DOI:
10.1039/C9SC02558H
(Edge Article)
Chem. Sci., 2019,
10, 7389-7398
Effector responsive hydroformylation catalysis†
Received
26th May 2019
, Accepted 15th June 2019
First published on 19th June 2019
Abstract
Herein, we report a supramolecular rhodium complex that can form dimeric or monomeric Rh-species catalytically active in hydroformylation, depending on the binding of effectors within the integrated DIM-receptor. X-ray crystal structures, in situ (high-pressure (HP)) spectroscopy studies, and molecular modelling studies show that in the absence of effectors, the preferred Rh-species formed is the dimer, of which two ligands coordinate to two rhodium metals. Importantly, upon binding guest molecules, -effectors-, to the DIM-receptor under hydroformylation conditions, the monomeric Rh-active species is formed, as evidenced by a combination of in situ HP NMR and IR spectroscopy studies and molecular modelling. As the monomeric complex has different catalytic properties from the dimeric complex, we effectively generate a catalytic system of which the properties respond to the presence of effectors, reminiscent of how the properties of proteins are regulated in nature. Indeed, catalytic and kinetic experiments show that both the selectivity and activity of this supramolecular catalytic system can be influenced in the hydroformylation of 1-octene using acetate as an effector that shift the equilibrium from the dimeric to monomeric species.
Introduction
Transition metal catalysis is a key technology for the development of efficient and sustainable synthetic protocols, and as such it has been an active research field for decades.1 In order to achieve new selectivity and reactivity, most research has been devoted to catalyst development. As the catalyst properties depend strongly on the ligands that coordinate to the metal, catalyst optimization mostly relies on ligand variation. Ligand modifications that are often applied and sometimes well understood in terms of effect on the selectivity and activity displayed by the catalyst, include steric2 and electronic2a,3 variations and the ligand bite angle.4 Interestingly, nature uses a much larger toolbox to construct catalytic systems that are generally far more active and selective than man-made catalysts. Enzymes are nature's catalysts and they convert the substrates in the active site next to a binding site. This binding site allows substrate preorganization, and as such control of catalyst properties via the second coordination sphere.5 Importantly, with the flourish of supramolecular approaches to build functional architectures including receptors,6 cages7 and artificial molecular machines,8 the exploration of enzyme inspired catalysis by precise control of substrate orientation has become an active field of research.9
Another big difference between synthetic and biological systems is that enzymes in their natural environment need to operate in a complex mixture of molecules, substrates and other enzymes and their reactivity and selectivity therefore needs to be regulated. Such a regulation can be achieved by cofactor regulation, i.e. a factor that is required for the activity of the enzyme, covalent functionalization of the enzyme, such as phosphorylation, or by allosteric binding of effectors.5 The effector controlled approach is based on noncovalent binding, which is important for controlling reactivity in more complex systems by feedback loops as it can increase as well as decrease reactivity. For this reason, it is interesting to design and study catalysts that can be controlled by effectors, i.e. supramolecular binding of molecules that otherwise do not interfere with the reaction. Mirkin, Kämer and co-workers reported an orthogonal metal–ligand coordination approach to create systems based on hemilabile coordination complexes.10 Chloride anions coordinate to the rhodium site thereby regulating the distance between two salphen complexes that operate as a binuclear Lewis acid catalyst in the “open state”. Considering the interest in effector controlled transition metal catalysis,11 we studied a supramolecular system that allows to control the activity and selectivity in hydroformylation by external factors. A new ligand coined OrthoDIMphos (L2), a regio-isomer of ParaDIMphos (L1) that also has a integrated binding site for carboxylate containing guests,12 forms dinuclear structures in which each ligand coordinates to two rhodium centers, even under hydroformylation conditions. By binding of a carboxylate guest molecule, the dimer breaks up to give mononuclear species only. As the monomer and dimer display different properties in alkene hydroformylation, we effectively generated an effector responsive catalyst system (Fig. 1).
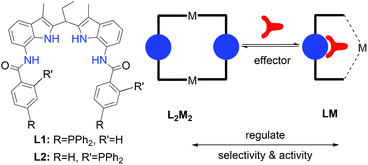 |
| | Fig. 1 The general concept of supramolecular tuning of the selectivity and activity via the regulation of the monomer–dimer catalyst equilibrium using effectors base on hydrogen bonding approach (NHs of the DIM-receptor shown in blue color). | |
Results and discussion
Synthesis of ligand OrthoDIMphos (L2)
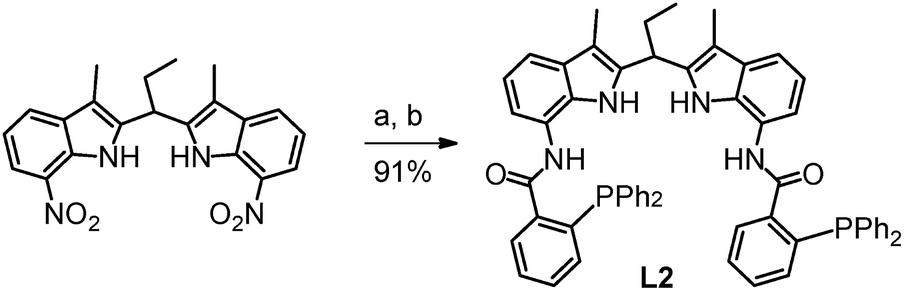
The ligand OrthoDIMphos (L2) was prepared following the same straightforward procedure as previously reported for the synthesis of ParaDIMphos (L1).12 Hydrogenation of 7,7′-dinitro-2,2′-diindolomethane led to the di-amine intermediate and subsequent condensation with 2-(diphenylphosphino)benzoic acid provided the new ligand in 91% overall yield (Schemes 1 and S3–S5†). The new ligand was fully characterized by 1H, 31P, and 13C NMR spectroscopy and HR-MS.
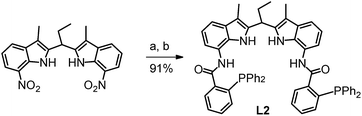 |
| | Scheme 1 Synthesis of OrthoDIMphos (L2): (a) 1 bar H2, Pd/C, THF/MeOH, RT; (b) 2-(diphenylphosphino)benzoic acid, 4-pyrrolidinopyridine, 4-dimethylaminopyridine, N,N′-diisopropylcarbodiimide, DCM, RT. | |
Characterization of the square planar dimeric complexes [Rh(L2)(CO)(BF4)]2 and [Rh(effector⊂L2)(CO)]2 as catalyst precursor
The rhodium complex based on OrthoDIMphos (L2) was formed by mixing the ligand with [Rh(nbd)2BF4]. Bubbling CO through a CH2Cl2 solution of OrthoDIMphos (L2) and [Rh(nbd)2BF4] (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) followed by slow diffusion of anti-solvent (cyclohexane) resulted in the formation of yellow crystals that were suitable for X-ray diffraction. The X-ray crystal structure shows the formation of centrosymmetric dimeric complexes ([Rh(L2)(CO)(BF4)]2Fig. 2 and S1†) in which two OrthoDIMphos (L2) ligands coordinate to two rhodium atoms. The rhodium complexes have a square planar geometry, with one ligand coordinating in a P–O chelating fashion and one with P, with the CO ligand in the fourth coordination site. The two DIM-receptors of the ligands in the dimeric complex are pointing in opposite directions and all NH hydrogen bond donors are free to bind guest molecules. Crystals were grown in the presence of acetate as guest, and the solid state structure indeed shows a similar dimeric structure but now with acetate guest binding to the DIM pocket (for details see ESI, Fig. S16†).
:1 ratio) followed by slow diffusion of anti-solvent (cyclohexane) resulted in the formation of yellow crystals that were suitable for X-ray diffraction. The X-ray crystal structure shows the formation of centrosymmetric dimeric complexes ([Rh(L2)(CO)(BF4)]2Fig. 2 and S1†) in which two OrthoDIMphos (L2) ligands coordinate to two rhodium atoms. The rhodium complexes have a square planar geometry, with one ligand coordinating in a P–O chelating fashion and one with P, with the CO ligand in the fourth coordination site. The two DIM-receptors of the ligands in the dimeric complex are pointing in opposite directions and all NH hydrogen bond donors are free to bind guest molecules. Crystals were grown in the presence of acetate as guest, and the solid state structure indeed shows a similar dimeric structure but now with acetate guest binding to the DIM pocket (for details see ESI, Fig. S16†).
