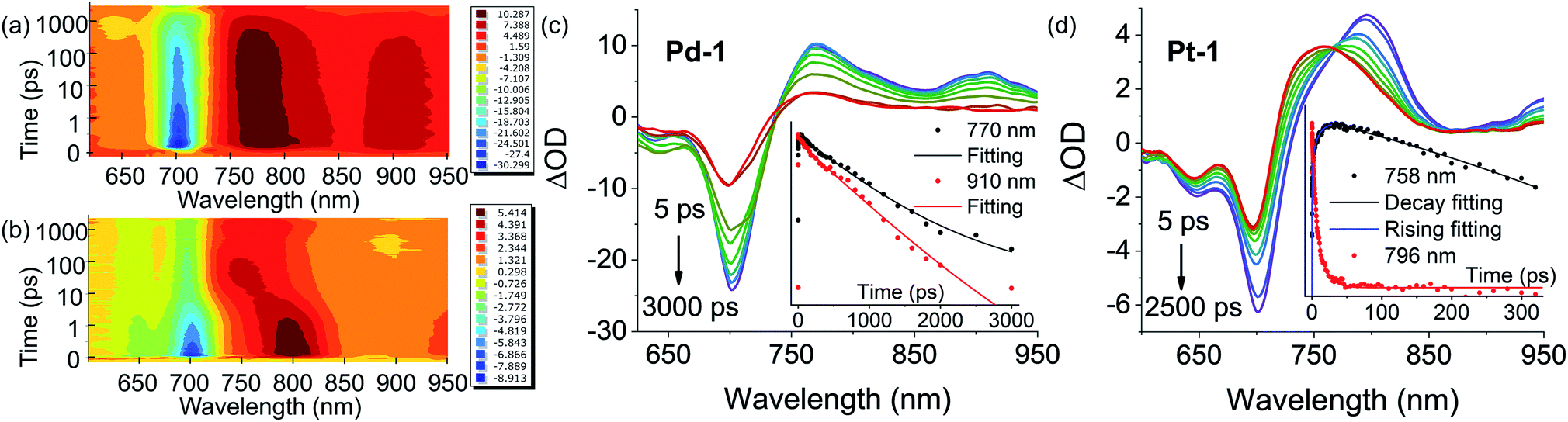 | ||
| Fig. 4 Contour plot of time-resolved absorption difference spectra of (a) Pd-1 and (b) Pt-1 for time delays up to 3000 ps in the range of 600–950 nm in toluene. Femtosecond transient absorption (fs-TA) difference spectra of (c) Pd-1 and (d) Pt-1 recorded at selected decay times in toluene at 298 K. Insert: ESA kinetic decay traces at the specified wavelengths and lifetimes: fitting with exponential decay for (c) Pd-1 (τ = 1281 ps, R2 = 0.996 at 770 nm; τ1 = 86.34 ps, τ2 = 1318 ps, and R2 = 0.996 at 910 nm) and (d) Pt-1 (τ = 3773 ps and R2 = 0.982 at 758 nm for curve decay at 758 nm; τ1 = 0.046 ps, τ2 = 0.78 ps, τ3 = 8.75 ps, and R2 = 0.995 for curve rising at 758 nm; τ1 = 0.20 ps, τ2 = 4.67 ps, τ3 = 16.34 ps, and R2 = 0.988 at 796 nm). | ||
More importantly, in the range of 10–100 ps, Pt-1 shows a new band around 758 nm accompanied with a simultaneous vanishing of ESA at 796 nm and a clear isosbestic point at 770 nm (Fig. 4d). As shown in Fig. 4d, a fast decay with tri-exponential lifetimes (0.20, 4.67 and 16.34 ps) at 796 nm decreases accompanied with a simultaneous appearance (0.046, 0.78 and 8.75 ps) of a new peak at 758 nm, followed by a single exponential decay lifetime of 3.77 ns comparable to the fluorescence lifetime of Pt-1 (Table S2†). This suggest a fast energy transfer (within 100 ps) from the intrinsic S1 state of Pt-1 to a dark state, which might quench emission. We assumed this to be a vibrational state, usually in accordance with the time range of the structural relaxation (10–100 ps). Thus the S1 excited state of Pt-1 is probably deactivated by this non-radiative vibrational state, which is ascribed to a more temperature- and vibration-dependent fluorescence (Fig. S77†).
