
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite- and degree-specific C–H oxidation on 5-methylcytosine homologues for probing active DNA demethylation†
Sam
Kavoosi‡
,
Babu
Sudhamalla‡
,
Debasis
Dey‡
,
Kirsten
Shriver
,
Simran
Arora
,
Sushma
Sappa
and
Kabirul
Islam
 *
*
Department of Chemistry, University of Pittsburgh, Pennsylvania 15260, USA. E-mail: kai27@pitt.edu
First published on 30th September 2019
Abstract
Ten-eleven translocation (TET) enzymes oxidize C–H bonds in 5-methylcytosine (5mC) to hydroxyl (5hmC), formyl (5fC) and carboxyl (5caC) intermediates en route to DNA demethylation. It has remained a challenge to study the function of a single oxidized product. We investigate whether alkyl groups other than methyl could be oxidized by TET proteins to generate a specific intermediate. We report here that TET2 oxidizes 5-ethylcytosine (5eC) only to 5-hydroxyethylcytosine (5heC). In biochemical assays, 5heC acts as a docking site for proteins implicated in transcription, imbuing this modification with potential gene regulatory activity. We observe that 5heC is resistant to downstream wild type hydrolases, but not to the engineered enzymes, thus establishing a unique tool to conditionally alter the stability of 5heC on DNA. Furthermore, we devised a chemical approach for orthogonal labeling of 5heC. Our work offers a platform for synthesis of novel 5-alkylcytosines, provides an approach to ‘tame’ TET activity, and identifies 5heC as an unnatural modification with a potential to control chromatin-dependent processes.
Introduction
Gene expression is regulated by chemical modifications of DNA such as the methylation of cytosine at carbon 5 (5mC).1 It has been reported that ten-eleven translocation (TET) enzymes iteratively oxidize C–H bonds in 5mC to generate 5-hydroxymethyl (5hmC), 5-formyl (5fC), and 5-carboxyl (5caC) cytosines using 2-ketoglutarate (2KG) and Fe(II) as co-substrates (Fig. 1A).2–4 5caC is subsequently excised and repaired to cytosine through base-excision repair (BER) pathway, providing a biochemical basis for active DNA demethylation (Fig. 1A).5 Growing evidence suggests that each individual intermediate can regulate gene expression.6 Among the oxidized species, 5hmC is the most abundant in embryonic stem cells as well as neuronal cells.7 Misregulation in 5hmC production is implicated in multiple cancers.8 These findings have spurred interest in developing analytical methods to gain mechanistic insights into the functions of 5hmC.9 However, heterogeneous distribution of the concomitant and fleeting intermediates in DNA pose a significant challenge to ascertain their specific role. Chemical means to specifically access and stabilize a particular intermediate would constitute a powerful approach to probe its potential role in gene expression and disease.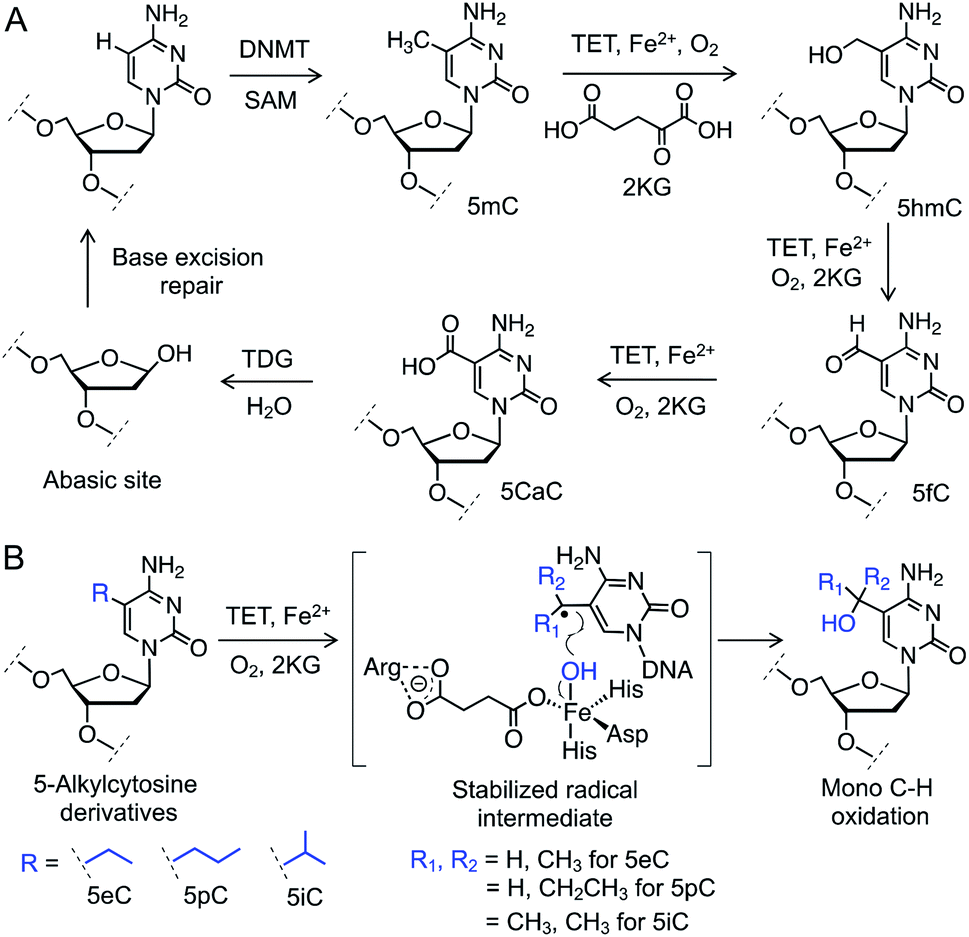 | ||
| Fig. 1 C–H oxidation by TET enzymes. (A) Schematic showing active DNA demethylation via TET-mediated successive C–H oxidation of 5mC to 5hmC, 5fC and 5caC followed by TDG catalyzed base hydrolysis and repair. (B) Structures of the 5-alkylcytosine derivatives synthesized and examined for TET activity. DNMT: DNA methyltransferase, SAM: S-adenosylmethionine. | ||
We envisioned employing 5mC homologues to control the degree of C–H hydroxylation by TET enzymes to access a specific intermediate (Fig. 1B). We reasoned that substituting the methyl group with an ethyl, a propyl or an isopropyl moiety would preferentially furnish a specific oxidized product by restricting the number of available C–H bonds for oxidation. Restricted rotation of the alkyl group in TET active site would also enhance the degree specificity of oxidation. Furthermore, given that TET-mediated C–H hydroxylation proceeds through a radical pathway,10 a secondary (from an ethyl or a propyl group) or a tertiary (from an isopropyl group) radical intermediate would have the benefit of enhanced hyperconjugative stabilization compared to their primary (from the methyl group) counterpart, potentially lowering the transition-state barrier for the proposed C–H activation (Fig. 1B).