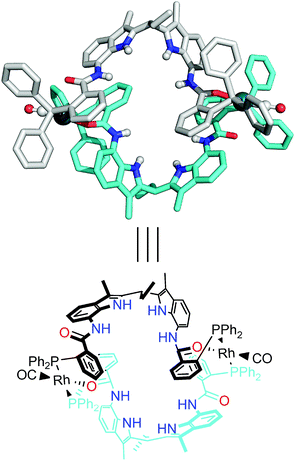 |
| | Fig. 2 X-ray crystal structure of the square planar dimeric complex [Rh(L2)(CO)(BF4)2]2. Two BF4− counterions, hydrogen atoms (except for NH) and disordered solvent molecules are omitted for clarity. For details see the ESI.† | |
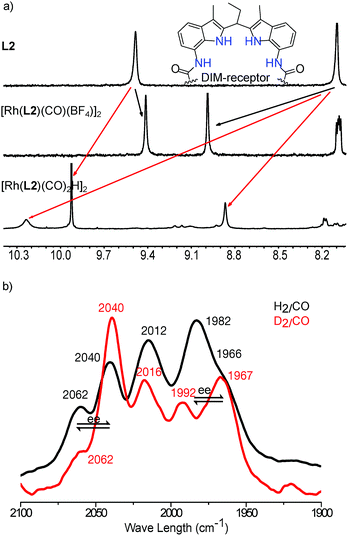
The complexes were also characterized in solution by NMR spectroscopy. 1H DOSY13 spectroscopy of the complexes in the absence of an effector in CD2Cl2 reveals a diffusion constant of logD of −9.27 m2 s−1, corresponding to a hydrodynamic radius of 10.2 Å, in line with the formation of the dimeric Rh-complex [Rh(L2)(CO)(BF4)]2 (Fig. S2†). Additional 31P{1H} (a doublet at 32.94 ppm, JRh–P = 167.8 Hz) and 1H NMR spectroscopy show the formation of one single dimeric species (the indole-NH and amide-NH signals at 9.05 ppm and 8.89 ppm with 1:1 ratio) , and the presence of a single set of signals indicates a fast exchange of the two ligands coordinating to Rh on the NMR time scale (Fig. S3–S12†).
NMR titration studies in CD2Cl2 using the dimeric Rh-complexes [Rh(L2)(CO)(BF4)]2 as the host and tetrabutylammonium benzoate as the guest show that the association constant for benzoate to the DIM-receptor is higher than 105 M−1, similar to that of the previously reported monomeric ParaDIMphos(L1)–Rh complex12 (Fig. S13 and S14†). 1H DOSY spectroscopy shows that the complex with guests bound has a similar diffusion constant of logD of −9.30 m2 s−1 as the free analogue, indicating that the dimer structure of the precursor [Rh(L2)(CO)(BF4)]2 stays intact when benzoate guests bind to the DIM-receptors (Fig. S15†).14
Characterization of the active species under hydroformylation conditions
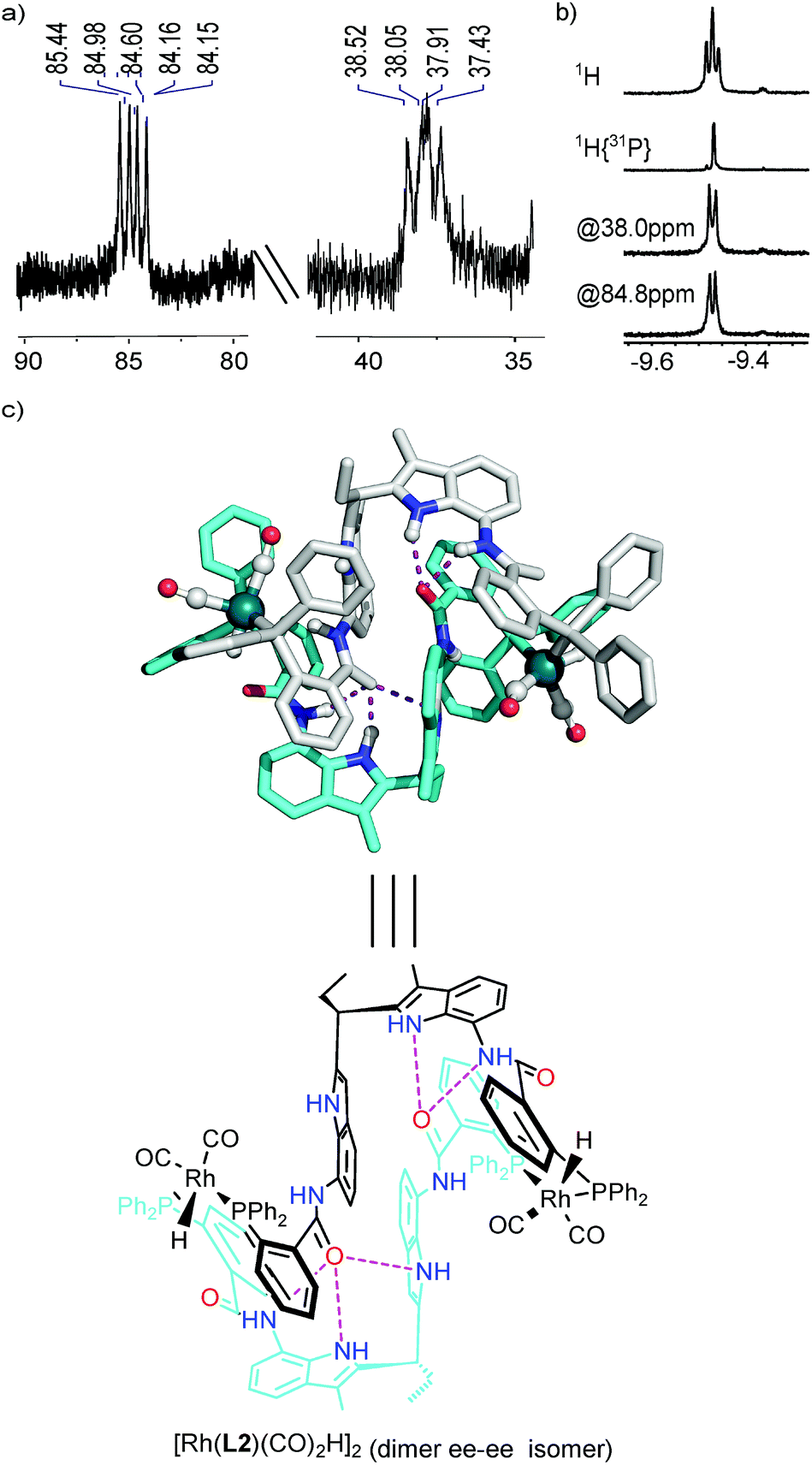
In order to characterize the rhodium complex under hydroformylation conditions, we performed in situ HP IR and NMR experiments. For this purpose, a 1:1 mixture of OrthoDIMphos (L2) and [Rh(acac)(CO)2] was mixed in CD2Cl2, and pressurized with 7 bar CO/H2 (1:1). Upon incubation of the sample at ambient temperature for about 10 min, complete formation of the active species was observed by 1H HP NMR spectroscopy. Various NMR experiments were performed to identify the rhodium complex formed under these conditions (Fig. 3, 4 and S17–S28†). The diffusion constant of the Rh-species under syngas conditions is comparable to that of the dimeric Rh-complex [Rh(L2)(CO)(BF4)]2 (logD of −9.27 vs. −9.20 m2 s−1, Fig. S17†), indicating that also under actual hydroformylation conditions a dimeric species is formed. Further NMR experimental data are also in line with a dimeric structure in which two ligands coordinate to two rhodium metals. The 31P{1H} NMR spectra shows two doublets of doublets at 84.8 ppm (JRh–P,P–P = 167.8, 90.9 Hz) and 38.0 ppm (JRh–P,P–P = 124.4, 93.5 Hz), indicating that the two phosphorus atoms coordinated to the Rh center are inequivalent on the NMR time scale (Fig. 3a, S18 and S19†). Moreover, phosphorus decoupled (1H{31P}) and selective phosphorus decoupled (1H{sel31P}) NMR spectroscopy reveals that the Rh-hydride couples to both phosphorus atoms at 84.8 ppm and 38.0 ppm with coupling constant of 29 Hz and 20 Hz, respectively (Fig. 3b, S20 and S21†). The phosphorus–phosphorus and phosphorus-hydride coupling constants are in line with a typical coordination