Computational studies
PdII and PtII complexes are well-known phosphorescent emitters due to the heavy-metal effect with large spin–orbit coupling (SOC) constants (ζSOC = 1504 and 4481 cm−1 for PdII and PtII).15 However, increasing examples demonstrated that incorporation of a heavy metal does not guarantee efficient ISC and the structure and the nature of ligands also play important roles.16 To understand the unusual fluorescence of PdII and PtII complexes, we investigated the electronic structures of 1–5, Pd-1 and Pt-1 using DFT and TDDFT calculations. Electron-donating substituents (NMe2 and OMe) destabilize both HOMO and LUMO in 2 and 4, while the electron-withdrawing substituent (cyano group) stabilizes them in 3 and 5 (Fig. S84†). TDDFT results indicate that the vertical π–π* excitations are localized in tripyrrin moiety (Fig. S84 and Table S4†) for 1 and 3–5. In 2, the charge transfers from the bridging phenyl group to the tripyrrin moiety, similar to photoinduced electron transfer (PET).14 Metal coordination stabilizes the LUMO and destabilizes the HOMO and thus narrows the HOMO–LUMO gap (Fig. S85†), which induces a red-shifted absorption. All the FMOs in Pd-1 and Pt-1 are ligand-centered.18 The natural localized molecular orbital (NLMO) analysis18b,19 reveals that the C-donor atom of the M–C bond shows sp3-like behavior, different from sp2-carbons in the benzene ring (Fig. S86†), which is suggestive of the σ-donating nature, analogous to the M–C bond in previously reported complexes of benziporphyrin and carbaporphyrin.8c,8e Formation of M–C bonds largely stabilizes the metal-centered bonding orbitals (Fig. S87†). The splitting (Δdd*) between the occupied and unoccupied d orbitals is found to be quite large for Pd-1 (5.78 eV) and Pt-1 (6.70 eV). This indicates that the non-radiative decay channels through the MC dd excited states could be excluded for Pd-1 and Pt-1.For Pd-1 and Pt-1, their S1 states are 6640 and 7292 cm−1 above the T1 state, 2135 and 59 cm−1 below the T2 state (Tables S5 and S6†). The calculated ISC rate constants using the MOMAP program20 are given in Fig. 5a and Table S7,† as well as the radiative and non-radiative rate constants of S1 states. Since the ΔEST are large (0.82 and 0.60 eV for Pd-1 and Pt-1), the spin–orbit coupling values are estimated to be 0.82 and 0.60 cm−1 for Pd-1 and Pt-1 (Table S7†). This leads to negligible ISC rate constants (3.66 × 105 and 1.87 × 105 s−1 for Pd-1 and Pt-1), much smaller than the radiative rate constants (8.11 × 106 and 5.13 × 106 s−1 for Pd-1 and Pt-1) and non-radiative rates (4.04 × 107 and 7.46 × 107 s−1 for Pd-1 and Pt-1). These results suggest that the small SOC, large ΔEST and the ligand-centered FMOs (Fig. S85†) are important factors for the non-population of the triplet state in PdII or PtII complexes.