Results and discussion
Synthesis of the alkylated oligonucleotide probes
To test our hypothesis for stereoelectronic control of TET activity, we designed a set of short oligonucleotides carrying cytidine analogues with alkyl groups at carbon 5 including 5-ethyl (5eC), 5-propyl (5pC) and 5-isopropyl (5iC) cytosines (Fig. 1B). We developed short synthetic schemes towards the corresponding phosphoramidites 1–3 (Fig. 2A). Although, synthesis of 1 and 2 is accomplished by modifying the available methods (Schemes S1 and S2†),11–13 access to novel phosphoramidite 3 required development of a new synthetic method. We employed metal-catalyzed cross-coupling reaction14 followed by hydrogenation to install the isopropyl group on unprotected cytidine as key step (Scheme S3†). Upon accessing all the phosphoramidite building blocks, we prepared a set of novel oligonucleotides containing the modified cytidines in a short palindromic sequence using solid-phase DNA synthesis technique (Fig. 2B, Table S1†).15 We also synthesized the corresponding 5mC-containing DNA to serve as a positive control in biochemical experiments. Integrity of the alkylated DNAs was confirmed by MALDI-MS (Fig. S1†).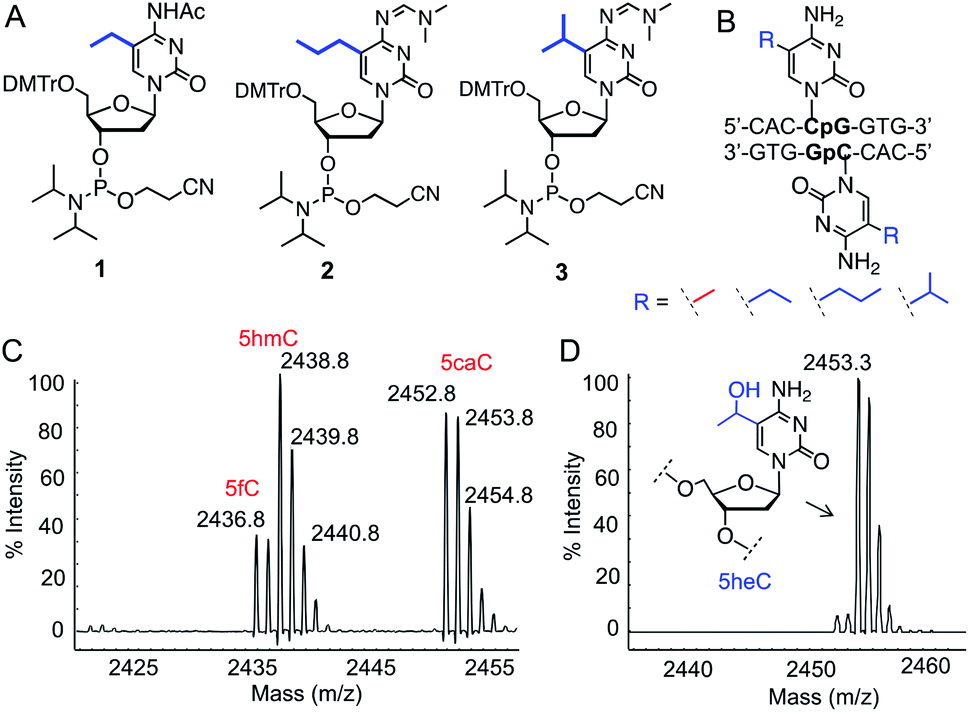 | ||
| Fig. 2 Activity of TET enzymes on 5-alkylcytosine containing DNAs. (A) Chemical structures of phosphoramidites 1, 2 and 3. (B) Sequence of double-stranded DNAs containing 5eC, 5pC or 5iC. (C) Formation of 5hmC, 5fC and 5caC from 5mC DNA by TET2 as evident from MALDI-MS spectra. (D) TET2 selectively generated 5-hydroxyethylcytosine (5heC) from 5eC DNA. | ||
Activity of TET2 towards alkylated oligonucleotides
We examined the activity of catalytic domain of TET2 on the synthetic oligonucleotides by analyzing the mass of each oxidized product.16 Consistent with earlier observations, TET2 mediated oxidation of double-stranded DNA containing 5mC led to a heterogeneous mixture of all the oxidized products 5hmC, 5fC and 5CaC (Fig. 2C and S2†). Under identical conditions, we observed that 5eC could be selectively oxidized to 5-hydroxyethylcytosine (5heC) by TET2 (Fig. 2D and S2†). In a time-dependent C–H hydroxylation assay, both 5mC and 5eC showed comparable activity (Fig. S3†). Further oxidation of 5heC was nearly undetectable (<2%). 5pC-containing DNA was marginally oxidized to 5-hydroxypropylcytosine (Fig. S4†). DNA with further extended modification such as 5iC remained inactive towards TET2 (Fig. S4†). The lack of oxidation on the bulky analogues (5pC and 5iC) is most likely due to the inability of TET2 to accommodate extended alkyl moiety in the active site.Successful oxidation of the 5-methylcytosine analogues by TET2 resonates with an earlier observation that lysine demethylases (KDMs), another class of Fe(II) and 2KG dependent hydroxylases, can act on ethyl and isopropyl lysine in histone peptides,17 suggesting flexibility in the substrate scope for this class of enzymes. It has been shown recently that TET2 can oxidize 5-vinylcytidine and 5-ethynylcytidine to produce fleeting epoxide and hydroxy-ethynyl intermediates, respectively.18 These cytidine analogues failed to deliver stable hydroxyl derivatives, as they lack the critical sp3 hybridized benzylic C–H bonds present in 5mC and 5eC.