geometry around rhodium, which is a mixture of coordination complexes with the ligands in equatorial–equatorial (ee) and equatorial–axial (ea) coordination fashion that exchanges rapidly on the NMR timescale (vide infra). The coupling constant of the hydride with Rh is 4.6 Hz, which is typical for these type of Rh–phosphine complexes.12,15 HP 1H NMR spectroscopy shows that also the DIM-receptor is no longer symmetrical (1:2:1 ratio instead of 2:2 ratio of the NH proton of the DIM-receptor for the square planar complex [Rh(L2)(CO)(BF4)]2 and the monomeric complex [Rh(L1)(CO)2H], Fig. 4a and S22–S24†). The distinct downfield shifts of the amide-NH and indole-NH signals of the DIM-receptors (Δδ 0.58–2.27 ppm and 1:2:1 ratio) indicate that these protons are involved in hydrogen bonding. As this is not observed for the monomeric complex based on L1 this suggests that under hydroformylation conditions this hydrogen bonding in the DIM-receptor is important for the formation of the dimeric structure. DFT calculations of the dimeric structure also show that in the energetically most favourable conformation the amide-carbonyl group of one ligand hydrogen bonds with the DIM-receptor of the adjacent ligand within the dimer complex (Fig. 3c, structures A–D at Fig. S25†).16
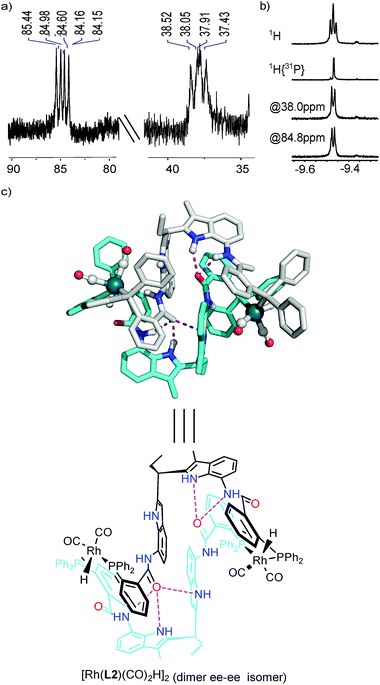 |
| | Fig. 3
In situ characterization of the major dimer rhodium complex. (a) 31P{1H} NMR; (b) HP 1H, 1H{31P} and 1H{sel31P} NMR of the Rh-hydride region; (c) DFT model of the neutral dimeric species D with ee–ee conformation (hydrogen bonds shown by purple dots). | |
 |
| | Fig. 4
In situ characterization of the major dimer Rh-active species. (a) 1H NMR of NH region of the DIM-receptor of the free ligand L2 (the upper spectra), the square planar dimeric complex [Rh(L2)(CO)(BF4)]2 (the middle spectra) and the trigonal bipyramidal dimeric active species [Rh(L2)2(CO)2H]2 (the bottom spectra); (b) in situ HP IR spectra of the dimeric active species under 20 bar of syngas. The ee coordination fashion is revealed by the shifts of the IR bands of the Rh–CO when changing the apical H to D. | |
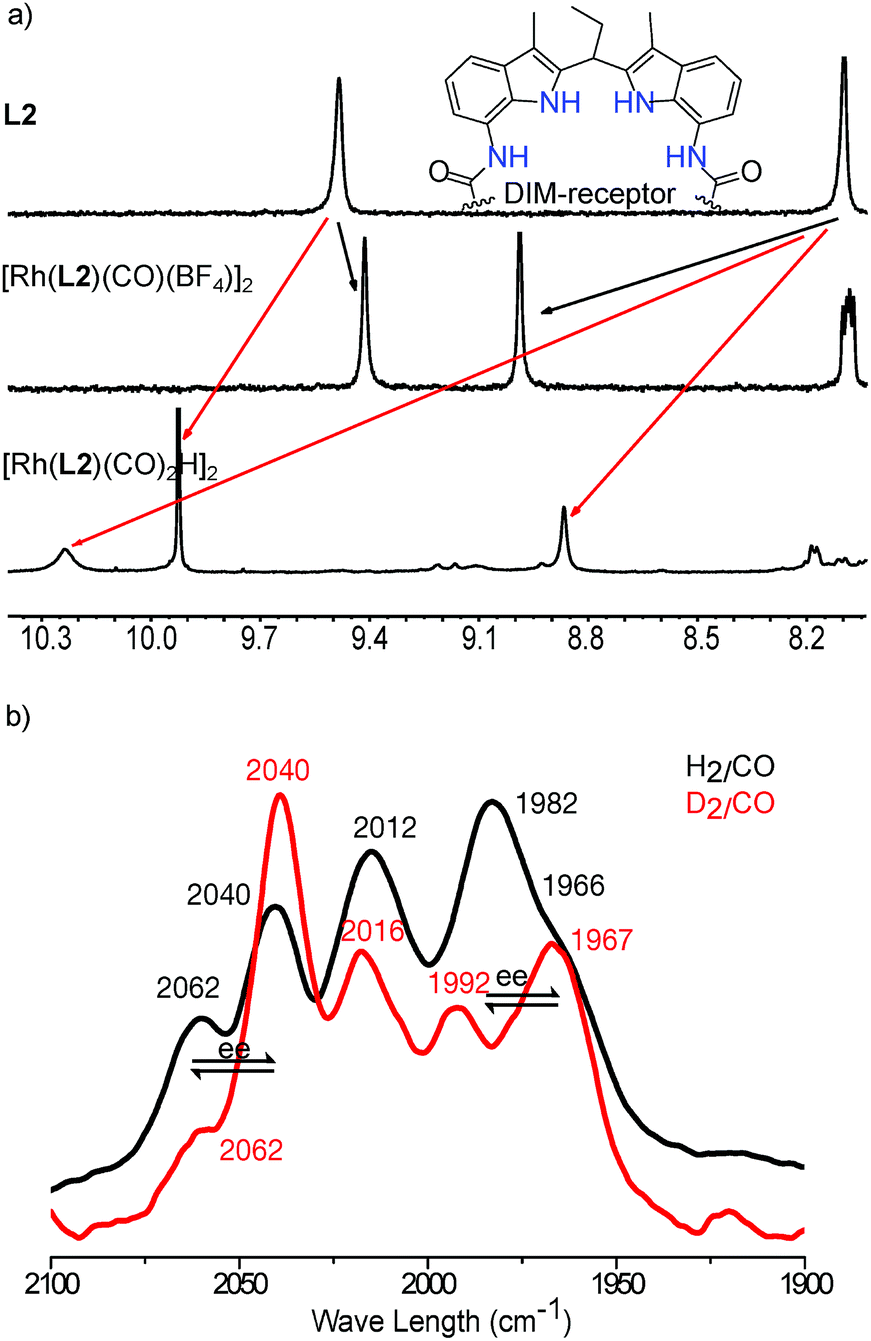
At room temperature a single set of phosphorus signals was obtained, indicating fast exchange between the complexes in various coordinations modes (ee–ea, ee–ee and ea–ea) on the NMR time scale. At low temperature these exchange processes slow down, and at −92 °C, three sets of signals are observed representing these different dimeric complexes (vide supra, Fig. S26†). To further investigate the coordination geometry around rhodium in the dimeric species, HP IR experiments were carried out using H2/CO or D2/CO (both 1:1). For a monomeric complex present as a mixture of the ee and ea coordination mode, four peaks in the carbonyl region can be expected, and for monomeric species [Rh(L1)(CO)2H]12 three peaks are observed as a result of overlap. For the dimer [Rh(L2)2(CO)2H]2 that is present as a mixture of various coordination modes (ee–ea, ee–ee and ea–ea) five peaks in the carbonyl region can be observed, again as a result of partial overlap (Fig. 4). The intensity and peak region difference (Δν 22 and 15 cm−1)17 indicate the presence of both ee and ea coordination species, as also predicted by DFT calculations (Fig. 4b, Table S1, Fig. S25 and 29,†vide supra).