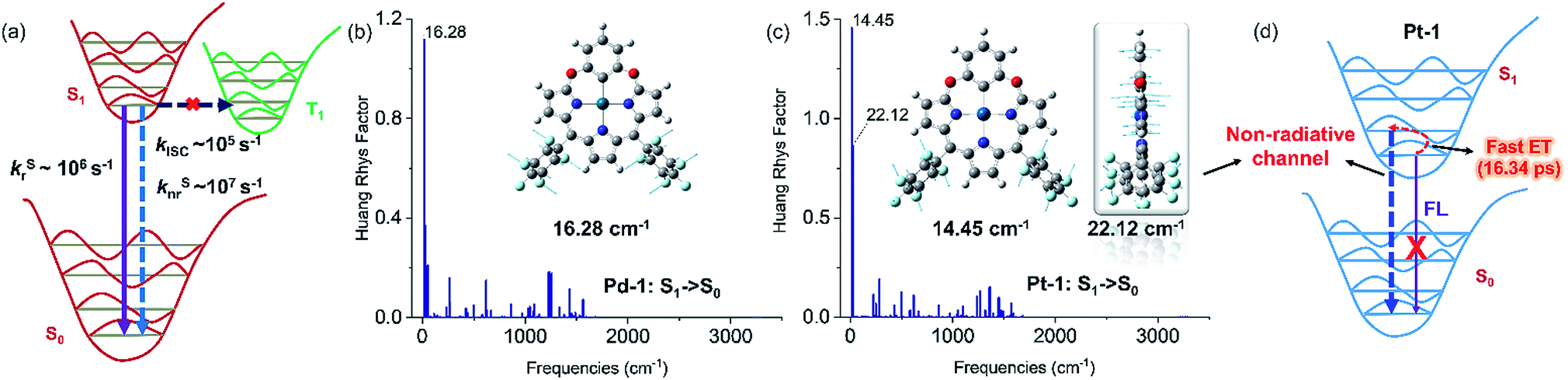 | ||
| Fig. 5 (a) General illustration of the low-lying singlet and triplet excited states of Pd-1 and Pt-1, and the calculated rate constants of different photophysical processes, in which the solid and dashed arrows represent radiative and non-radiative processes, respectively. Huang–Rhys factors for energy conversion between the S1 and S0 states of (b) Pd-1 and (c) Pt-1, as well as the displacement vectors of important vibrational modes with the largest value of Huang–Rhys factor.17 (d) Illustration of fluorescence quenching by the non-radiative channel in Pt-1. | ||
To understand the S1 → S0 process, we analyzed the Huang–Rhys factors as well as the displacement vectors of the vibrational modes with the largest Huang–Rhys factors (Fig. 5b, c and S88 details in the ESI†).17 The most important non-radiative channels for Pd-1 and Pt-1 can be assigned to the torsional motion of the pentafluorophenyl group (with frequencies of 16.28 and 14.45 cm−1 in Fig. 5b and c). Pt-1 has an additional non-negligible non-radiative channel (Fig. 5c), which is the out-plane vibration of the tripyrrin plane with a frequency of 22.12 cm−1. This could be correlated to the dark state assigned to the structural relaxation observed in fs-TA for Pt-1 in Fig. 4. Moreover, this out-plane vibration of tripyrrin plane can also be associated with the 1 → 0 band in the emission spectra according to the simulation of vibrationally resolved spectra (Fig. S89 and details in ESI†). This additional non-radiative channel leads to a larger non-radiative rate constant (7.46 × 107 s−1, in Table S7†), compared with Pd-1 (4.04 × 107 s−1), and quenches the luminescence of Pt-1 by a fast energy transfer (16.34 ps) in the luminescent S1 state (Fig. 5d).
NIR bioimaging
As a proof-of-concept application, we herein introduced Pd-1 and Pd-3 into nanochannels of mesoporous silica nanoparticles, which endows fluorescent PdII complexes with excellent colloidal stability, low cytotoxicity, desirable blood circulation time and passive tumor-targeting ability by the enhanced permeability and retention (EPR) effects.21 We prepared and characterized NIR fluorescent nanocapsules (Pd-1-MSN-F127 and Pd-3-MSN-F127), by encapsulating Pd-1 or Pd-3 into mesoporous channels of silica nanoparticles following previous reports in the literature (Fig. 6a, S90 and S91†).22 As expected, Pd-1-MSN-F127 and Pd-3-MSN-F127 display strong NIR emission centered at 717 and 771 nm in water solution, upon excitation at 600 or 700 nm (Fig. S92–S95†). After verifying the high cell viability (Fig. S96†), we performed living cell imaging by incubation of Pd-1-MSN-F127 (∼10 μM) and Pd-3-MSN-F127 (∼10 μM) in living HeLa cells. As shown in Fig. S97† and 6b, distinct NIR fluorescence (700–000 nm) precisely co-localized with commercial LysoTracker Green DND-26 Dye (the Pearson coefficients were 0.74 and 0.80 for Pd-1 and Pd-3 respectively, Fig. S98†), suggesting the capability of Pd-1 and Pd-3 as NIR bioprobes for living cell imaging.