Regiospecificity of 5eC oxidation by TET2
In order to determine the regioselectivity of oxidation on the ethyl group (–CH2– vs. –CH3), we implemented 5-CH3CD2-cytidine containing DNA (Fig. 3A). Oxidation of –CD2– would result in the addition of 15 atomic mass units whereas oxidation of the –CH3 group would lead to an increase of 16 mass units. For this purpose, we developed a four-step protocol to 5-CH3CD2-cytidine phosphoramidite 4 and incorporated it into a DNA sequence identical to that of 5eC (Fig. 3B, C and S5, Scheme S4†). Upon exposure to TET2, 5-CH3CD2-cytosine DNA underwent smooth hydroxylation at the heavy methylene (–CD2–) site as discernible from the increase of 15 mass units in MALDI spectrum (Fig. 3D). The observation that –CD2–, and not the terminal –CH3, is oxidized by TET2 is congruent with a radical-based mechanism leading to a stable benzylic radical intermediate at –CD2– site.19 Degree-specific oxidation of 5eC to 5heC is likely due to the restricted conformational freedom of the ethyl group inside TET2 pocket, as structural and computational studies have shown that iterative oxidation of 5mC requires free rotation along the C5–CH3 bond to project the methyl hydrogens towards oxo-ferryl [Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] species.20,21 Taken together, our data show that TET2 is capable of oxidizing 5mC homologue 5eC in a site- and degree-specific manner to furnish 5heC exclusively.
O] species.20,21 Taken together, our data show that TET2 is capable of oxidizing 5mC homologue 5eC in a site- and degree-specific manner to furnish 5heC exclusively.
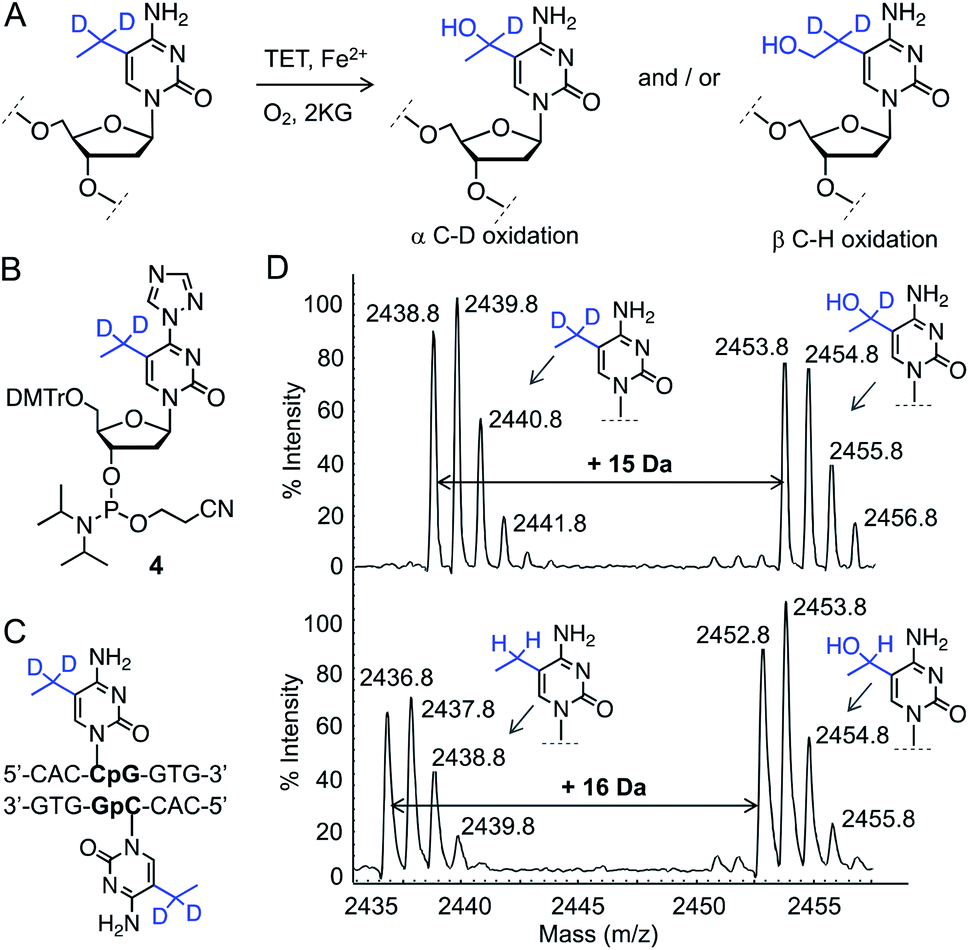 | ||
| Fig. 3 Characterization of TET2 activity on 5eC. (A) Reaction scheme showing two plausible 5heC positional isomers generated from 5eC. (B and C) Structures of 5-CH3CD2-cytidine phosphoramidite 4 and its corresponding DNA. (D) MALDI-MS spectra of TET2-mediated oxidation of deuterated 5eC (top panel). Addition of 15 daltons indicates oxidation of α-carbon carrying deuterium atoms. Oxidation of 5-CH3CH2-cytidine (5eC) by TET2 leads to increase of 16 daltons (lower panel). | ||