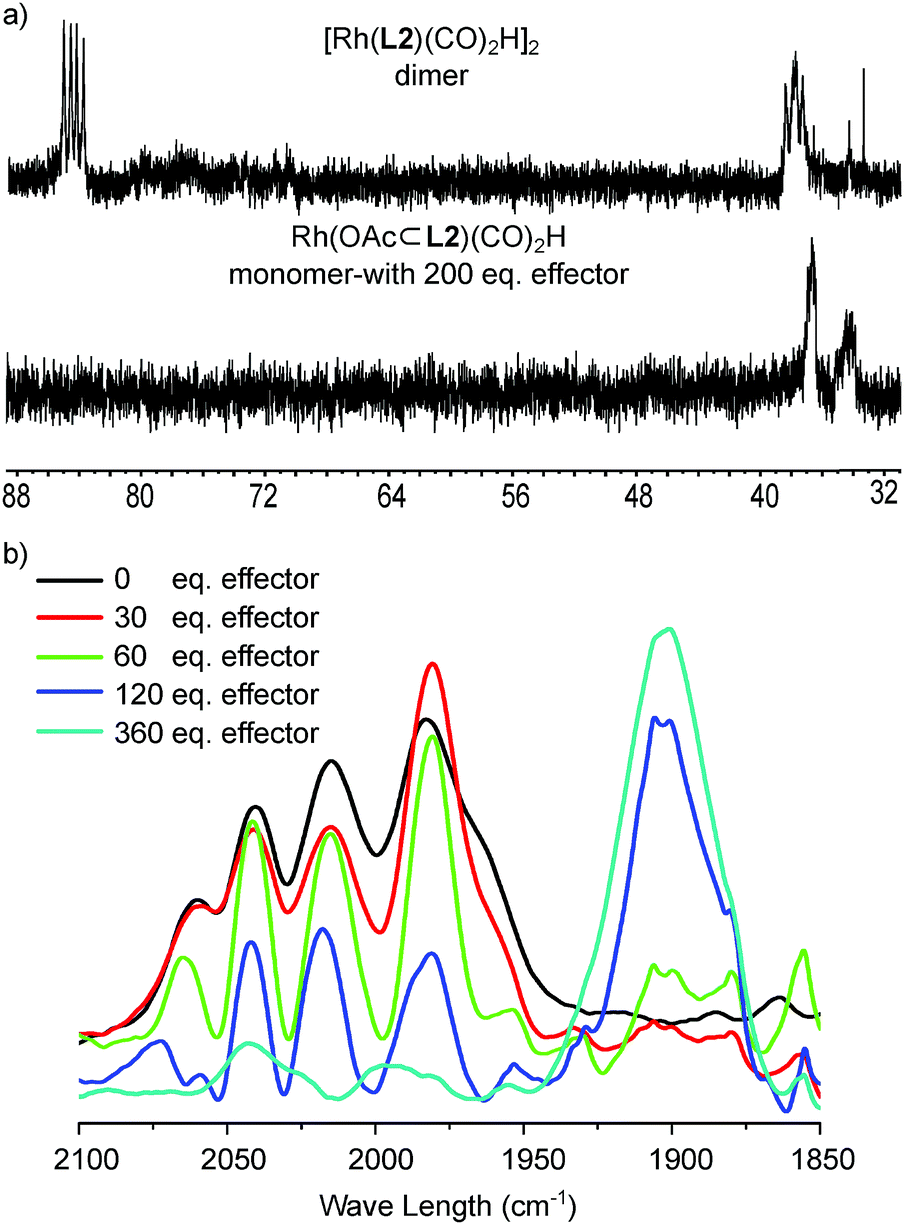
Besides the major dimeric Rh-species, Rh-hydride signals of a minor Rh-species was observed by 1H NMR (less than 10%, Fig. S20, S27 and S28†).181H DOSY spectroscopy shows that the signal is of a species that has a diffusion constant comparable to the monomeric ParaDIMphos–Rh complex (logD of −9.10 vs. −9.13 m2 s−1, Fig. S17 and S28†), confirming that this species involves a monomeric Rh complex. VT NMR spectroscopy also shows that the monomer/dimer ratio changes in favour of the dimer at lower temperatures, indicating an enthalpy driven process in line with hydrogen bonds partaking in the dimerization process.
Controlling dimer–monomer equilibrium by effector binding under hydroformylation conditions
As the dimeric structure of the hydride species is stabilized by hydrogen bonding of the carbonyl-O groups of the ligand with the DIM-receptors, the binding of competing carboxylate containing effectors would affect the relative stability of the dimer compared to the monomeric Rh-species. We therefore investigated the effect of the binding of effectors on the monomer/dimer equilibrium (Fig. 5 and S30–S35†). For this purpose, a mixture of OrthoDIMphos (L2), [Rh(acac)(CO)2], acetic acid and triethylamine (1:1:400:400 ratio, [Rh] = 1 mM) was dissolved in CH2Cl2 and pressurized with 20 bar CO/H2 (1:1). In situ HP IR spectra of this mixture shows the formation of a new rhodium species that displays one broad peak in the Rh-carbonyl region (1902 cm−1), instead of five peaks observed for the dimeric rhodium complex. Interestingly, the IR band at 1902 cm−1 is quite unusual as the Rh-carbonyl IR bands for these type of complexes are generally found in the region of 1950–2100 cm−1.1f,12,19 This suggests strong back donation from the Rh center to the carbonyl ligands, or it may shift as a result of hydrogen bonding with HNEt3+ that is also present in solution. We therefore performed DFT (BP86-D3/def2-SV(P)) calculations to understand the experimental spectra (Fig. 5b and S30–S35†).20 The calculated IR spectra of the monomeric Rh-species E [Rh(OAc⊂L2)(CO)2H] with acetate binding to the DIM-receptor shows two Rh-carbonyl and one Rh–H bands merged at 2035 cm−1. This absorption is shifted with respect to the experimentally observed band at 1902 cm−1. Instead, the calculated IR spectra of the monomeric Rh-species E′ [Rh(OAc⊂L2)(CO)2H]·(HNEt3+)2, with two HNEt3+ present that form hydrogen bonds with the Rh–CO, shows two Rh-carbonyl bands merged in a broad peak at 1900 cm−1, close to the peak found in the experiment. The Rh–H band calculated to be at 2030 cm−1 was not observed experimentally, as is rather common for these type of rhodium(I)hydrido species.1e,19 Similar IR shifts were observed for complexes based on our previously reported bisphosphite DIMPhos ligands.12d Experiments with other effectors, such as 3-butenoate, benzoate and 2-vinyl benzoate, show the similar spectra (Fig. 5a). This shows that the equilibrium between the dimeric and monomeric species can be controlled by the binding of different carboxylate containing effectors in the DIM-receptor.
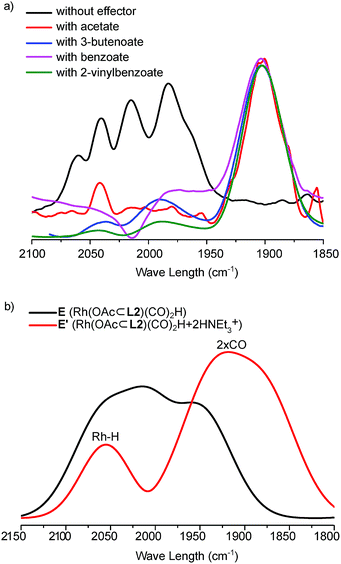 |
| | Fig. 5
In situ characterization of the monomeric Rh-active species via effector binding to the DIM-receptor. (a) In situ HP IR spectra of the monomeric Rh-species with different effectors (200 equivalents of effectors with respect to the DIM-receptor); (b) calculated IR spectra (BP86-D3/def2-SV(P)) of the active monomeric [Rh(OAc⊂L2)(CO)2H] species with acetate as effector binding to the DIM-receptor. | |
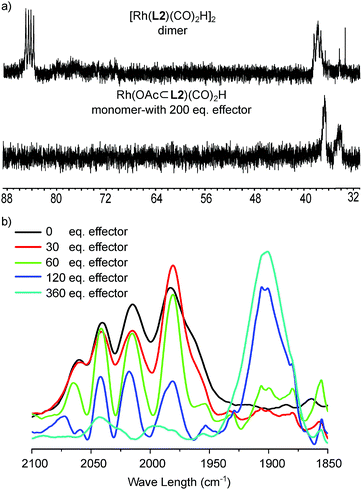
Further evidence for the ability to control the monomer/dimer equilibrium by effector binding came from NMR spectroscopy studies (Fig. 6a and S36–S43†). The 31P{1H} NMR spectrum of the complex in the presence of acetate shows two broad peaks at 36.7 and 34.4 ppm separated by 2.3 ppm, which is very different from the two doublets of doublets at 84.8 and 38.0 ppm assigned to the dimeric complexes (Fig. 6a and S38†), as also predicted by DFT (BP86/DZP) calculated NMR spectra21 (Fig. S39–S43†).