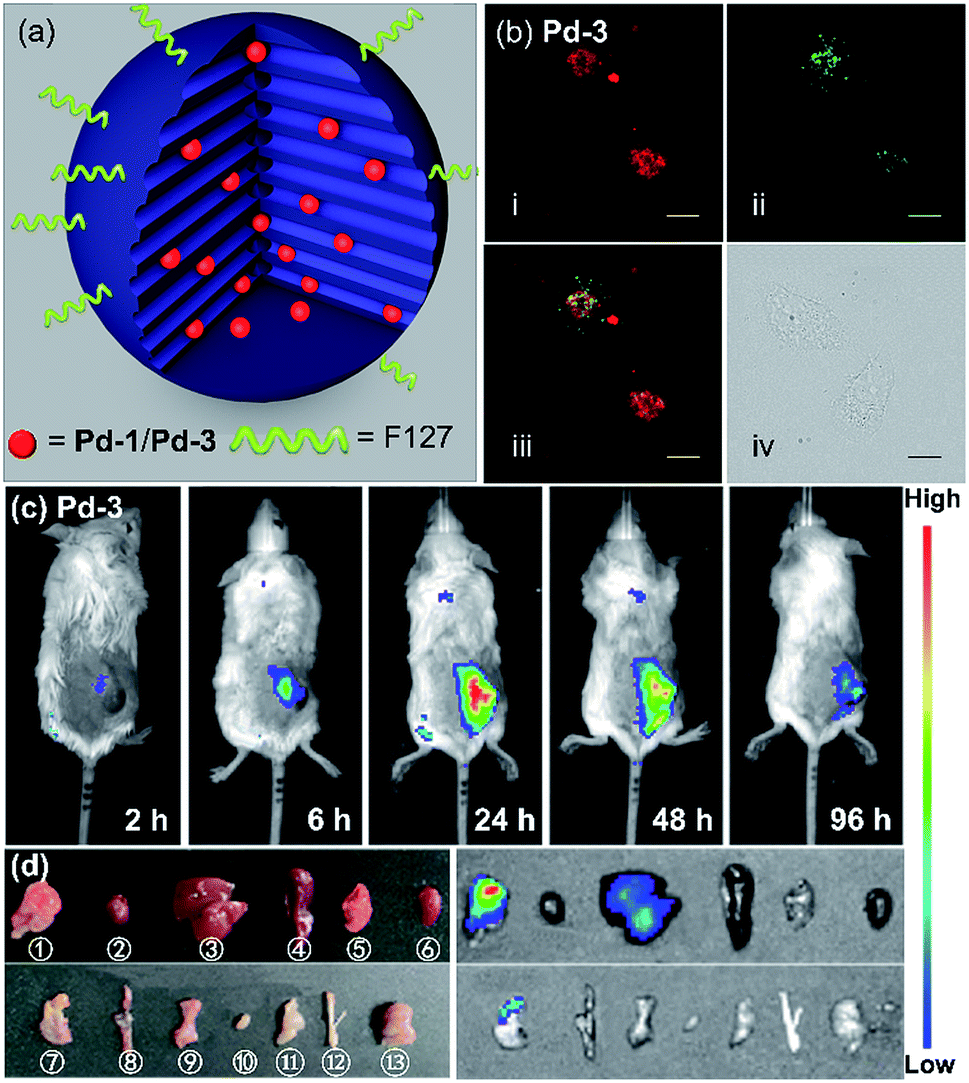 | ||
| Fig. 6 (a) Sketch of the Pd-1 and Pd-3 in a single NIR-MSN-F127. (b) Confocal fluorescence image of (i) living HeLa cells with Pd-3-MSN-F127 by the em > 776 nm channel under laser excitation at 700 nm; (ii) LysoTracker Green DND-26 excited at 488 nm, em = 525/50 channel; (iii) merged confocal fluorescence images of (i) and (ii); and (iv) differential interference contrast (DIC) images. Images of Pd-3-MSN-F127 (incubated for 12 h) are presented with fake color; scale bar presents 20 μm. (c) Fluorescence imaging of 4T1 tumor-bearing mice after intravenous injection of Pd-3-MSN-F127 at different time points. (d) Photograph (left) and fluorescence imaging (right) of main organs of 4T1 tumor-bearing mice at 96 h p.i. of Pd-3-MSN-F127. ①–⑬ stand for tumor, heart, liver, spleen, lung, kidney, stomach, intestine, pancreas, bladder, muscle, bone, and brain, respectively (λex = 710 nm, λem = 776 nm). | ||
To demonstrate the application in in vivo bioimaging, we chose 4T1 tumor-bearing mice as our model. Each mouse was intravenously injected via the tail vein with Pd-1-MSN-F127 or Pd-3-MSN-F127 (100 μL, ∼10 μM) and subjected to fluorescence imaging with 710 nm excitation and 776 nm emission at different time points. The luminescence signal was weak in 6 h (Fig. 6c and S99a†). After 24 h, we observed strong in vivo fluorescence, mainly accumulated specifically at the tumor site, as shown in Fig. 6c and S99a.† The mouse was sacrificed and dissected after 96 h post injection. Ex vivo fluorescence imaging as shown in Fig. S99b† and 6d displayed strong NIR fluorescence signals in the tumor and low intensity in the liver and stomach, indicating that Pd-1-MSN-F127 or Pd-3-MSN-F127 accumulated specifically at the tumor and was probably excreted through enterohepatic circulation and the digestive system. The sharp contrast of the tumor versus the remaining organs and tissues could be attributed to the strong fluorescence of Pd-1-MSN-F127 or Pd-3-MSN-F127, their good stability and effective accumulation in tumors. Although more details about the long-term fate and clearance pathways remain unclear at the present stage, Pd-1-MSN-F127 or Pd-3-MSN-F127 are promising NIR candidate probes for in vivo bioimaging applications such as tumor diagnosis and imaging-guided surgery.
To further construct stimuli-responsive probes, we used Pd-2, which has weak fluorescence due to PET, to prepare a pH sensitive NIR probe (Pd-2-MSN-F12) by encapsulation of Pd-2 into mesoporous silica nanoparticles. Pd-2-MSN-F127 localized in the lysosome with a Pearson coefficient of 0.74 (Fig. 7a and S100†). From pH 5 to 10, Pd-2-MSN-F127 exhibits a remarkable fluorescence increase of 4 fold and shows good reversibility (Fig. 7b–d, S100 and S101†). This could be assigned to the protonation of NMe2 which inhibits the PET process and turns on the NIR fluorescence (Fig. 7b). To study the pH-sensitivity in living cells, we monitor the endogenous pH changes using the antibiotic nigericin as an H+/K+ ionophore to induce H+ efflux.23 As shown in Fig. 7e, the intracellular NIR fluorescence intensity decreases as the pH is increased from 6 to 10 (Fig. 7f), similar to pH sensitive NIR emission in aqueous solution (Fig. 7c). These results demonstrate the capability of Pd complexes as a molecular platform to further design physiological stimuli-responsive NIR probes.
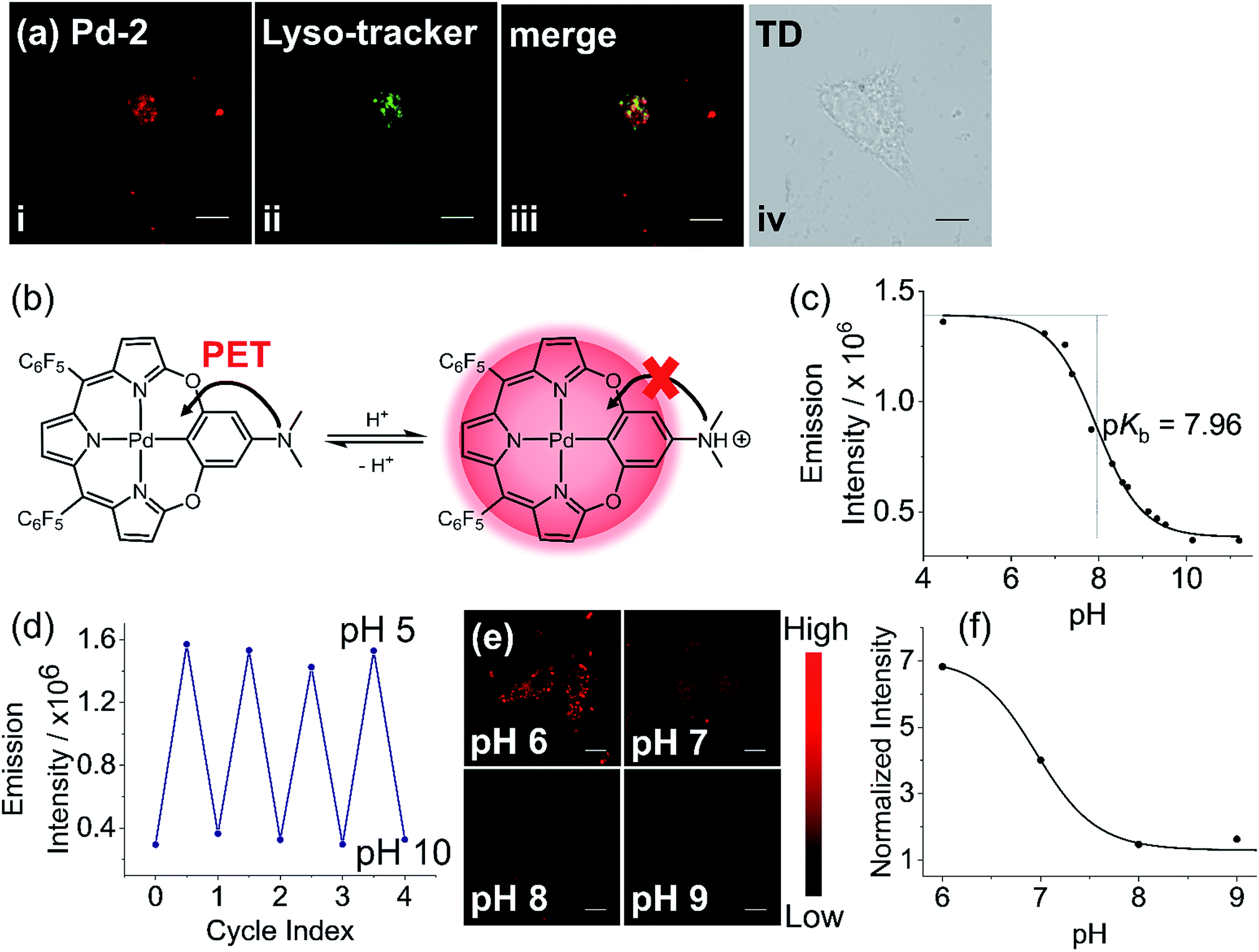 | ||
| Fig. 7 (a) Confocal fluorescence image of (i) living HeLa cells with Pd-2-MSN-F127 by em > 776 nm channel under laser excitation at 700 nm; (ii) LysoTracker Green DND-26 excited at 488 nm, em = 525/50 channel; (iii) merged confocal fluorescence images of (i) and (ii); and (iv) differential interference contrast (DIC) images. (b) Schematic view of pH sensitivity of Pd-2 due to the switching on/off of photoinduced electron transfer (PET) by protonation. (c) Emission intensity of Pd-2-MSN-F127versus the pH values in the pH range 5–11. (d) Plot showing the reversible switching on/off of the emission of Pd-2-MSN-F127 between pH 5 and 10. (e) Fluorescence imaging of Pd-2-MSN-F127 at different pH values in pH range 6–9. (f) Dependence of normalized average intensity of fluorescence per cell on pH. Images of Pd-2-MSN-F127 (incubated for 12 h) are presented with fake color; scale bar represents 20 μm. | ||
Conclusions
In summary, we designed and synthesized a series of new tetradentate macrocyclic benzitripyrrin C^N^N^N ligands (1–5) and the palladium(II) and platinum(II) complexes Pd-1–3 and Pt-3. Coordination of PdII or PtII ion leads to a bathochromic shift of absorption and emission compared to the corresponding ligands, indicating that the metal chelating affects the ground- and excited-state structures. Importantly, chelating the PdII ion produces strong NIR fluorescence while no phosphorescence is observed, which is unusual for the previously reported PdII porphyrinoids or palladacycles. Femtosecond transient absorption spectra of Pd-1 and Pt-1 showed the intraligand (1IL) charge transfer process and the nanosecond transient absorption spectra suggest no triplet states are populated. This could be attributed to a small SOC, arising from large singlet-triplet energy gaps and the ligand-centered FMOs, as suggested by the TDDFT calculation results. Additionally, the weak fluorescence of Pt-1 can be ascribed to the non-radiative channel of the out-plane vibration of the tripyrrin plane, different from Pd-1. More importantly, we also demonstrated the strong NIR emission of Pd complexes (Pd-1 and Pd-3) by encapsulation into mesoporous silica nanoparticles is advantageous compared to other phosphorescent metal complexes in in vitro or in vivo bioimaging because of much less oxygen interference. Finally, Pd-2 was employed as a pH sensitive probe intracellularly, demonstrating the potential of these Pd complexes as a molecular scaffold to design stimuli-responsive materials. Therefore, this work reveals the potential application of NIR fluorescent Pd complexes and promotes the development of Pd chemistry in biomedicine.Ethical statement
All animal experiments were implemented in accordance with the Animal Management Rules of the Ministry of Health of the People's Republic of China (Document no. 55, 2001) and were approved and according to the guidelines from Laboratory Animal Ethics Committee of Peking University First Hospital (Peking University, China) (Approval ID: J201859).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Scientific Foundation of China (NSFC) (21571007, 21621061, 21778002, 21861162008) and the National Key Basic Research Support Foundation of China (NKBRSFC) (2015CB856301) for financial support. This work was supported by the High-performance Computing Platform of Peking University.Notes and references
- (a) G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef; (b) R. R. Zhang, A. B. Schroeder, J. J. Grudzinski, E. L. Rosenthal, J. M. Warram, A. N. Pinchuk, K. W. Eliceiri, J. S. Kuo and J. P. Weichert, Nat. Rev. Clin. Oncol., 2017, 14, 347–364 CrossRef CAS PubMed.
- (a) W. Stummer, U. Pichlmeier, T. Meinel, O. D. Wiestler, F. Zanella, H. J. Reulen and A. L.-G. S. Group, Lancet Oncol., 2006, 7, 392–401 CrossRef CAS PubMed; (b) V. Pansare, S. Hejazi, W. Faenza and R. K. Prud'homme, Chem. Mater., 2012, 24, 812–827 CrossRef CAS PubMed; (c) G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef CAS PubMed; (d) S. Tsuboi, S. Yamada, Y. Nakane, T. Sakata, H. Yasuda and T. Jin, ECS J. Solid State Sci. Technol., 2017, 7, R3093–R3101 CrossRef; (e) G. Chen, Y. Zhang, C. Li, D. Huang, Q. Wang and Q. Wang, Adv. Healthcare Mater., 2018, 7, 1800497 CrossRef PubMed; (f) Y. Duan and B. Liu, Adv. Mater., 2018, 30, 1802394 CrossRef PubMed; (g) Q. Miao and K. Pu, Adv. Mater., 2018, 30, 1801778 CrossRef PubMed; (h) Y. Cai, Z. Wei, C. Song, C. Tang, W. Han and X. Dong, Chem. Soc. Rev., 2019, 48, 22–37 RSC; (i) F. Ding, Y. Fan, Y. Sun and F. Zhang, Adv. Healthcare Mater., 2019, 8, 1900260 CrossRef PubMed; (j) J. Li, H. Duan and K. Pu, Adv. Mater., 2019, 1901607, DOI:10.1002/adma.201901607; (k) S. Zhu, R. Tian, A. L. Antaris, X. Chen and H. Dai, Adv. Mater., 2019, 31, 1900321 CrossRef PubMed.
- (a) S. He, J. Song, J. Qu and Z. Cheng, Chem. Soc. Rev., 2018, 47, 4258–4278 RSC; (b) F. Ding, Y. Zhan, X. Lu and Y. Sun, Chem. Sci., 2018, 9, 4370–4380 RSC.
- (a) Y. Ning, S. Chen, H. Chen, J.-X. Wang, S. He, Y.-W. Liu, Z. Cheng and J.-L. Zhang, Inorg. Chem. Front., 2019, 6, 1962–1967 RSC; (b) Y. Ning, S. Cheng, J. X. Wang, Y. W. Liu, W. Feng, F. Li and J.-L. Zhang, Chem. Sci., 2019, 10, 4227–4235 RSC.
- (a) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS; (b) K. K. W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed; (c) C. N. Ko, G. D. Li, C. H. Leung and D. L. Ma, Coord. Chem. Rev., 2019, 381, 79–103 CrossRef CAS; (d) K. Q. Qiu, Y. Chen, T. W. Rees, L. N. Ji and H. Chao, Coord. Chem. Rev., 2019, 378, 66–86 CrossRef CAS.
- J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2571 CrossRef CAS PubMed.
- (a) P. K. Chow, C. Ma, W. P. To, G. S. Tong, S. L. Lai, S. C. Kui, W. M. Kwok and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 11775–11779 CrossRef CAS PubMed; (b) P. K. Chow, W. P. To, K. H. Low and C.-M. Che, Chem.–Asian J., 2014, 9, 534–545 CrossRef CAS PubMed; (c) P. K. Chow, G. Cheng, G. S. M. Tong, C. Ma, W. M. Kwok, W. H. Ang, C. Y. Chung, C. Yang, F. Wang and C.-M. Che, Chem. Sci., 2016, 7, 6083–6098 RSC; (d) T. Fleetham, G. Li and J. Li, Adv. Mater., 2017, 29, 1601861 CrossRef PubMed; (e) Z. Q. Zhu, K. Klimes, S. Holloway and J. Li, Adv. Mater., 2017, 29, 1605002 CrossRef PubMed; (f) Q. Wan, W. P. To, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 3089–3093 CrossRef CAS PubMed; (g) G. Li, Z.-Q. Zhu, Q. Chen and J. Li, Org. Electron., 2019, 69, 135–152 CrossRef CAS.
- (a) H. Furuta, H. Maeda and A. Osuka, Chem. Commun., 2002, 1795–1804, 10.1039/b200525p; (b) J. Harvey, Coord. Chem. Rev., 2003, 247, 1–19 CrossRef CAS; (c) M. Stepien and L. Latos-Grazynski, Acc. Chem. Res., 2005, 38, 88–98 CrossRef CAS PubMed; (d) T. D. Lash, Acc. Chem. Res., 2016, 49, 471–482 CrossRef CAS PubMed; (e) T. D. Lash, Chem. Rev., 2017, 117, 2313–2446 CrossRef CAS PubMed.
- G. S. Tong, P. K. Chow and C.-M. Che, Angew. Chem., Int. Ed., 2010, 49, 9206–9209 CrossRef CAS PubMed.
- (a) J.-F. Wang, Y. Yao, Y. Ning, Y.-S. Meng, C.-L. Hou, J. Zhang and J.-L. Zhang, Org. Chem. Front., 2018, 5, 1877–1885 RSC; (b) C.-L. Hou, Y. Yao, D. Wang, J. Zhang and J.-L. Zhang, Org. Chem. Front., 2019, 6, 2266–2274 RSC.
- (a) B. W. Pedersen, L. E. Sinks, T. Breitenbach, N. B. Schack, S. A. Vinogradov and P. R. Ogilby, Photochem. Photobiol., 2011, 87, 1077–1091 CrossRef CAS PubMed; (b) W. P. To, Y. Liu, T. C. Lau and C.-M. Che, Chem.–Eur. J., 2013, 19, 5654–5664 CrossRef CAS PubMed; (c) T. V. Esipova, H. J. Rivera-Jacquez, B. Weber, A. E. Masunov and S. A. Vinogradov, J. Am. Chem. Soc., 2016, 138, 15648–15662 CrossRef CAS PubMed; (d) Z. Xun, Y. Zeng, J. Chen, T. Yu, X. Zhang, G. Yang and Y. Li, Chem.–Eur. J., 2016, 22, 8654–8662 CrossRef CAS PubMed.
- (a) M. L. Deda, M. Ghedini, I. Aiello, T. Pugliese, F. Barigelletti and G. Accorsi, J. Organomet. Chem., 2005, 690, 857–861 CrossRef CAS; (b) V. Lakshmi, W.-Z. Lee and M. Ravikanth, Dalton Trans., 2014, 43, 16006–16014 RSC; (c) A. Kumar, S. Kumar, T. Chatterjee and M. Ravikanth, ChemistrySelect, 2016, 1, 94–100 CrossRef CAS; (d) S. Riese, M. Holzapfel, A. Schmiedel, I. Gert, D. Schmidt, F. Wurthner and C. Lambert, Inorg. Chem., 2018, 57, 12480–12488 CrossRef CAS PubMed.
- H. Weissman, E. Shirman, T. Ben-Moshe, R. Cohen, G. Leitus, L. J. Shimon and B. Rybtchinski, Inorg. Chem., 2007, 46, 4790–4792 CrossRef CAS PubMed.
- (a) A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385–2393 RSC; (b) E. M. Stennett, M. A. Ciuba and M. Levitus, Chem. Soc. Rev., 2014, 43, 1057–1075 RSC; (c) D. Escudero, Acc. Chem. Res., 2016, 49, 1816–1824 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of photochemistry, CRC press, 2006 Search PubMed.
- (a) G. S. Tong, P. K. Chow, W. P. To, W. M. Kwok and C.-M. Che, Chem.–Eur. J., 2014, 20, 6433–6443 CrossRef CAS PubMed; (b) K. T. Chana, G. S. M. Tong, W.-P. Toa, C. Yanga, L. Dub, D. L. Phillipsb and C.-M. Che, Chem. Sci., 2016, 8, 2352–2364 RSC; (c) Y. Bai, J. Rawson, S. A. Roget, J.-H. Olivier, J. Lin, P. Zhang, D. N. Beratan and M. J. Therien, Chem. Sci., 2017, 8, 5889–5901 RSC; (d) Y. Yao, H.-Y. Yin, Y. Ning, J. Wang, Y.-S. Meng, X. Huang, W. Zhang, L. Kang and J.-L. Zhang, Inorg. Chem., 2019, 58, 1806–1814 CrossRef CAS PubMed.
- Q. Peng, D. Fan, R. Duan, Y. Yi, Y. Niu, D. Wang and Z. Shuai, J. Phys. Chem. C, 2017, 121, 13448–13456 CrossRef CAS.
- (a) T. Lu and F. Chen, Acta Chim. Sin., 2011, 69, 2393–2406 CAS; (b) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed; (c) M. Xiao and T. Lu, J. Adv. Phys. Chem., 2015, 4, 111–124 CrossRef CAS.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef CAS.
- (a) Z. G. Shuai, Q. Peng, Y. L. Niu and H. Geng, MOMAP, a molecular materials property prediction package, revision 0.2.004, Tsinghua University, Beijing, China, 2014, http://www.shuaigroup.net/ Search PubMed; (b) Y. Niu, W. Li, P. Qian, G. Hua and Z. Shuai, Mol. Phys., 2018, 116, 1–13 CrossRef.
- (a) M. P. Melancon, M. Zhou and C. Li, Acc. Chem. Res., 2011, 44, 947–956 CrossRef CAS PubMed; (b) A. Z. Wang, R. Langer and O. C. Farokhzad, Annu. Rev. Med., 2012, 63, 185–198 CrossRef CAS PubMed; (c) D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- S. Mattiello, A. Monguzzi, J. Pedrini, M. Sassi, C. Villa, Y. Torrente, R. Marotta, F. Meinardi and L. Beverina, Adv. Funct. Mater., 2016, 26, 8447–8454 CrossRef CAS.
- (a) J. R. Casey, S. Grinstein and J. Orlowski, Nat. Rev. Mol. Cell Biol., 2010, 11, 50–61 CrossRef CAS PubMed; (b) Y. Yue, F. Huo, S. Lee, C. Yin and J. Yoon, Analyst, 2016, 142, 30–41 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1912139–1912141. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04044g |
| This journal is © The Royal Society of Chemistry 2019 |