Recognition of 5heC by 5hmC ‘reader’
We next investigated if 5heC could mimic 5hmC in biochemical assays. It has been shown that UHRF2 binds 5hmC through its SRA domain by flipping the modified base allowing access to chromatin-modifying enzymes (Fig. 4A).22 We asked whether ‘reader’ modules dedicated to binding 5hmC could recognize 5heC.23 To access 5heC-containing DNA, we developed a synthetic scheme towards 5heC phosphoramidite 5 involving electrophilic addition of acetaldehyde to protected 5-bromouridine as a key step (Fig. 4B, Scheme S5†). In order to examine the interaction between UHRF2 and 5heC, we synthesized 5′-carboxyfluorescein (FAM) labeled DNA using 5 (Fig. S6, Table S1†).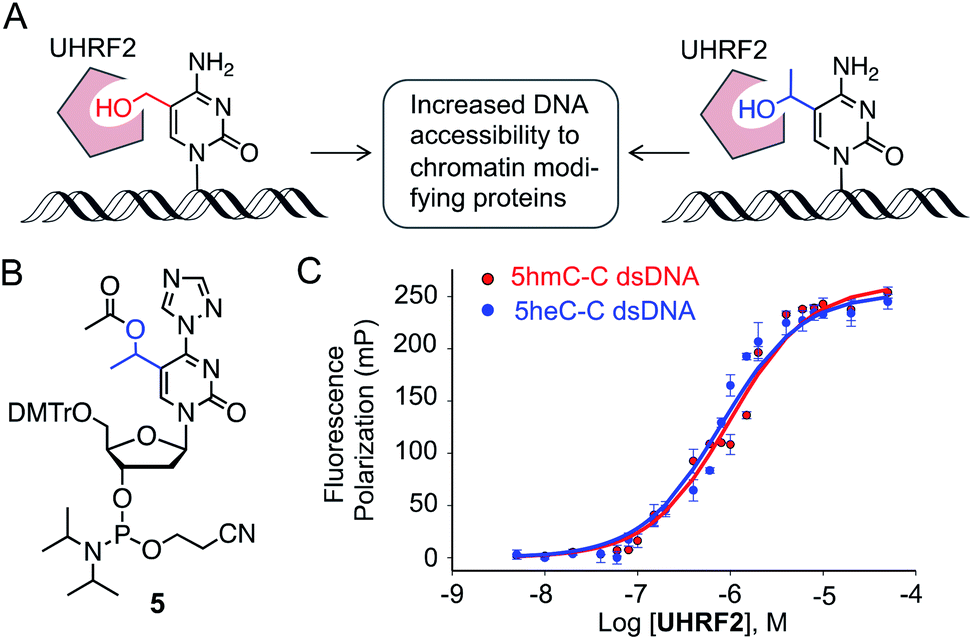 | ||
| Fig. 4 Binding of 5heC by 5hmC ‘reader’ protein. (A) UHRF2 binds to 5hmC (and potentially to 5heC) and interacts with a range of proteins for enhanced chromatin accessibility. (B) Structure of 5heC phosphoramidite 5. (C) Fluorescence anisotropy measurement showing that UHRF2 binds double-stranded DNA carrying either 5hmC or 5heC with similar affinity. | ||
We first measured the affinity between UHRF2-SRA domain and 5hmC-carrying DNA in fluorescence polarization (FP) assay and obtained a Kd of 0.97 ± 0.12 μM, consistent with an earlier report (Fig. 4C).24 Furthermore, the interaction between 5heC DNA and the reader domain was strong (Kd = 0.75 ± 0.13 μM), confirming that UHRF2-SRA could recognize 5heC with avidity comparable to that of 5hmC. A structural study has revealed that UHRF2-SRA domain carries a spacious hydrophobic pocket and can potentially bind a group bulkier than 5hmC,24 corroborating well with our experimental data. Collectively, the binding data support our hypothesis that 5heC could act as docking site for appropriate ‘reader’ proteins to elicit biological functions similar to 5hmC.