 |
| | Fig. 6
In situ characterization of the monomeric Rh-active species via effector binding to the DIM-receptor. (a) 31P{1H} NMR spectra of the dimeric and monomeric Rh-active species; (b) in situ regulation of the equilibrium between the dimeric and monomeric Rh-active species with varying amounts of effectors. | |
In order to show that the equilibrium between the dimeric and monomeric species can be regulated by the binding of an effector, we next performed a titration study monitored by in situ HP IR spectroscopy. For this experiment, the dimeric rhodium complex was prepared under hydroformylation conditions (20 bar H2/CO), and subsequently various amounts of acetates as effectors (acetic acid:triethylamine, 1:1 ratio, 30–360 equivalents to the DIM-receptor) were injected into the HP IR autoclave under syngas pressure. As expected, the HP IR spectra show the gradual transformation with increasing amounts of acetate of the spectrum with five bands typical for the dimeric complex, to the large broad band associated to the monomeric complex (Fig. 6b). This experiment confirms that the equilibrium between the dimer and the monomer can be regulated by the binding of the effector under catalytic conditions.
To further support that this dimer–monomer equilibrium can be controlled by effector binding, the dimeric and monomeric rhodium complexes were modelled at BP86-D3/def2-SV(P) level, and the energies were computed using single point calculation at B3LYP-D3/TZVP/COSMO(DCM) level (Fig. 7, S44 and Table S4†). Since the entropy is difficult to calculate accurately for large supramolecular systems, the enthalpy energy was used to compare the stability of various structures. These DFT calculations show that the dimeric rhodium complexes in ee–ee, ee–ea and ea–ea coordination fashion are 1.2–5.2 kcal mol−1 more stable than the monomeric complex in which the phosphorus ligands are coordinated in the ee mode (Fig. S44†). In the calculated structure of the dimeric complex the two phosphorus atoms of each ligand coordinate to two different Rh atoms, as observed in the X-ray structures of the related complexes. The two phosphorus atoms are different as only one has the carbonyl-O function partially hydrogen bonded in the DIM-receptor of the adjacent ligand (the N–O distances of 2.937–3.035 Å), in line with experimental observations in the NMR spectra. According to these calculations, these dimeric complexes are more stable than the monomeric analogue due to the formation of these hydrogen bonds. When acetate as effector binds to the DIM-receptor, the monomeric rhodium complex, in either ee or ea coordination mode, is more stable than the dimeric complex by ca. 25 kcal mol−1.22 Thus, combined experimental and theoretical evidence shows that under hydroformylation conditions the complexes are in the dimeric form, unless a sufficient amount of acetate containing effector is present to bind to the DIM-receptor.
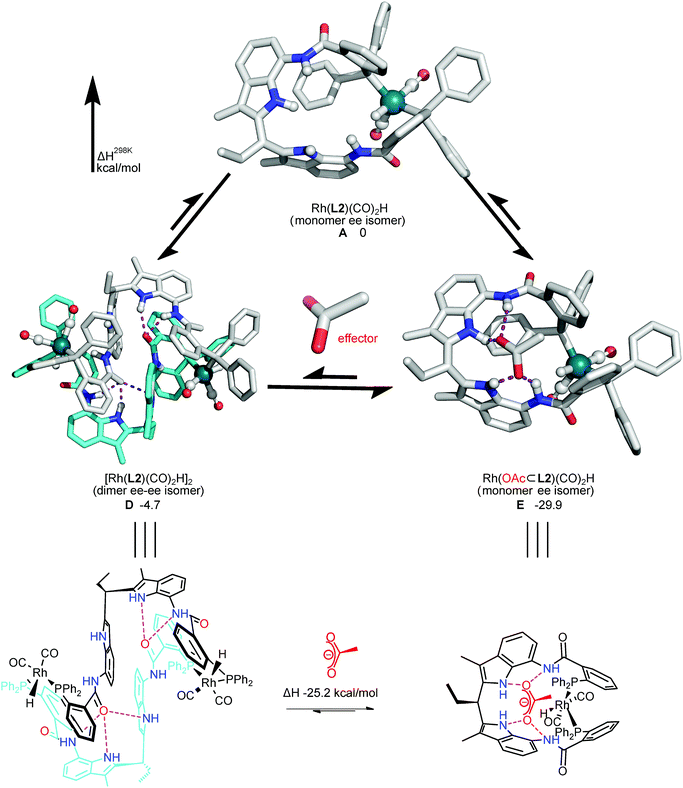 |
| | Fig. 7 DFT optimized structures of the key dimer and monomer rhodium complexes controlled by acetate binding as effector (BP86-D3/def2-SV(P)//B3LYP-D3/def2-TZVP/COSMO(DCM)). The ee isomer of the monomeric trigonal bipyramidal Rh-species (A), the ee–ee isomer of the dimeric trigonal bipyramidal Rh-species (D), the ee isomer of the monomeric trigonal bipyramidal Rh-species [Rh(OAc⊂L2)(CO)2H] with acetate bound in the DIM-receptor (E). H298: enthalpy at 298 K (relative to the monomeric active Rh-species (A) in kcal mol−1). Hydrogen bonds are shown in purple dots. The charge of the effector is −1. Details see the ESI.† | |
Control over selectivity and reactivity in hydroformylation catalysis using the dimer–monomer equilibrium
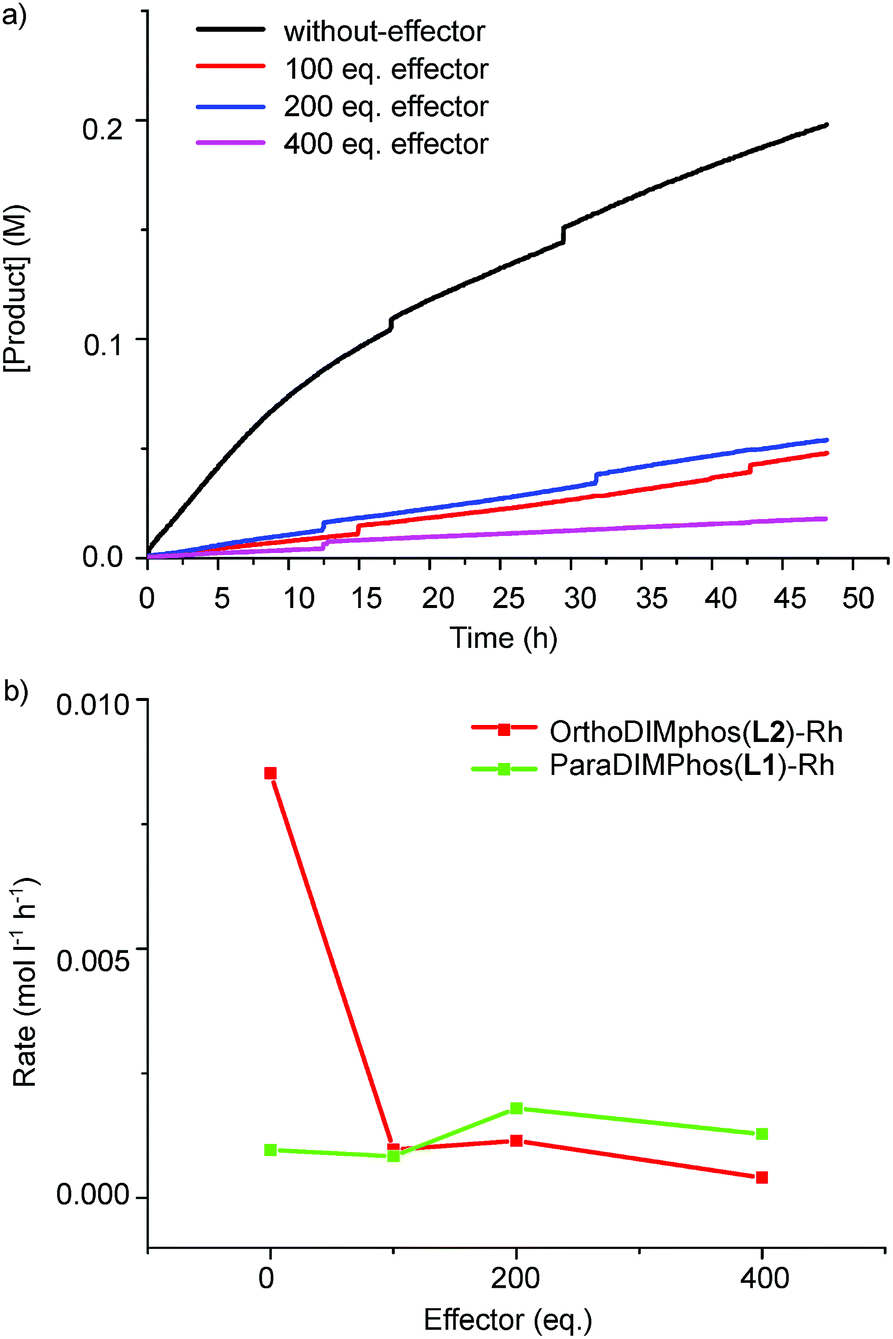
We next studied if the dimeric and monomeric complexes also display different selectivity and reactivity in the hydroformylation of 1-octene (Scheme 2, Fig. 8, 9 and S45–S48, Tables S2 and S3†). All the experiments were performed in an AMTEC SPR16 under identical conditions of pressure and temperature while recording the gas-uptake curves. The selectivity and conversion were determined by gas chromatography and NMR analysis using an internal standard. We used acetate as effector to shift the equilibrium from dimer to monomer, in order to limit the steric effects of the binding of the effector on the catalysis.23 In the absence of acetate effector, the dimeric rhodium complex converted 1-octene with a linear/branched product ratio of 4.2 (81% linear aldehyde and 19% branched aldehyde). In the presence of the acetate effector (100–400 equivalents), the monomer rhodium complex is the dominant species present and this complex converts 1-octene with a linear/branched product ratio of 1.8, which is two fold increase of the branched isomer product (19% vs. 37%). Next to the selectivity, the reactivity displayed by the monomeric complex is also very different. The recorded gas-uptake curves have been analyzed and for the OrthoDIMphos based catalyst system, the reaction rate is about ten times faster in absence than in presence of the acetate effector, in line with the conversion (Fig. 8, 9b, c and S45–S48†). Importantly, these results reveal that the dimeric and monomeric active species display different activity and selectivity in the hydroformylation of 1-octene. Thus, the catalyst properties can be regulated by the effector binding to the DIM-receptor.
 |
| | Scheme 2 Hydroformylation of 1-octene via effector controlled catalysis. | |
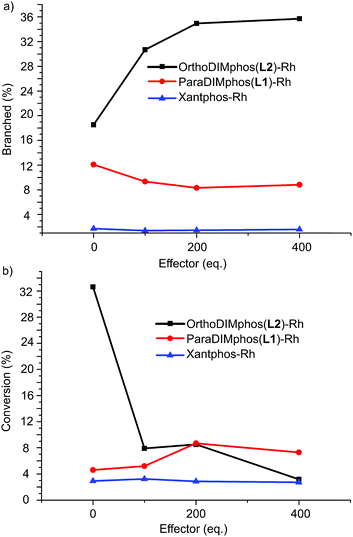 |
| | Fig. 8 (a) The selectivity and (b) conversion vs. the amounts of effectors applied in the hydroformylation of 1-octene for various catalytic systems. Reaction conditions: [substrate] = 0.6 M, [Rh] = 1 mM, acetate effector (acetic acid:TEA 1:1 ratio, 0–0.4 M), solvent dichloromethane, 20 bar of syngas, reaction temperature 25 °C, reaction time of 48 h. The error is less than 5%. | |
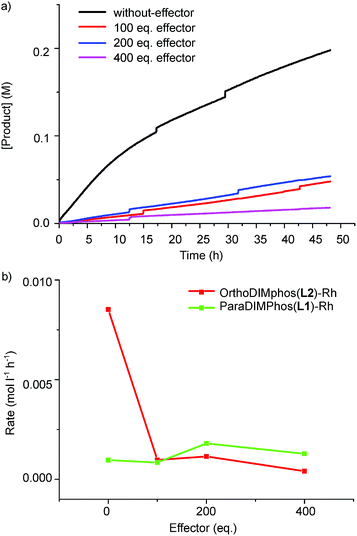 |
| | Fig. 9 (a) Aldehyde product concentration vs. time using the OrthoDIMphos–Rh catalyst system; and (b) reaction rate at 2% conversion vs. the amount of effectors. Reaction conditions: [substrate] = 0.6 M, [Rh] = 1 mM, acetate effector (acetic acid:TEA 1:1 ratio, 0–0.4 M), solvent dichloromethane, 20 bar of syngas, reaction temperature 25 °C, reaction time of 48 h. The error is less than 5%. | |
As control experiments we studied the effect of the acetate binding to the ParaDIMphos–rhodium12 catalyst, which is only present in the monomeric form (Fig. 8, Tables S2 and S3†). As expected, the presence of acetate has limited influence on the selectivity and activity (l/b ca. 10, conversion 4–8%). A similar control experiment using rhodium Xantphos24 as catalyst also shows that the presence of 0–400 equivalents of acetate does not influence the selectivity and activity (l/b ca. 60, conversion 3%, Fig. 8). In line with that, analysis of the kinetic data of the Xantphos and ParaDIMphos based rhodium complexes show that the activity does not change in the presence of acetate, i.e. acetic acid and triethylamine of 1:1 ratio. Importantly, these control experiments also reveal that the triethylammonium carboxylate salts generated do not influence the activity and selectivity via the interaction with the catalytic center. Instead, the modification of the catalyst conformation via effector binding to the second coordination sphere plays dominated role in the regulation of the activity and selectivity. Thus, these experiments demonstrate that the OrthoDIMphos–Rh catalyst represents an unique responsive catalyst system as the activity and selectivity can be tuned by the addition of acetate as effector.
Conclusions
In this paper, we report a new ligand, OrthoDIMphos (L2), that forms dimeric complexes [Rh(L2)]2 when coordinated to rhodium, as evidenced by the two X-ray crystal structures and spectroscopic studies in solution. The dimeric structure under hydroformylation conditions is favoured by hydrogen bonding interactions between the carbonyl-O groups of the ligand and the DIM-receptors. As binding of guests competes with this hydrogen bonding, the presence of carboxylate containing effectors in solution results in the formation of monomeric complexes, and as such these guests are effectors that control the dimer–monomer equilibrium of these complexes. As the monomeric and dimeric complexes have different properties in the hydroformylation of 1-octene, this catalytic system represents an effector responsive hydroformylation catalyst. Both the regioselectivity and reactivity are different when the reaction is carried out with catalyst in monomeric or dimeric state (l/b 4.2 to 1.8, rate 0.008 to 0.0004 mol (l−1 h−1)). As such, we demonstrate in this paper the first example of the control of the selectivity and reactivity in the hydroformylation reaction using effectors to change the active species from dimer to monomer conformation. This type of regulatory mechanism is rather common in nature, but very rare for synthetic systems. These systems can be useful for catalysis in more complex mixtures of catalysts and substrates, and experiments along these lines are currently carried out in our laboratories.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge Jan Meine Ernsting and Dr Andreas W. Ehlers for helping with High-Pressure NMR studies and many fruitful discussions. S.-T. Bai thanks the China Scholarship Council for a PhD fellowship (CSC student number 201506010269) and University of Amsterdam for financial support. VS and BdB thank the NWO-Shell Computational sciences for energy research initiative (project 13CSER003) and the RPA Sustainable Chemistry of the University of Amsterdam.
Notes and references
-
(a)
B. Breit and L. Diab, Hydroformylation and Related Carbonylation Reactions of Alkenes, Alkynes, and Allenes, Elsevier Ltd., 2014, vol. 4 Search PubMed
 ;
(b)
P. W. N. M. van Leeuwen, R. Lazzaroni, R. Settambolo, A. Caiazzo, P. C. J. Kamer, J. N. H. Reek, C. P. Casey, G. T. Whiteker, C. Claver, S. Castillón, E. Fernández, J. Herwig, R. Fischer and P. Arnoldy, Rhodium catalyzed hydroformylation, Springer Netherlands, 1st edn, 2002 CrossRef ;
(c)
M. L. Clarke and J. J. R. Frew, in Organometallic Chemistry, The Royal Society of Chemistry, 2009, vol. 35, pp. 19–46 Search PubMed ;
(d)
A. A. Oswald, D. E. Hendriksen, R. V Kastrup and E. J. Mozeleski, in Homogeneous Transition Metal Catalyzed Reactions, American Chemical Society, 1992, vol. 230, pp. 395–418 Search PubMed ;
(e) J. Klosin and C. R. Landis, Acc. Chem. Res., 2007, 40, 1251–1259 CrossRef CAS PubMed ;
(f) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed ;
(g) F. Agbossou, J. Carpentier and A. Mortreux, Chem. Rev., 1995, 95, 2485–2506 CrossRef CAS ;
(h) P. C. J. Kamer, P. W. N. M. Van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS PubMed ;
(i)
P. W. N. M. van Leeuwen, Homogeneous Catalysis, Springer Netherlands, Dordrecht, 1st edn, 2004, vol. 30 CrossRef ;
(j)
B. Breit, in Metal Catalyzed Reductive C–C Bond Formation, ed. M. J. Krische, Springer Berlin Heidelberg, Berlin, Heidelberg, Heidelberg, 2007, pp. 139–172 Search PubMed .
;
(b)
P. W. N. M. van Leeuwen, R. Lazzaroni, R. Settambolo, A. Caiazzo, P. C. J. Kamer, J. N. H. Reek, C. P. Casey, G. T. Whiteker, C. Claver, S. Castillón, E. Fernández, J. Herwig, R. Fischer and P. Arnoldy, Rhodium catalyzed hydroformylation, Springer Netherlands, 1st edn, 2002 CrossRef ;
(c)
M. L. Clarke and J. J. R. Frew, in Organometallic Chemistry, The Royal Society of Chemistry, 2009, vol. 35, pp. 19–46 Search PubMed ;
(d)
A. A. Oswald, D. E. Hendriksen, R. V Kastrup and E. J. Mozeleski, in Homogeneous Transition Metal Catalyzed Reactions, American Chemical Society, 1992, vol. 230, pp. 395–418 Search PubMed ;
(e) J. Klosin and C. R. Landis, Acc. Chem. Res., 2007, 40, 1251–1259 CrossRef CAS PubMed ;
(f) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed ;
(g) F. Agbossou, J. Carpentier and A. Mortreux, Chem. Rev., 1995, 95, 2485–2506 CrossRef CAS ;
(h) P. C. J. Kamer, P. W. N. M. Van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS PubMed ;
(i)
P. W. N. M. van Leeuwen, Homogeneous Catalysis, Springer Netherlands, Dordrecht, 1st edn, 2004, vol. 30 CrossRef ;
(j)
B. Breit, in Metal Catalyzed Reductive C–C Bond Formation, ed. M. J. Krische, Springer Berlin Heidelberg, Berlin, Heidelberg, Heidelberg, 2007, pp. 139–172 Search PubMed .
-
(a)
Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed ;
(b) C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS ;
(c) C. A. Fleckenstein and H. Plenio, Chem. Soc. Rev., 2010, 39, 694–711 RSC ;
(d) T. L. Brown and K. J. Lee, Coord. Chem. Rev., 1993, 128, 89–116 CrossRef CAS ;
(e) M. L. Clarke and M. Heydt, Organometallics, 2005, 24, 6475–6478 CrossRef CAS ;
(f) A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC ;
(g) A. A. Oswald, D. E. Hendriksen, R. V. Kastrup, K. Irikura, E. J. Mozeleski and D. A. Young, Phosphorous Sulfur Relat. Elem., 1987, 30, 237–240 CrossRef CAS ;
(h) S. Würtz and F. Glorius, Acc. Chem. Res., 2008, 41, 1523–1533 CrossRef PubMed ;
(i) R. F. Heck, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS ;
(j) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1859 RSC .
-
(a) L. A. van der Veen, M. D. K. Boele, F. R. Bregman, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, J. Fraanje, H. Schenk and C. Bo, J. Am. Chem. Soc., 1998, 120, 11616–11626 CrossRef CAS ;
(b)
M. L. Clarke and J. J. R. Frew, in Organometallic Chemistry, The Royal Society of Chemistry, Cambridge, 2009, vol. 35, pp. 19–46 Search PubMed ;
(c) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed ;
(d) J. F. Hartwig, Inorg. Chem., 2007, 46, 1936–1947 CrossRef CAS PubMed ;
(e) R. Burch, Acc. Chem. Res., 1982, 15, 24–31 CrossRef CAS .
-
(a) P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2770 CrossRef CAS PubMed ;
(b) L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, CATTECH, 2002, 6, 116–120 CrossRef CAS ;
(c) P. W. N. M. van Leeuwen, P. C. J. Kamer, L. A. van der Veen and J. N. H. Reek, Chin. J. Chem., 2001, 19, 1–8 CrossRef CAS ;
(d) M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC ;
(e) P. C. J. Kamer, P. W. N. M. Van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS PubMed .
- For some general papers on regulation of proteins see:
(a) I. Bahar, C. Chennubhotla and D. Tobi, Curr. Opin. Struct. Biol., 2007, 17, 633–640 CrossRef CAS PubMed ;
(b) J. H. Shi, J. Sivaraman and J. X. Song, J. Virol., 2008, 82, 4620–4629 CrossRef CAS PubMed ;
(c) S. Raboni, S. Bettati and A. Mozzarelli, Cell. Mol. Life Sci., 2009, 66, 2391–2403 CrossRef CAS PubMed ;
(d) N. J. Marianayagam, M. Sunde and J. M. Matthews, Trends Biochem. Sci., 2004, 29, 618–625 CrossRef CAS PubMed ;
(e) E. U. Woehl and M. F. Dunn, Coord. Chem. Rev., 1995, 144, 147–197 CrossRef CAS ;
(f) T. H. Kim, P. Mehrabi, Z. Ren, A. Sljoka, C. Ing, A. Bezginov, L. Ye, R. Pomès, R. Scott Prosser and E. F. Pai, Science, 2017, 355, eaag2355 CrossRef PubMed ;
(g)
R. L. Stein, in Kinetics of Enzyme Action, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, vol. 2, pp. 199–217 CrossRef ;
(h)
S. D. Copley, in Comprehensive Natural Products II, Elsevier, 2010, pp. 9–46 Search PubMed ;
(i)
N. F. König and N. Kulak, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2015, pp. 1–15 Search PubMed ;
(j) B. Nagel, H. Dellweg and L. M. Gierasch, Pure Appl. Chem., 1992, 64, 143–168 CAS .
-
(a) N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed ;
(b) P. Dydio, D. Lichosyt and J. Jurczak, Chem. Soc. Rev., 2011, 40, 2971–2985 RSC ;
(c) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed ;
(d) N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed ;
(e) L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161–3188 CrossRef CAS PubMed .
- For reviews see:
(a) D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS ;
(b) A. V Davis, R. M. Yeh and K. N. Raymond, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4793–4796 CrossRef PubMed ;
(c) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed ;
(d) D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 RSC ;
(e) A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed ;
(f) T. R. Cook and P.
J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed ;
(g) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed .
- For reviews and selected contributions:
(a) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC ;
(b) E. R. Kay and D. A. Leigh, Angew. Chem., Int. Ed., 2015, 54, 10080–10088 CrossRef CAS PubMed ;
(c) W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed ;
(d) R. S. Forgan, J. P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed ;
(e) B. L. Feringa, J. Org. Chem., 2007, 72, 6635–6652 CrossRef CAS PubMed ;
(f) K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377–1400 CrossRef CAS PubMed ;
(g) S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC ;
(h) C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS PubMed ;
(i) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC ;
(j) D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174–179 CrossRef CAS PubMed .
- For reviews and selected contributions, see:
(a) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC ;
(b) M. Vaquero, L. Rovira and A. Vidal-Ferran, Chem. Commun., 2016, 52, 11038–11051 RSC ;
(c) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed ;
(d) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC ;
(e) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC ;
(f) S. S. Nurttila, P. R. Linnebank, T. Krachko and J. N. H. Reek, ACS Catal., 2018, 8, 3469–3488 CrossRef CAS PubMed ;
(g) E. Lindbäck, S. Dawaigher and K. Warmark, Chem.–Eur. J., 2014, 20, 13332–13481 CrossRef PubMed ;
(h) S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941–1943 CrossRef CAS PubMed ;
(i) A. Bauer, F. Westkämper, S. Grimme and T. Bach, Nature, 2005, 436, 1139–1140 CrossRef CAS PubMed ;
(j) S. C. Coote and T. Bach, J. Am. Chem. Soc., 2013, 135, 14948–14951 CrossRef CAS PubMed ;
(k) T. Šmejkal and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 3946–3949 CrossRef PubMed ;
(l) L. J. Jongkind, X. Caumes, A. P. T. Hartendorp and J. N. H. Reek, Acc. Chem. Res., 2018, 51, 2115–2128 CrossRef CAS PubMed .
-
(a) C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618–1629 CrossRef CAS PubMed ;
(b) N. C. Gianneschi, M. S. Masar and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825–837 CrossRef CAS PubMed ;
(c) H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS PubMed ;
(d) I. O. Fritsky, R. Ott, H. Pritzkow and R. Krämer, Chem.–Eur. J., 2001, 7, 1221–1231 CrossRef CAS PubMed ;
(e) I. O. Fritsky, R. Ott and R. Krämer, Angew. Chem., Int. Ed., 2000, 39, 3255–3258 CrossRef CAS ;
(f) N. C. Gianneschi, P. A. Bertin, S. T. Nguyen, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 10508–10509 CrossRef CAS PubMed ;
(g) N. C. Gianneschi, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 1644–1645 CrossRef CAS PubMed ;
(h) M. S. Masar, N. C. Gianneschi, C. G. Oliveri, C. L. Stern, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 10149–10158 CrossRef CAS PubMed .
- For contributions in effector controlled switch on/of (enantioselective/encapsulation) transition metal catalysis, see:
(a) M. Vaquero, L. Rovira and A. Vidal-Ferran, Chem. Commun., 2016, 52, 11038–11051 RSC ;
(b) G.-H. Ouyang, Y.-M. He, Y. Li, J.-F. Xiang and Q.-H. Fan, Angew. Chem., Int. Ed., 2015, 54, 4334–4337 CrossRef CAS PubMed ;
(c) I. Mon, D. A. Jose and A. Vidal-Ferran, Chem.–Eur. J., 2013, 19, 2720–2725 CrossRef CAS PubMed ;
(d) L. Rovira, H. Fernández-Pérez and A. Vidal-Ferran, Organometallics, 2016, 35, 528–533 CrossRef CAS ;
(e) L. J. Jongkind, J. A. A. W. Elemans and J. N. H. Reek, Angew. Chem., Int. Ed., 2019, 58, 2696–2699 CrossRef CAS PubMed ;
(f) P. Dydio, C. Ruby, T. Gadzikwa, M. Lutz and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 17176–14179 CrossRef CAS PubMed ;
(g) L. Théveau, R. Bellini, P. Dydio, Z. Szabo, A. van der Werf, R. Afshin Sander, J. N. H. Reek, C. Moberg, L. T. Théveau, R. Bellini, P. Dydio, Z. Szabo, A. Van Der Werf, R. A. Sander, J. N. H. Reek and C. Moberg, Organometallics, 2016, 35, 1956–1963 CrossRef ;
(h) P. W. N. M. van Leeuwen, D. Rivillo, M. Raynal and Z. Freixa, J. Am. Chem. Soc., 2011, 133, 18562–18565 CrossRef CAS PubMed ;
(i) A. C. H. Jans, A. Gómez-Suárez, S. P. Nolan and J. N. H. Reek, Chem.–Eur. J., 2016, 22, 14836–14939 CrossRef CAS PubMed .
-
(a) P. Dydio and J. N. H. Reek, Angew. Chem., Int. Ed., 2013, 52, 3878–3882 CrossRef CAS PubMed ;
(b) P. Dydio, R. J. Detz and J. N. H. Reek, J. Am. Chem. Soc., 2013, 135, 10817–10828 CrossRef CAS PubMed ;
(c) P. Dydio, W. I. Dzik, M. Lutz, B. de Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2011, 50, 396–400 CrossRef CAS PubMed ;
(d) P. Dydio, R. J. Detz, B. de Bruin and J. N. H. Reek, J. Am. Chem. Soc., 2014, 136, 8418–8429 CrossRef CAS PubMed .
- C. S. Johnson, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef CAS .
- Acetic acid was deprotonated by acac and the Hacac was observed by 1H NMR at ca. 15 pmm when mixing [Rh(acac)(CO)2], L2 and acetic acid in CD2Cl2.
- S. H. Chikkali, J. I. van der Vlugt and J. N. H. H. Reek, Coord. Chem. Rev., 2014, 262, 1–15 CrossRef CAS .
- J. E. Del Bene, S. A. Perera and R. J. Bartlett, J. Phys. Chem. A, 1999, 103, 8121–8124 CrossRef CAS .
- Experimentally, we observe IR bands shifts when changing the apical H to D. Compared with HPIR spectra using H2/CO, the IR bands 2062 cm−1 and 1982 cm−1 of the Rh–CO absorption shift to the low frequency region 2040 cm−1 and 1967 cm−1 with 22 and 15 cm−1, respectively, upon performing HPIR experiment using D2/CO.
- The minor active species is identified by 1H NMR. According to DOSY spectra, the diffusion constant is comparable to the monomeric species [Rh(L1)(CO)2H], indicating a monomeric structure. Unfortunately, the phosphorus NMR signals is not identified for this species. Noted that it needs four days of acquisition to obtain phosphorus NMR spectra with satisfactory signal-to-noise ratio for the monomeric species with effector binding to the DIM-receptor. It suggest that even longer time needed to obtain the phosphorus signals for such small amounts of species.
- For typical metal-carbonyl species IR bands, see:
(a) Y. S. Varshavskii, T. G. Cherkasova, I. S. Podkorytov, A. A. Korlyukov, V. N. Khrustalev and A. B. Nikol'skii, Russ. J. Coord. Chem., 2005, 31, 121–131 CrossRef CAS ;
(b) D. Evans, J. A. Osborn and G. Wilkinson, J. Chem. Soc. A, 1968, 3133–3142 RSC ;
(c) K. Nozaki, T. Matsuo, F. Shibahara and T. Hiyama, Organometallics, 2003, 22, 594–600 CrossRef CAS .
- For theory of DFT calculations on the metal-carbonyl IR bands, see:
(a) P. Deglmann and F. Furche, J. Chem. Phys., 2002, 117, 9535–9538 CrossRef CAS ;
(b) P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511–518 CrossRef CAS .
- For theory of DFT calculations on phosphorus chemicals shifts and coupling constants, see:
(a) M. Kollwitz and J. Gauss, Chem. Phys. Lett., 1996, 260, 639–646 CrossRef CAS ;
(b) S. K. Wolff and T. Ziegler, J. Chem. Phys., 1998, 109, 895–905 CrossRef CAS ;
(c) G. Schreckenbach and T. Ziegler, J. Phys. Chem., 1995, 99, 606–611 CrossRef CAS .
- DFT calculations show that the monomeric species with effector binding to the DIM-receptor is much more stable than the dimeric conformers. However, experimentally we need adequate amounts of effectors to drive this process. On one hand, the effectors generated are also under equilibrium and thus the concentration of the carboxylate anions might be not that high as the input. On the other hand, HP IR titration experiments indeed confirm that the effector binding to the DIM-receptor is the driving force for converting the dimeric to monomeric species. We also tried to use tetrabutylammonium acetate as the effector to regulate the equilibrium. Unfortunately, we did not observe the expected IR band at 1902 cm−1 and also no aldehydes were detected when adding substrate into the autoclave. These failed experiments suggest that the strong acetate anions or the tetrabutylammonium salts might inhibit the active species formation.
-
(a) S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC ;
(b) Q.-Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed .
-
(a) M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz and J. Fraanje, Organometallics, 1995, 14, 3081–3089 CrossRef CAS ;
(b) Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890–1901 RSC .
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, Fig S1–S52, Scheme S1, Tables S1–S4. CCDC 1877078 and 1877079. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02558h |
| ‡ Present address: Complex Systems in Synthesis & Catalysis, University of Strasbourg, CNRS, ISIS UMR 7006, 8 Rue Gaspard Monge, F-67000 Strasbourg, France. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Vivek
Sinha
a,
Alexander M.
Kluwer
b,
Pim R.
Linnebank
a,
Zohar
Abiri
b,
Paweł
Dydio‡
a,
Vivek
Sinha
a,
Alexander M.
Kluwer
b,
Pim R.
Linnebank
a,
Zohar
Abiri
b,
Paweł
Dydio‡