Hydrolytic resistance of 5heC towards 5hmC ‘erasers’
We next sought to assess the stability of 5heC on synthetic DNA. Active demethylation could occur via deamination of 5hmC to 5-hydroxymethyluridine (5hmU) by activation-induced cytidine deaminase (AID)/APOBEC family of deaminases, followed by TDG-mediated base excision repair (BER) (Fig. S7†).25,26 To evaluate whether these hydrolytic enzymes could act on 5eC and 5heC in tandem, we synthesized short DNA substrates bearing a central XpG motif (X represents a 5-modified cytidine) and a 5′-FAM unit (Fig. S8 and S9, Table S1†). To analyze the hydrolyzed intermediates, we employed a coupled assay where the AID/APOBEC-catalyzed deaminated products were duplexed with complementary strand to generate U:G mismatch for TDG-mediated base-excision followed by strand cleavage under alkaline condition (Fig. 5A). The fluorescently labeled cleaved DNA fragments were visualized on a denaturing gel.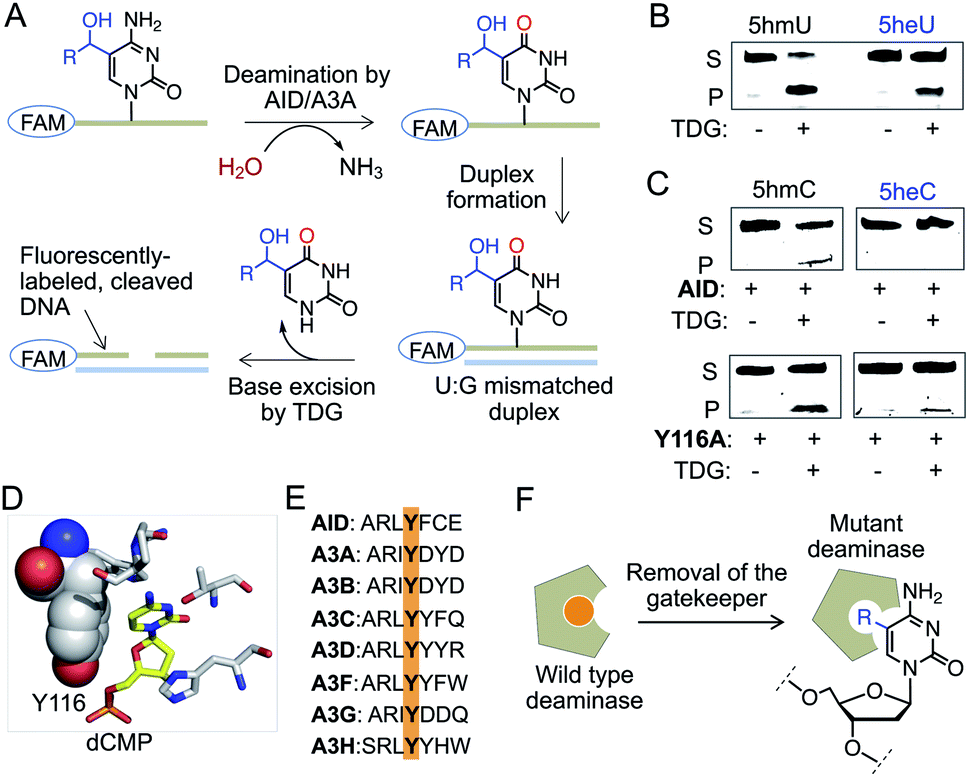 | ||
| Fig. 5 Stability of 5heC against hydrolytic enzymes. (A) Schematic showing coupled assay involving deamination and base excision of FAM-labeled DNA. (B) TDG can excise both 5hmU and 5heU duplexed DNAs carrying a U:G mismatch as confirmed by in-gel fluorescence. (C) Activity of wild type AID and its Y116A mutant towards DNA strands carrying either 5hmC (R = H) or 5heC (R = CH3) modifications. Deaminated DNAs were duplexed to create U:G mismatch prior to TDG-mediated base excision and strand cleavage. (D) Crystal structures of human AID (PDB: 5w0u). Y116 resides closely to substrate cytidine in the active site. (E) Sequence alignment showing that Y116 is conserved in human AID and APOBEC family of deaminases. (F) The tyrosine is likely acting as a gatekeeper residue that can be mutated to smaller amino acid to accommodate modified cytidine. S: substrate DNA; P: product DNA; dCMP: deoxycytidine monophosphate. | ||
We first confirmed TDG activity on synthetic DNAs with T:G and 5hmU:G mismatch, which generated DNA fragments upon base excision (Fig. 5B and S10†). Next, we examined whether TDG could excise 5-ethyluridine (5eU) and 5-hydroxy-ethyluridine (5heU) from DNA. For this purpose, we synthesized 5eU and 5heU phosphoramidites, 6 and 7, respectively and incorporated each into a DNA with sequence identical to that of 5hmU (Schemes S6 and S7, Fig. S8†). TDG was indeed able to cleave both 5eC and 5heU, albeit with lower efficiency compared to their methyl counterparts (Fig. 5B and S10†). Encouraged by these results, we assembled the entire erasure pathway by incubating AID and APOBEC 3A (A3A), one at a time, with each synthesized DNA containing 5mC, 5eC, 5hmC or 5heC (Fig. S9†). The putative deaminated products (T from 5mC, 5eU from 5eC, 5hmU from 5hmC and 5heU from 5heC) were subsequently duplexed to generate a U-G mismatch and subjected to base excision by TDG (Fig. 5A). We noticed that wild type AID and A3A disfavored 5hmC but not 5mC as substrate and generated a low level of deaminated product (Fig. 5C and S10†). Consistently, these enzymes failed to deaminate 5eC and 5heC (Fig. 5C and S10†).
Our results are along the lines of earlier reports that cytosine analogues carrying substituents at carbon 5 are poor substrates of AID and APOBECs.27–29 We analyzed the crystal structure and sequence of several human deaminases, and identified a conserved tyrosine that resides closely to the target cytosine in the active sites of AID and A3A (Fig. 5D and E, S10†).30–32 This tyrosine is likely acting as a gatekeeper residue for discrimination against 5hmC. We reasoned that substituting the tyrosine with alanine would expand the pocket to accommodate a bulky substrate (Fig. 5F).33 We generated Y116A and Y130A mutants of AID and A3A, respectively, and tested in the coupled assay. Remarkably, the mutants acquired increased deaminase activity towards 5hmC, and 5eC and 5heC, as evident from the formation of shorter fluorescently labeled fragments only in the presence of TDG (Fig. 5C and S10†). These results demonstrate that 5eC and 5heC do not undergo deamination by wild type deaminases such as AID and A3A, likely because of their compromised binding to the active site.
A second pathway for active demethylation involves direct excision of 5fC and 5caC by TDG (Fig. 1A).5,34 Electron-withdrawing formyl and carboxyl groups in 5fC and 5caC, respectively, weaken the base pairing ability in double-stranded DNA and contribute to their selective recognition and excision by TDG.35,36 To examine if further oxidation of 5heC to 5-acetylcytidine could provide an electronic feature similar to 5fC and 5caC, we oxidized 5heC in a short DNA using KRuO4 (Fig. 6A and S11†).37 Indeed, duplexed DNA carrying 5-acetylcytidine motif underwent smooth base excision by TDG, much like its 5fC counterpart (Fig. 6B and C). The sample lacking KRuO4 did not undergo cleavage, confirming that 5heC is not a direct substrate of TDG likely because it does not offer an electron poor pyrimidine ring. Collectively, these results confirmed that 5heC is refractory towards both deamination and base excision, unlike its natural congener 5hmC which is rapidly oxidized and eliminated from DNA. Importantly, the engineered deaminases with expanded active site deaminated 5heC for subsequent TDG-mediated base excision, thus offering a biochemical approach to conditionally alter the stability of 5heC in DNA.
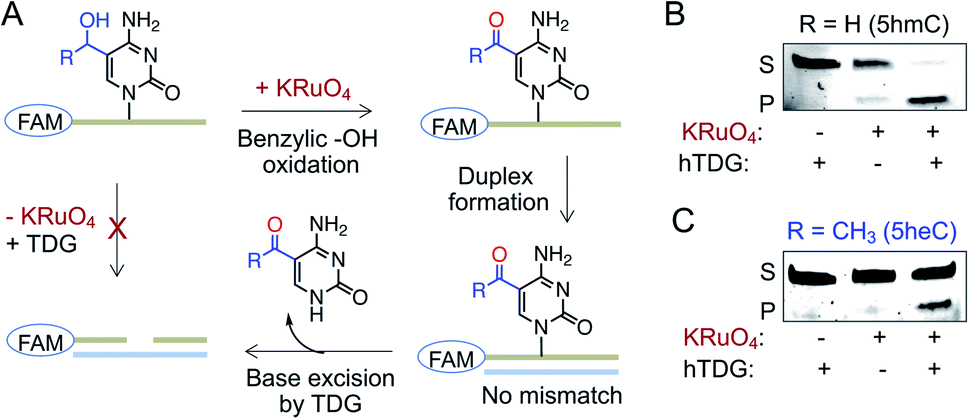 | ||
| Fig. 6 Activity of TDG towards 5heC. (A) Schematic showing an assay involving oxidation of 5hmC (R = H) or 5heC (R = CH3) on FAM-labeled DNA, duplex formation and base excision by TDG. (B and C) TDG can excise 5hmC (B) and 5heC (C) only in the presence of KRuO4 as confirmed by in-gel fluorescence. S: substrate DNA; P: product DNA. | ||
Selective functionalization of 5heC
Finally, we sought to functionalize 5heC selectively over 5hmC for selective labeling and enrichment from biological milieu. A particular chemoenzymatic approach includes β-glucosyltransferase (β-GT) mediated azido-glucosylation of 5hmC with 6-azidoglucose-UDP, followed by ligation with appropriate reporter molecule (Fig. 7A).38,39 We observed robust glucosylation, but not azido-glucosylation, of 5heC by β-GT (Fig. 7B and S12†).38,39 We realized that such difference in β-GT activity could constitute a strategy for selective protection of 5hmC with 6-azidoglucose-UDP in the presence of 5heC, keeping it available for further derivatization. Next, to functionalize 5heC, we adopted a two-step chemical approach involving treatment of 5heC DNA with KRuO4 followed by oxime formation with biotin hydroxylamine-based aldehyde reactive probe (ARP) (Fig. 7A). The reaction was analyzed using biotin antibody in a dot blot assay. We observed successful product formation as evident by strong chemiluminescent signal only in the presence of KRuO4 (Fig. 7C). In summary, our results offer a practical method for orthogonal labeling of 5heC-containing DNA first by masking 5hmC with 6-azidoglucose followed by oxidative biotinylation of 5heC.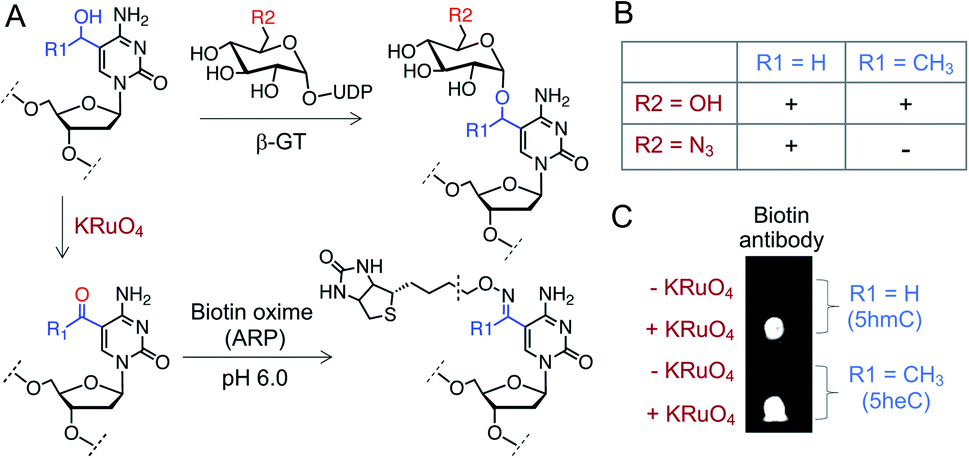 | ||
| Fig. 7 Selective labeling of 5heC. (A) Schematic showing a tandem oxidation and biotinylation approach to functionalize 5hmC and 5heC. (B) β-GT can transfer both glucose (R2 = OH) and N3-glucose (R2 = N3) to 5hmC (R1 = H) but only glucose to 5heC (R1 = CH3). (C) Dot-blot assay with anti-biotin antibody confirms tandem KRuO4 oxidation and oxime formation on 5hmC and 5heC. ARP: aldehyde reactive probe (Fig. S12† for structure). | ||
Conclusion
In this work, we have tailored an unnatural cytosine modification suitable for probing the DNA demethylation pathway. We first develop synthetic methods to install alkylated cytidine analogues on oligonucleotide segments. With these DNA probes, we show that TET2 could oxidize 5eC in site- and degree-specific manner to offer the mono-hydroxylated product via benzylic C–H activation. Such selective C–H functionalization is advantageous in studying transcriptional role of 5hmC in the guise of 5heC, as oxidation of 5mC leads to all the three intermediates with varied distribution. The rationale for a potential gene regulatory activity of 5heC came from our binding experiments that showed 5heC could act as a platform for chromatin-associated proteins such as UHRF2 and likely other 5hmC ‘readers’ for targeting transcriptional complexes. Finally, we examined the activity of human deaminases AID and A3A, and glycosylase TDG towards 5heC and presented convincing evidence that 5heC is not a substrate for these enzymes that act downstream of TETs to complete the DNA demethylation pathway. These results are particularly important as they establish that 5heC could stay as a stable modification in DNA. Finally, we devised a unique approach for orthogonal labelling of 5heC in DNA for selective enrichment. To harness the potential of 5heC in controlling DNA demethylation in cell, we anticipate incorporating 5heC into genomic DNA; we will employ SAM synthetase to access ethyl-SAM in cells cultured with ethionine which will be coupled with the DNA methyltransferase variant to incorporate 5eC in genomic DNA.40,41 Such ‘one-pot’ cellular assay to install 5eC and subsequently 5heC in chromosomal DNA by tapping into the relevant biosynthetic machinery within a cell will be pivotal in examining the gene regulatory potential of 5eC and 5heC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Pittsburgh, the National Science Foundation (MCB-1817692) and the National Institutes of Health (R01GM123234) for financial support; Dr D. Chakraborty and members of our laboratory for critical reading and editing of the manuscript. Support for MALDI-TOF MS instrumentation was provided by a grant from the National Science Foundation (CHE-1625002).Notes and references
- E. Li and Y. Zhang, Cold Spring Harbor Perspect. Biol., 2014, 6, a019133 CrossRef.
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 CrossRef CAS PubMed.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef CAS PubMed.
- Y. F. He, B. Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen, L. Li, Y. Sun, X. Li, Q. Dai, C. X. Song, K. Zhang, C. He and G. L. Xu, Science, 2011, 333, 1303–1307 CrossRef CAS PubMed.
- C. X. Song and C. He, Trends Biochem. Sci., 2013, 38, 480–484 CrossRef CAS PubMed.
- M. Munzel, D. Globisch and T. Carell, Angew. Chem., Int. Ed., 2011, 50, 6460–6468 CrossRef PubMed.
- M. Ko, Y. Huang, A. M. Jankowska, U. J. Pape, M. Tahiliani, H. S. Bandukwala, J. An, E. D. Lamperti, K. P. Koh, R. Ganetzky, X. S. Liu, L. Aravind, S. Agarwal, J. P. Maciejewski and A. Rao, Nature, 2010, 468, 839–843 CrossRef CAS PubMed.
- M. J. Booth, E. A. Raiber and S. Balasubramanian, Chem. Rev., 2015, 115, 2240–2254 CrossRef CAS PubMed.
- X. Lu, B. S. Zhao and C. He, Chem. Rev., 2015, 115, 2225–2239 CrossRef CAS PubMed.
- J. L. Ruth and D. E. Bergstrom, J. Org. Chem., 1978, 43, 2870–2876 CrossRef CAS.
- M. J. Robins and P. J. Barr, J. Org. Chem., 1983, 48, 1854–1862 CrossRef CAS.
- D. Kotandeniya, C. L. Seiler, J. Fernandez, S. S. Pujari, L. Curwick, K. Murphy, S. Wickramaratne, S. Yan, D. Murphy, Y. Y. Sham and N. Y. Tretyakova, Chem. Commun., 2018, 54, 1061–1064 RSC.
- L. Lercher, J. F. McGouran, B. M. Kessler, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2013, 52, 10553–10558 CrossRef CAS.
- B. Sudhamalla, D. Dey, M. Breski and K. Islam, Anal. Biochem., 2017, 534, 28–35 CrossRef CAS PubMed.
- L. Hu, Z. Li, J. Cheng, Q. Rao, W. Gong, M. Liu, Y. G. Shi, J. Zhu, P. Wang and Y. Xu, Cell, 2013, 155, 1545–1555 CrossRef CAS PubMed.
- R. J. Hopkinson, L. J. Walport, M. Munzel, N. R. Rose, T. J. Smart, A. Kawamura, T. D. Claridge and C. J. Schofield, Angew. Chem., Int. Ed., 2013, 52, 7709–7713 CrossRef CAS PubMed.
- U. Ghanty, J. E. DeNizio, M. Y. Liu and R. M. Kohli, J. Am. Chem. Soc., 2018, 140, 17329–17332 CrossRef CAS PubMed.
- M. Bachman, S. Uribe-Lewis, X. Yang, H. E. Burgess, M. Iurlaro, W. Reik, A. Murrell and S. Balasubramanian, Nat. Chem. Biol., 2015, 11, 555–557 CrossRef CAS PubMed.
- L. Hu, J. Lu, J. Cheng, Q. Rao, Z. Li, H. Hou, Z. Lou, L. Zhang, W. Li, W. Gong, M. Liu, C. Sun, X. Yin, J. Li, X. Tan, P. Wang, Y. Wang, D. Fang, Q. Cui, P. Yang, C. He, H. Jiang, C. Luo and Y. Xu, Nature, 2015, 527, 118–122 CrossRef CAS PubMed.
- H. Hashimoto, J. E. Pais, N. Dai, I. R. Correa Jr, X. Zhang, Y. Zheng and X. Cheng, Nucleic Acids Res., 2015, 43, 10713–10721 CrossRef CAS PubMed.
- C. G. Spruijt, F. Gnerlich, A. H. Smits, T. Pfaffeneder, P. W. Jansen, C. Bauer, M. Munzel, M. Wagner, M. Muller, F. Khan, H. C. Eberl, A. Mensinga, A. B. Brinkman, K. Lephikov, U. Muller, J. Walter, R. Boelens, H. van Ingen, H. Leonhardt, T. Carell and M. Vermeulen, Cell, 2013, 152, 1146–1159 CrossRef CAS PubMed.
- M. Iurlaro, G. Ficz, D. Oxley, E. A. Raiber, M. Bachman, M. J. Booth, S. Andrews, S. Balasubramanian and W. Reik, Genome Biol., 2013, 14, R119 CrossRef PubMed.
- T. Zhou, J. Xiong, M. Wang, N. Yang, J. Wong, B. Zhu and R. M. Xu, Mol. Cell, 2014, 54, 879–886 CrossRef CAS PubMed.
- S. Cortellino, J. Xu, M. Sannai, R. Moore, E. Caretti, A. Cigliano, M. Le Coz, K. Devarajan, A. Wessels, D. Soprano, L. K. Abramowitz, M. S. Bartolomei, F. Rambow, M. R. Bassi, T. Bruno, M. Fanciulli, C. Renner, A. J. Klein-Szanto, Y. Matsumoto, D. Kobi, I. Davidson, C. Alberti, L. Larue and A. Bellacosa, Cell, 2011, 146, 67–79 CrossRef CAS PubMed.
- J. U. Guo, Y. Su, C. Zhong, G. L. Ming and H. Song, Cell, 2011, 145, 423–434 CrossRef CAS PubMed.
- C. S. Nabel, H. Jia, Y. Ye, L. Shen, H. L. Goldschmidt, J. T. Stivers, Y. Zhang and R. M. Kohli, Nat. Chem. Biol., 2012, 8, 751–758 CrossRef CAS.
- G. Rangam, K. M. Schmitz, A. J. Cobb and S. K. Petersen-Mahrt, PLoS One, 2012, 7, e43279 CrossRef CAS PubMed.
- E. K. Schutsky, C. S. Nabel, A. K. F. Davis, J. E. DeNizio and R. M. Kohli, Nucleic Acids Res., 2017, 45, 7655–7665 CrossRef CAS PubMed.
- Q. Qiao, L. Wang, F. L. Meng, J. K. Hwang, F. W. Alt and H. Wu, Mol. Cell, 2017, 67, 361–373 CrossRef CAS PubMed.
- I. J. Byeon, J. Ahn, M. Mitra, C. H. Byeon, K. Hercik, J. Hritz, L. M. Charlton, J. G. Levin and A. M. Gronenborn, Nat. Commun., 2013, 4, 1890 CrossRef PubMed.
- S. M. Shandilya, M. F. Bohn and C. A. Schiffer, Virology, 2014, 471–473, 105–116 CrossRef CAS PubMed.
- B. Kavli, G. Slupphaug, C. D. Mol, A. S. Arvai, S. B. Peterson, J. A. Tainer and H. E. Krokan, EMBO J., 1996, 15, 3442–3447 CrossRef CAS.
- A. Maiti and A. C. Drohat, J. Biol. Chem., 2011, 286, 35334–35338 CrossRef CAS PubMed.
- L. Zhang, X. Lu, J. Lu, H. Liang, Q. Dai, G. L. Xu, C. Luo, H. Jiang and C. He, Nat. Chem. Biol., 2012, 8, 328–330 CrossRef CAS.
- Q. Dai, P. J. Sanstead, C. S. Peng, D. Han, C. He and A. Tokmakoff, ACS Chem. Biol., 2016, 11, 470–477 CrossRef CAS PubMed.
- M. J. Booth, M. R. Branco, G. Ficz, D. Oxley, F. Krueger, W. Reik and S. Balasubramanian, Science, 2012, 336, 934–937 CrossRef CAS.
- C. X. Song, T. A. Clark, X. Y. Lu, A. Kislyuk, Q. Dai, S. W. Turner, C. He and J. Korlach, Nat. Methods, 2011, 9, 75–77 CrossRef PubMed.
- A. Szwagierczak, S. Bultmann, C. S. Schmidt, F. Spada and H. Leonhardt, Nucleic Acids Res., 2010, 38, e181 CrossRef.
- R. Wang, K. Islam, Y. Liu, W. Zheng, H. Tang, N. Lailler, G. Blum, H. Deng and M. Luo, J. Am. Chem. Soc., 2013, 135, 1048–1056 CrossRef CAS PubMed.
- C. Dalhoff, G. Lukinavicius, S. Klimasauskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, synthesis and characterizations of compounds, supplementary data, figures and references. See DOI: 10.1039/c9sc02629k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |