
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBis(catecholato)silanes: assessing, rationalizing and increasing silicon's Lewis superacidity†
Deborah
Hartmann
,
Marcel
Schädler
and
Lutz
Greb
 *
*
Anorganisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 275, 69120 Heidelberg, Germany. E-mail: greb@uni-heidelberg.de
First published on 17th June 2019
Abstract
Although bis(catecholato)silanes have been known for several decades, their substantial Lewis acidity is not yet well described in the literature. Herewith, the synthesis and characterization of multiple substituted bis(catecholato)silanes and their triethylphosphine oxide, fluoride and chloride ion adducts are reported. The Lewis acidity of bis(catecholato)silanes is assessed by effective (Gutmann–Beckett, catalytic efficiency), global (theoretical and relative experimental fluoride (FIA) and chloride (CIA) ion affinities) and intrinsic (electrophilicity index) scaling methods. This comprehensive set of experimental and theoretical results reveals their general Lewis acidic nature and provides a consistent Lewis acidity trend for bis(catecholato)silanes for the first time. All experimental findings are supported by high-level DLPNO-CCSD(T) based thermochemical data and the Lewis acidity is rationalized by complementary chemical bonding analysis tools. Against the common belief that inductive electron withdrawal is the most important criterion for strong Lewis acidity, the present work highlights the decisive role of π-back bonding effects in aromatic ring systems to enhance electron deficiency. Thus, bis(perbromocatecholato)silane is identified and synthesized as the new record holder for silicon Lewis superacids.
Introduction
Lewis acids play a prominent role in all domains of chemistry, such as (bio)organic, organometallic, (bio)inorganic, polymer and materials science.1 They are of particular relevance for application in frustrated Lewis pair chemistry.2 Especially, Lewis acids that exceed the fluoride ion affinity (FIA) of SbF5, so-called Lewis superacids (LSAs), are of current interest.4 Promising LSAs based on boron,5 aluminum,4a,6 or cationic group 15 elements7 have been demonstrated in recent years. Very strong Lewis acids based on neutral silicon species are much less developed. The Lewis acidity at silicon can be enhanced by three strategies: (1) strong electron withdrawing substituents,8 (2) incorporation of silicon in strained cycles9 or (3) transient activation via Lewis base coordination.10 For example, the use of perfluoroaryl substituents in (C6F5)nSiF4−n was reported by Frohn and Dilman.11 Hoge demonstrated the C2F5 group as a robust substituent for the generation of strong silicon Lewis acids.13 Although bis(catecholato)silanes have been known for several decades, their Lewis acidity was realized only in a recent seminal work by Tilley.14 Therein, bis(perfluorocatecholato)silane Si(catF)2 (Fig. 1) was demonstrated as a catalyst for the hydrosilylation of electron-poor aldehydes. Encouraged by the fact that aromatic chloro substituents have a weaker π-overlap with aromatic systems than fluorides, our group addressed bis(perchlorocatecholato)silane Si(catCl)2.3 According to its computed and experimental FIA in comparison to other neutral silanes, Si(catCl)2 represented the first silane exceeding the affinity of SbF5, thus being the first neutral silicon Lewis superacid. Many well-defined pentacoordinate12,15 and hexacoordinate16 complexes based on silicon catecholates have been characterized much earlier. Pentacoordinate anionic bis(catecholato)silicates were used as hydride or allyl donors to carbonyls,17 and more recently for the photoredox catalytic generation of alkyl radicals.18 The related tris(catecholato) silicate dianions have in fact been known for almost 100 years and serve as a prime example for hexacoordination of silicon.19 Obviously, the Lewis acidity of the underlying catecholatosilanes is decisive for the stability and reactivity of all their adducts. However, a general account of the Lewis acidity of bis(catecholato)silanes is missing in the literature.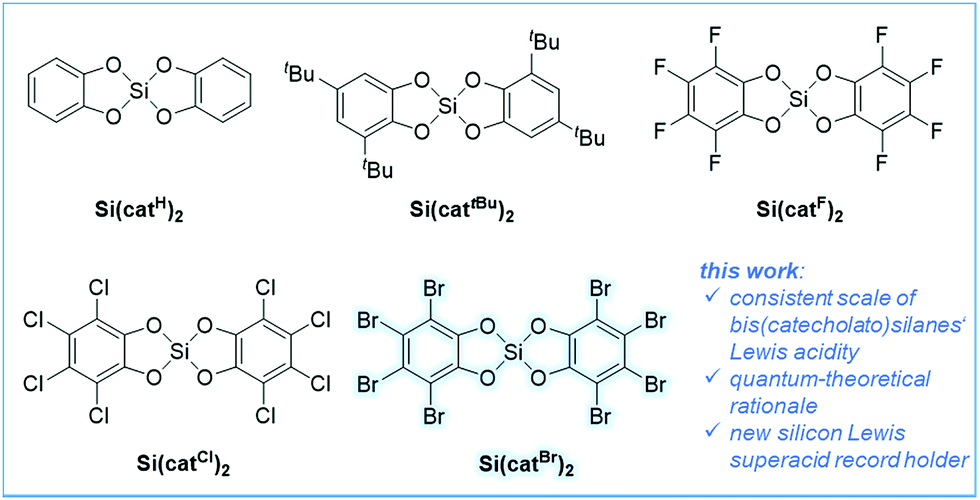 | ||
| Fig. 1 Bis(catecholato)silanes Si(catX)2 are synthesized and assessed for their Lewis acidity in this work, and the herein introduced new Lewis superacid record holder is Si(catBr)2; derivatives with X = Cl, Br were isolated as CH3CN-bis-adducts. | ||
The present work provides a systematic experimental and theoretical study of the Lewis acidity of bis(catecholato)silanes. Even the weakest derivative Si(catH)2 is uncovered as sufficiently Lewis acidic to form an anionic chlorosilicate. More important, the limit of Lewis acidity with neutral silicon species is pushed beyond that of the previous LSA record holder Si(catCl)2. All experimental findings and the origin of the Lewis acidity of bis(catecholato)silanes are rationalized by state-of-the-art quantum theoretical tools.
Results and discussion
Synthesis of bis(catecholato)silanes
The syntheses of variably substituted bis(catecholato)silanes Si(catX)2 (Fig. 1) were performed by a standard procedure (Scheme 1). Two equivalents of the respective catechols in acetonitrile were reacted with HSiCl3 for 1 h at room temperature, followed by the release of gaseous by-products (H2, HCl). The reaction was continued for 12 h at 40 °C, leading to the precipitation of the desired bis(catecholato)silanes as acetonitrile adducts from the reaction mixture. Washing of the precipitates with acetonitrile and CH2Cl2 was performed to remove residual HCl and unreacted catechol. In the case of X = H, 3,5-tBu and F, complete release of CH3CN was possible upon exposure to high-vacuum conditions, whereas for X = Cl, Br two equivalents of CH3CN remained in the samples. Based on this method, the derivatives Si(catX)2(·2CH3CN) (X = H, 3,5-tBu, F, Cl, Br) were accessible in good yields at multigram scale (76–89%). The pronounced affinity towards acetonitrile gave the first hints of the exceptional Lewis acidity of Si(catCl)2 and Si(catBr)2. The poor solubility of the derivatives Si(catX)2(·2CH3CN) in non-donor solvents prevented full NMR spectroscopic analysis of the free Lewis acids. However, NMR spectroscopy in donor solvents, mass spectrometry and elemental analysis confirmed the composition Si(catX)2 or Si(catX)2·2CH3CN for the formed precipitates and were in agreement with previous literature reports.3,14,15i,20 The new Lewis acid Si(catBr)2 was additionally characterized by X-ray diffraction as a bis-adduct with diethyl ether (see ESI†). Depending on the reaction conditions, bis(catecholato)silanes have been proposed as either monomeric,15i,16c,20a,21 oligomeric20a,22 or polymeric.23 However, facile depolymerization of the oligomers was believed to occur in the presence of Lewis bases.20a,24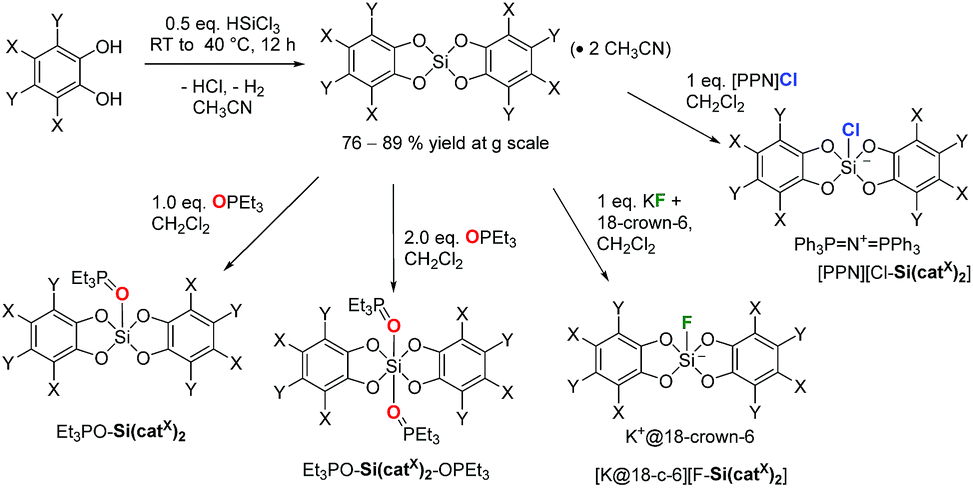 | ||
| Scheme 1 The synthesis of the herein described species (X/Y = H, F, Cl, Br or H/tBu). | ||
Although the presence of oligomers or aggregates in the formed precipitates cannot be ruled out herein, it is believed that the presence of acetonitrile as the reaction medium stabilizes the monomeric Si(catX)2 species via coordination, in particular for the halogenated derivatives (X = F, Cl, Br). Most convincingly, the addition of various neutral or anionic donors (vide infra) to the obtained powders led to the exclusive formation of donor→Si(catX)2 species without the detection of any side products. The remainder of this work exploits the defined donor→Si(catX)2 adducts to assess the inherent Lewis acidity of the Si(catX)2 species by effective, global and intrinsic scaling methods.4b
Preparation of adducts and the scaling of the Lewis acidity of bis(catecholato)silanes
The Lewis acidity of a compound can be inspected by the spectroscopic response of a suitable probe molecule bound to the acceptor side – the so-called effective scaling method.4b As was discussed previously, the ν(CN) IR-stretching mode of CH3CN-adducts does not provide a meaningful scaling – and was not considered herein.4b Although the Gutmann–Beckett (GB) method25 also has its shortcomings due to its dependence on steric effects or “Pearson hardness”, it enables a reasonable scaling of the Lewis acidity within one class of compounds.4b Hence, the triethylphosphine oxide adducts of Si(catX)2 were prepared by the addition of varying amounts of Et3PO (0.5 to 3.0 eq.) in CD2Cl2. Depending on the amount of added Et3PO, two major species were identified: the mono-adduct Et3PO–Si(catX)2 and the bis-adduct Et3PO–Si(catX)2–OPEt3, further verified by X-ray structural analysis (vide infra). For the non-halogenated Lewis acids Si(catH)2 and Si(cattBu)2, a single averaged 31P-NMR signal was visible, which continuously shifted upon increasing the amount of Et3PO, indicating a fast exchange between the different adducts and free Et3PO at the NMR time scale (see Fig. 2a for Si(catH)2 and Fig. S1† for Si(cattBu)2). For the halogenated Lewis acids Si(catX)2 (X = F, Cl, Br), distinct signals for the mono- (δ ≈ 86 ppm) and bis-adducts (δ ≈ 73 ppm) were observable, indicating stronger binding (see Fig. 2b for Si(catBr)2 and Fig. S2/S3† for Si(catF)2/Si(catCl)2). In addition, a minor species (δ ≈ 70 ppm) was visible for the halogenated Lewis acids, proposed as a coordination product of a third Et3PO in the second coordination sphere of the cis bis-adduct Et3PO–Si(catX)2–OPEt3 (see ESI†).26 The chemical shifts of the 31P-NMR signals of the mono-adducts Et3PO–Si(catX)2 were chosen as the probe for the GB method (see Fig. S4†). According to the induced shift relative to the free Et3PO, a ranking for Si(catX)2 of X = 3,5-tBu < H ≪ F < Cl < Br was obtained (Table 1).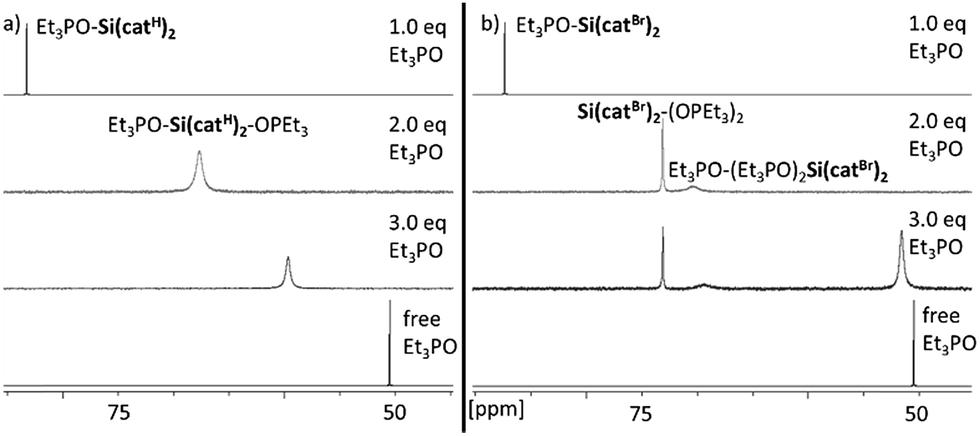 | ||
| Fig. 2 Changes in the 31P-NMR spectra for samples of (a) Si(catH)2 and 1.0–3.0 eq. of Et3PO, and (b) Si(catBr)2 and 1.0–3.0 eq. of Et3PO in CD2Cl2. | ||
This is an interesting finding, as it supported the hypothesis of increasing Lewis acidity with decreasing π-backbonding capability of the substituents X in the catechols. Indeed, the σm Hammett parameters for the respective substituents X (Table 1, last column) are in line with the observed Lewis acidity trend.27
The mono-fluoride adducts of the bis(catecholato)silanes [K@18-crown-6][F-Si(catX)2] were obtained by the addition of 1 eq. KF and 18-crown-6 to Si(catX)2(·2CH3CN) in CH2Cl2 at room temperature and subsequent precipitation with pentanes. The fluorosilicates were characterized by heteronuclear NMR spectroscopy, ESI-MS and (except for X = tBu) X-ray diffraction. The 19F-NMR chemical shifts are all around −133 ppm and the 29Si-NMR chemical shifts of around −90 ppm lie in the expected range for pentacoordinate silicates (Table 2). Interestingly, the 1JSiF coupling constants (189–195 Hz) correlate with the Lewis acidity trend obtained by the GB method, and indicate an increased covalent interaction between silicon and fluoride (as is further supported in the computational section). The 19F-NMR spectra of trigonal-bipyramidal [F-Si(cattBu)2]− with an unsymmetrically substituted catechol revealed two trans isomers (95%) which are in mutual exchange by Berry pseudo-rotation and 5% of the cis form (see Fig. S5†).28
| [Z-Si(catX)2]− | δ 19F-NMR (Z = F) | δ 29Si-NMR (Z = F) | 1 J SiF [Hz] (Z = F) | δ 29Si-NMR (Z = Cl) |
|---|---|---|---|---|
| Si(cattBu)2 | −131.6; −133.7, −133.9 | −104.8 | 188.9 | −91.8 |
| Si(catH)2 | −133.1 | −104.8 | 191.1 | −91.6 |
| Si(catF)2 | −133.5 | −101.6 | 194.8 | −87.1 |
| Si(catCl)2 | −132.7 | −105.1 | 195.2 | −90.4 |
| Si(catBr)2 | −132.3 | −107.0 | 195.2 | −92.5 |
The respective chlorosilicates [PPN][Cl-Si(catX)2] were accessible by reacting 1 eq. bis(triphenylphosphoranylidene)ammonium chloride ([PPN]Cl) with Si(catX)2(·2CH3CN) in CH2Cl2 for 24 h at room temperature and subsequent precipitation with pentanes. The adduct formation was verified by heteronuclear NMR spectroscopy and in the case of [PPN][Cl-Si(catH)2] also by X-ray diffraction. The respective 29Si-NMR chemical shifts lie in the range of pentacoordinate silanes (see Table 2) and are in agreement with the computed 29Si-NMR shifts (see Table S4†). Intriguingly, those compounds belong to the rare class of anionic chlorosilicates. Due to the large solvation free energy of the chloride ion, it usually needs strong silicon Lewis acids to stabilize the respective chlorosilicates in solution, as was demonstrated for the first time only in 2014 with perfluoroethyl-substituted silanes.29 Remarkably, the acceptor strengths of even the weakest bis(catecholato)silanes, Si(catH)2 and Si(cattBu)2, are sufficient to achieve this goal.
With the range of halide adducts, the relative solution phase fluoride ion affinities (FIAsol) and chloride ion affinities (CIAsol) were addressed by competition experiments. The five Lewis acids were grouped into two sets: X = H, 3,5-tBu, F (set 1) and X = F, Cl, Br (set 2). Within one set, all possible combinations of 1 eq. fluoride adduct [F-Si(catX)2]− and 1 eq. of another Lewis acid, Si(catY)2(·2CH3CN), were mixed in CD2Cl2, and the ratio of the respective fluoride adducts [F-Si(catX)2]− and [F-Si(catY)2]− was obtained by 19F-NMR peak integration (after a minimum of 36 h at rt, ESI Section 1.2.6†). The assignment of the peaks was accomplished by comparison with the known 19F-NMR spectra of the pure compounds [F-Si(catX)2]−. In addition, a prominent third singlet appeared between the expected two 19F-NMR signals of the fluoride adducts [F-Si(catX)2]− and [F-Si(catY)2]−. The origin of this signal was identified as the product [F-Si(catX)(catY)]− resulting from a catecholato exchange reaction. Indeed, identical products formed statistically after mixing of equimolar amounts of two fluoride adducts [F-Si(catX)2]−/[F-Si(catY)2]−. Catecholato scrambling in anionic hypercoordinate silicon catecholates was proposed by Woollins et al., but not further verified.28 Herein, evidence of the process was provided by 19F/29Si-NMR spectroscopy, ESI-MS and in the case of [K@18-crown-6][F-Si(catF)(catBr)] by X-ray structural analysis. Despite this catecholato scrambling, the competition experiments allowed for a meaningful interpretation of the relative FIAsol. In particular, consideration of the results of the back and forth reaction of a specific pair provided sound conclusions. For example, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [F-Si(catF)2]− and Si(catBr)2·2CH3CN led to a distribution of 17% of [F-Si(catF)2]−, 30% of [F-Si(catBr)2]− and 53% of heteroleptic [F-Si(catBr)(catF)]−. In the respective back reaction (a combination of [F-Si(catBr)2]− and Si(catF)2), almost no fluoride exchange occurred (only 4% of [F-Si(catF)2]− formed after 36 h), besides 41% of the catecholate scrambling product. Based on the product distributions in the mixtures for all considered combinations (back and forth), a FIAsol trend of tBu < H ≪ F < Cl < Br (Table 3) was obtained. Strikingly, this trend is in agreement with the results of the GB method. Analogous competition reactions were performed with the chloride adducts [PPN][Cl-Si(catX)2], yielding the same relative order for the CIAsol (see Table S2†).
:1 mixture of [F-Si(catF)2]− and Si(catBr)2·2CH3CN led to a distribution of 17% of [F-Si(catF)2]−, 30% of [F-Si(catBr)2]− and 53% of heteroleptic [F-Si(catBr)(catF)]−. In the respective back reaction (a combination of [F-Si(catBr)2]− and Si(catF)2), almost no fluoride exchange occurred (only 4% of [F-Si(catF)2]− formed after 36 h), besides 41% of the catecholate scrambling product. Based on the product distributions in the mixtures for all considered combinations (back and forth), a FIAsol trend of tBu < H ≪ F < Cl < Br (Table 3) was obtained. Strikingly, this trend is in agreement with the results of the GB method. Analogous competition reactions were performed with the chloride adducts [PPN][Cl-Si(catX)2], yielding the same relative order for the CIAsol (see Table S2†).
| [F-Si(catX)2]− + Si(catY)2 | [F-Si(catX)2]− | [F-Si(catY)2]− | [F-Si(catX) (catY)]− | rel. FIAsol |
|---|---|---|---|---|
| a 0.05 M of [K@18-c-6][F-Si(catX)2] and 0.05 M Si(catY)2 in CD2Cl2, room temp., min. 36 h equilibration time, for Y = Cl and Br, the CH3CN adducts were used. | ||||
| X = H, Y = tBu | 65 | <1 | 35 | H > tBu |
| X = tBu, Y = H | <1 | 70 | 30 | |
| X = H, Y = F | 20 | 76 | 4 | F > H |
| X = F, Y = H | >99 | <1 | 0 | |
| X = tBu, Y = F | <1 | 72 | 28 | F > tBu |
| X = F, Y = tBu | >99 | <1 | <1 | |
| X = F, Y = Cl | 21 | 27 | 52 | Cl > F |
| X = Cl, Y = F | 49 | 8 | 43 | |
| X = F, Y = Br | 17 | 30 | 53 | Br > F |
| X = Br, Y = F | 55 | 5 | 40 | |
| X = Cl, Y = Br | 20 | 29 | 51 | Br > Cl |
| X = Br, Y = Cl | 58 | 4 | 38 | |
Description of solid-state structures
Multiple X-ray diffraction analyses of pentacoordinate and hexacoordinate neutral and anionic donor→Si(catX)2 species were performed. In the following, the structural parameters of relevance will be discussed. Those are in particular the donor atom-Si and the Si–Ocat bond lengths. In addition, the distortion of trigonal-bipyramidal (tbp) to square-pyramidal (sp) (in the present case, more precisely, rhomboid-pyramidal) along the so-called Berry coordinate will be compared for all pentacoordinate species.By crystallization of mixtures of Si(catX)2 with less than 1.0 eq. of Et3PO in CH2Cl2/pentanes, the mono-adducts Et3PO–Si(catX)2 for X = H, F and Br were obtained (Fig. 3a for X = Br). The Si–Ocat bond lengths are similar for all the adducts and lie in the typical range of pentacoordinate silicon catecholates.24 The donor–acceptor Si–O5 bond lengths, as well as the phosphorus–oxygen bond lengths of the coordinated phosphine oxides (P–O5), do not correlate with the induced 31P-NMR chemical shifts of the GB method (Table 4). For the Et3PO–Si(catF)2 adduct, two independent donor–acceptor pairs are present in the unit cell, for which the Si–O5 and the P–O5 bond lengths in fact differ by 0.02 Å. These observations emphasize the inability to discuss Lewis acidities in terms of solid-state structural parameters due to the small curvature of the potential energy surface at its minimum in donor–acceptor bonding.30 By crystallization of Si(catX)2 combined with 2.0 eq. of Et3PO in CH2Cl2/pentanes, single crystals of the bis-adducts Et3PO–Si(catX)2–OPEt3 for X = H, F, Cl and Br were obtained (Fig. 3b, for X = Br). All complexes show an almost ideal octahedral coordination sphere with an inversion center at silicon (Ci point group). As expected for hexacoordinate silicon structures, the Si–Ocat bond lengths are longer than the respective ones in the corresponding pentacoordinate species. The Si–O3 bond lengths are also longer than the ones in the Et3PO mono-adducts. Again, they do not correlate with the obtained Lewis acidity trend. The P–O3 bond lengths are shorter than in the Et3PO–Si(catX)2 mono-adducts in all cases. The anionic fluorosilicates [K@18-crown-6][F-Si(catX)2] were crystallized by vapor diffusion of pentanes into saturated solutions of dichloromethane (Fig. 3c for X = F). The coordination geometries around silicon lie between tbp and sp, with the fluoride anions consequently occupying the apical position in the sp structures, and the equatorial positions in the tbp arrangements. [K@18-crown-6][F-Si(catF)2] was crystallized in two polymorphs A and B: in polymorph A (Fig. 3c), the [K@18-crown-6]+ unit is coordinated to the apical fluoride F1 at silicon, whereas in polymorph B (Fig. 3d), one cationic unit is coordinated to a fluoro substituent at catechol and another unit has the closest contact to the π-system of another catechol, overall forming a coordination polymer. The product of the catecholato scrambling, [K@18-crown-6][F-Si(catF)(catBr)], shows two different [K@18-crown-6]+ units, one bridging two anions by K–F1 interaction, and one unit stacked between two O2C6F4 rings (only one of the two independent cationic units is shown in Fig. 3e). The Si–F bond lengths compared for all fluorosilicates are again not in correlation with the experimental solution phase Lewis acidities (Table 4). The strongest impact on the Si–F bond length seems to be interaction with the potassium. Structures in which the Si–F is coordinated to potassium ([F-Si(catF)2]− polymorph A, [F-Si(catCl)2]− and [F-Si(catBr)2]−) generally show longer Si–F bonds than structures wherein the [K@18-crown-6]+ is coordinated elsewhere ([F-Si(catH)2]− and [F-Si(catF)2]− polymorph B). Single crystals of the anionic chlorosilicate [PPN][Cl-Si(catH)2] were obtained by overlayering a dichloromethane solution with pentanes (Fig. 3f). The coordination geometry around silicon is strongly distorted towards sp, with the chloride in the apical position. The Si–Cl bond length is rather on the short side (2.104 Å) in comparison to the few other structurally characterized chlorosilicates (2.100–2.218 Å).29,31 The distortion of all pentacoordinate species from tbp to sp can be discussed by reference to a simple topology parameter (TP) along the so called Berry coordinate (a brief definition of the TP can be found in the ESI).32 TP = 1 means an ideal tbp and TP = 0 an ideal sp structure. A clear distinction between the “weak” Si(catH)2 and the stronger halogenated derivatives can be found. In agreement with earlier observations, the pentacoordinate halogenated derivatives are much more distorted towards the sp form (Table 4).33 Therein, a stabilization of the sp form, which is usually higher in energy, is attributed to the diminished electron density at the mutually repulsing oxygen atoms in the basal plane.12 This is in line with the stronger π-back donation of the free electron pairs to the aromatic ring systems in the halogenated derivatives. However, no clear trend is observed within the class of halogenated derivatives, due to the predominant effect of crystal packing forces and the low tbp–sp deformation energy.34
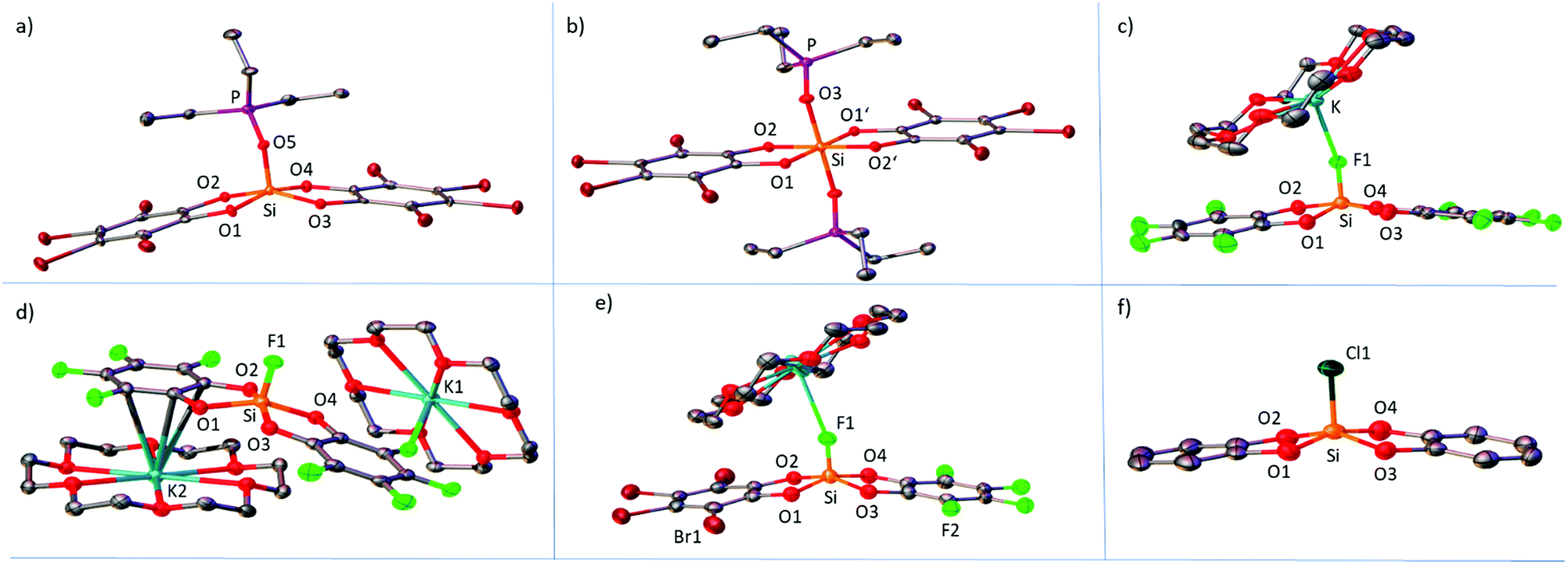 | ||
| Fig. 3 Representative solid-state molecular structures of (a) Et3PO–Si(catBr)2, (b) Et3PO–Si(catBr)2–OPEt3, and (c) [K@18-crown-6][F-Si(catF)2], polymorph A; (d) selection of the unit cell of [K@18-crown-6][F-Si(catF)2], polymorph B, (e) [K@18-crown-6][F-Si(catBr)(catF)], and (f) [PPN][Cl-Si(catH)2], cation omitted for clarity (ellipsoids at 50% probability level, hydrogen atoms omitted for clarity). | ||
| Et3PO–Si(catX)2 | X = H | X = F (unit 1) | X = F (unit 2) | X = Br | (Et3PO)2–Si(catX)2 | X = H | X = F | X = Cl3 | X = Br |
|---|---|---|---|---|---|---|---|---|---|
| Si–O5 | 1.709(2) | 1.688(2) | 1.694(2) | 1.702(4) | Si–O3 | 1.863(2) | 1.823(2) | 1.832(1) | 1.836(2) |
| P–O5 | 1.560(2) | 1.544(2) | 1.557(2) | 1.550(3) | P–O3 | 1.526(2) | 1.523(2) | 1.533(1) | 1.535(2) |
| Si–O1 | 1.745(2) | 1.727(2) | 1.746(2) | 1.726(3) | Si–O1 | 1.754(1) | 1.767(2) | 1.766(1) | 1.763(2) |
| Si–O2 | 1.705(2) | 1.733(2) | 1.726(2) | 1.734(3) | Si–O2 | 1.757(1) | 1.768(1) | 1.763(1) | 1.766(2) |
| Si–O3 | 1.737(2) | 1.721(2) | 1.727(2) | 1.713(3) | |||||
| Si–O4 | 1.709(2) | 1.731(2) | 1.725(2) | 1.723(3) | |||||
| O1–Si–O2 | 89.6(1) | 89.5(1) | 89.2(1) | 89.3(2) | O1–Si–O2 | 90.7(1) | 90.8(1) | 90.6(1) | 90.4(1) |
| O3–Si–O4 | 89.8(1) | 89.7(1) | 90.1(1) | 90.3(2) | O1–Si–O2′ | 89.3(1) | 89.3(1) | 89.4(1) | 89.6(1) |
| O1–Si–O4 | 87.0(1) | 86.2(1) | 85.4(1) | 87.1(2) | O2–Si–O3 | 88.5(1) | 88.3(1) | 88.9(1) | 89.0(1) |
| O2–Si–O3 | 87.0(1) | 85.0(1) | 85.6(1) | 84.5(2) | O2′–Si–O3 | 91.4(1) | 91.8(1) | 91.1(1) | 91.0(1) |
| O1–Si–O3 | 170.3(1) | 154.2(1) | 156.8(1) | 158.5(2) | |||||
| O2–Si–O4 | 140.2(1) | 158.3(1) | 155.7(1) | 156.3(2) | |||||
| TP (% tbp) | 0.50 | 0.02 | 0.02 | 0.04 |
| [F-Si(catX)2]− | X = H12 | X = F (Poly A) | X = F (Poly B) | X = Cl3 | X = Br | X = F/Br | [Cl-Si(catX)2]− | X = H |
|---|---|---|---|---|---|---|---|---|
| Si–F1 | 1.600(8) | 1.600(1) | 1.609(2) | 1.606(1) | 1.610(2) | 1.604(4) | Si–Cl1 | 2.104(2) |
| Si–O1 | 1.767(8) | 1.731(1) | 1.742(2) | 1.731(1) | 1.751(3) | 1.736(4) | Si–O1 | 1.711(4) |
| Si–O2 | 1.746(9) | 1.745(1) | 1.720(2) | 1.738(1) | 1.724(3) | 1.731(4) | Si–O2 | 1.735(4) |
| Si–O3 | 1.686(8) | 1.728(1) | 1.746(2) | 1.739(1) | 1.743(3) | 1.739(5) | Si–O3 | 1.720(3) |
| Si–O4 | 1.720(8) | 1.741(1) | 1.718(2) | 1.738(1) | 1.734(3) | 1.732(4) | Si–O4 | 1.736(4) |
| O1–Si–O2 | 88.9(1) | 89.4(1) | 89.6(1) | 89.1(1) | 89.0(1) | 89.0(2) | O1–Si–O2 | 89.9(2) |
| O3–Si–O4 | 89.5(1) | 89.3(1) | 89.6(1) | 89.0(1) | 89.0(1) | 89.1(2) | O3–Si–O4 | 89.0(2) |
| O1–Si–O4 | 86.4(1) | 85.5(1) | 85.4(1) | 85.7(1) | 84.4(1) | 85.5(2) | O1–Si–O4 | 85.7(2) |
| O2–Si–O3 | 88.0(1) | 86.3(1) | 85.7(1) | 85.6(1) | 87.4(1) | 85.8(2) | O2–Si–O3 | 85.2(2) |
| O1–Si–O3 | 133.1(5) | 149.8(1) | 162.3(1) | 153.7(1) | 160.9(1) | 155.2(2) | O1–Si–O3 | 151.4(2) |
| O2–Si–O4 | 171.2(5) | 161.8(1) | 148.2(1) | 156.5(1) | 148.6(1) | 155.1(2) | O2–Si–O4 | 159.1(2) |
| TP (% tbp) | 0.64 | 0.20 | 0.24 | 0.05 | 0.21 | 0.00 | 0.12 |
Comparison of bis(catecholato)silanes in chloride abstraction and catalytic hydrodefluorination
As an additional Lewis acidity scaling method, the reactions of Si(catX)2 towards trityl chloride in CD2Cl2 were investigated. The weaker Lewis acids Si(catH)2 and Si(cattBu)2 did not induce significant ionization of trityl chloride, whereas with the halogenated derivatives, strongly colored solutions and clear NMR signals of the trityl cation developed. Although isolation of the formed salts was not possible, the amount of trityl cation in solution was easily determined by 1H-NMR signal integration (Table 5 and Fig. S6†). Whereas the previous scaling methods and computations (see below) indicated a larger CIA for Si(catCl)2vs.Si(catF)2, in the chloride abstraction experiments Si(catF)2 and Si(catCl)2·2CH3CN displayed similar efficiency. This observation might be explained by the poor solubility of Si(catCl)2·2CH3CN in the absence of a suitable donor and/or the competing adduct formation with acetonitrile. Importantly, the highest ratio of chloride abstraction was clearly observed with the new Lewis acid Si(catBr)2·2CH3CN.| Si(catX)2(·2CH3CN) + Ph3CCl ⇌ [Ph3C][Cl-Si(catX)2] | |
|---|---|
| X = | % of Ph3C+ |
| F | 62 |
| Cl | 60 |
| Br | 83 |
To compare the catalytic efficiencies of the halogenated Lewis acids, the reaction profile of the hydrodefluorination reaction (3 mol% Si(catX)2) of 1-adamantylfluoride with 2 eq. of Et3SiH in CD3CN at 75 °C) was followed (Fig. 4). The efficiencies for Si(catF)2 and Si(catCl)2 were similar, which again might be caused by the poorer solubility of Si(catCl)2·2CH3CN. The early cessation of the reaction progress, the need for catalyst loadings of min. 10 mol% to complete the reaction and the observation of decomposition products via19F-NMR spectroscopy all indicated low turnover number (TON) for X = F, Cl. In contrast, the catalytic performance of Si(catBr)2 was markedly superior, both in terms of estimated TON and turnover frequency (TOF). Although the full mechanistic and kinetic analysis of this reaction is beyond the scope of the present contribution, these results clearly reveal the efficiency and robustness of Si(catBr)2 for future catalytic applications. It might be surprising that the Si(catX)2 catalysts show poorer performance in this hydrodefluorination reaction compared to boranes like B(C6F5)3 or 9-BBN – which are rated less Lewis acidic by FIA or GB.35 However, two points are of note here: (1) Lewis acidity is ill-defined for parametrization along a single dimension. It is governed by three (partially) independent variables: charge density, orbital energies/localization and sterics.4b The parameters of Lewis acidity that result in high FIA or GB numbers do not necessarily impart high catalytic efficiency. This efficiency is often governed by orbital mixing terms, which profit from localized LUMO shapes and low lying LUMO energies (soft Lewis acidity). (2) The FIA and GB numbers reflect thermodynamic parameters, whereas catalytic efficiencies are also influenced by the reaction kinetics.
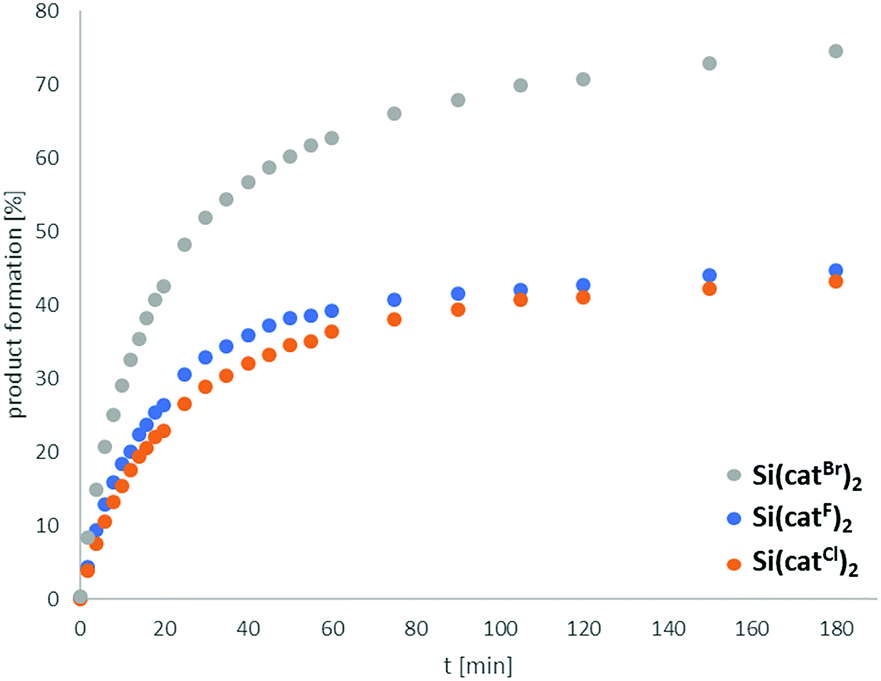 | ||
| Fig. 4 Catalytic hydrodefluorination reaction of 1-adamantylfluoride with 2 eq. Et3SiH and 3 mol% Si(catX)2 in CD3CN (0.26 M) at 75 °C. Reaction progress obtained by 19F-NMR peak integration against C6F6 as an internal standard. | ||
Computational results
To obtain a meaningful theoretical scaling of the Lewis acidities of the species Si(catX)2 (X = H, F, Cl, Br), the gas and solution phase FIA and CIA were computed at the accurate DLPNO-CCSD(T)/aug-cc-pVQZ//PW6B95-D3(BJ)/def2-TZVPP level of theory via the isodesmic reaction enthalpies. Previous benchmark studies revealed the PW6B95-D3(BJ)/def2-TZVPP level of theory as ideal for geometry optimization for this class of substances.3 First, the dissociation free energies for CH3CN from Si(catCl)2·2CH3CN were computed. Loss of the first CH3CN was found to be endergonic (ΔG = 12 kJ mol−1), but the dissociation of the second CH3CN was exergonic (ΔG = −31 kJ mol−1). Consequently, dissociation of both CH3CN units should occur under diluted conditions, and a discussion of the FIA trends based on the free Lewis acids should therefore be reasonable. To our delight, the FIA and CIA of the free Lewis acids Si(catX)2 with and without solvent correction were in agreement with our experimental findings (X = H ≪ F < Cl < Br, Table 6, FIA, for CIA see Table S3†). The computations further disclosed the perbrominated Lewis acid Si(catBr)2as by far the strongest of all bis(catecholato)silanes, which represents the new record holder of reachable Lewis superacidity with neutral silanes. The same trend was obtained for the respective chloride ion affinities (see Table S3†). To provide a rationale for the origin of the high affinity of bis(catecholato)silanes, stepwise structural variations starting from SiH4 were performed in silico and the effect on the FIA was investigated. The FIA of SiH4 was arbitrarily set to zero. The nature of bonding between silicon and the fluoride ion in the respective fluorosilicates was concomitantly inspected by energy decomposition analyses (EDA, see Table 6).36 The exchange of the hydrides in SiH4 for methoxy groups in Si(OMe)4 induced an increase of the FIA by 35 kJ mol−1. This is caused by a stronger positive polarization of the silicon center, leading to a more pronounced Coulomb interaction with the negative fluoride ion (Table 6, orange shade). Because of the extra space provided to the free electron pairs at the oxygen atoms via delocalization in Si(OPh)4, the FIA increases significantly. This is a consequence of diminished Pauli repulsion in the respective adduct, strengthening the overall interaction between the fluoride and the silane (Table 6, blue shade). The geometric effect in the spiro compounds Si(catX)2 again increases the FIA significantly. The ring closure reduces the formation energy in comparison to Si(OPh)4 and increases the absolute amounts of Coulomb and orbital interaction in the fluoride adducts (Table 6, purple shade). The halogenation of the catechol backbone does not change the ratio of electrostatic to orbital contributions in the Si–F bonding, but increases both of their absolute values simultaneously.| a Geometries at PW6B95-D3/def2-TZVPP. b DLPNO-CCSD(T)/aug-cc-pVQZ, values in brackets corrected for solvation (COSMO-RS/CH2Cl2). c BP86-D/TZ2P. d kcal mol−1. e PBE0-D3/def2-TZVPP. f % of all pz(O) NLMO at Si. g Second order perturbation energy of pz(O) to π*(CC) in aromatic ring [kcal mol−1]. h PBE0-D3/def2-TZVPP electron densities, values in atomic units. iI cc-pVQZ basis set (see ESI). j Isolated as acetonitrile adducts. |
|---|
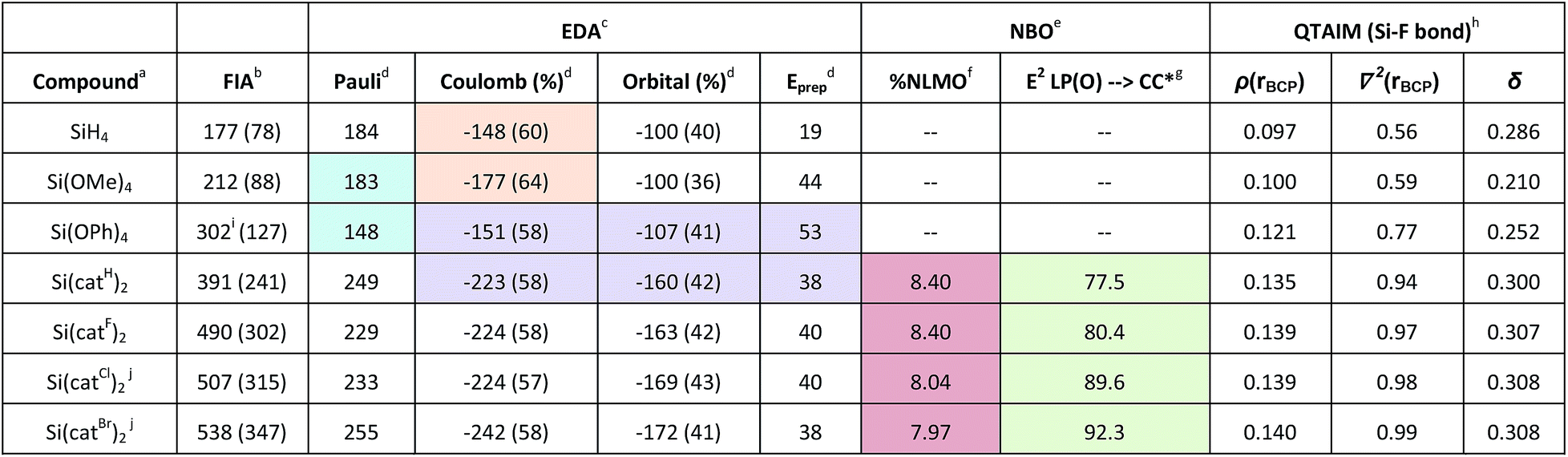
|
To rationalize the dependence of the FIA on the substituents X in the catechol backbone, the electronic structures of the [F-Si(catX)2]− anions were analyzed further by natural bond orbital (NBO) methods.37 In particular, the pz-type natural localized molecular orbitals (NLMOs) at oxygen were inspected. The delocalization tails of the NLMOs at oxygen towards silicon decrease with X in the order H ≥ F > Cl > Br (expressed by the % of participation at silicon for this NLMO, Table 6, red shade). At the same time, the second order perturbation energy of the free pz(O) NBO towards the aromatic π*-CC orbitals increases in the same order (Table 6, green shade). This finding is in agreement with the hypothesis of decreased π-electron density in the order X = H > F > Cl > Br, which is empirically manifested by the increasing Hammett constants of the substituents X. When substituting the aromatic rings with weaker π-donors, but still suitable σ-acceptors, the free electron pairs at oxygen favorably delocalize in the aromatic ring systems instead of competing with the fluoride ion at silicon to occupy accessible space, which would lead to a weaker Si–F bond and a weaker Lewis acid. The strong inductive effect of the electronegative oxygen atoms remains unaffected by the groups X and guarantees the overall strong positive polarization of the silicon center.
Analysis of all fluoridosilicates by the quantum theory of atoms in molecules (QTAIM) was performed next. It generally revealed closed shell ionic interactions for the silicon–fluoride bonds (Table 6).38 The Laplacian of the electron density ∇2 at the bond critical point of the Si–F bonds increases with increasing Lewis acidity – which is characteristic for enforced ionic bonding.38b,39 However, in addition, the electron density ρ and the delocalization index δ between silicon and fluoride increase according to the same trend. Both observations reveal enforced covalent bonding upon enhancement of FIA/Lewis acidity. The simultaneous increase of ionic and covalent bonding effects – orthogonal not antipodal – has been described only recently in the context of the silicon–oxygen bond.40 It is also operative in this case, providing the silicon bis(catecholates) with their Lewis acidity.
To evaluate an intrinsic scale for the Lewis acidity of all studied silanes, the global electrophilicity index (GEI) was computed.41 As can be seen in Fig. S11,† the GEI yields a good correlation with the FIA within the class of bis(catecholato)silanes and thus provides a shortcut tool for a rapid preliminary assessment. However, it fails for the comparison with the other classes of silanes which have been investigated computationally during this work.
Conclusions
We herein provide the first general account of the Lewis acidity of bis(catecholato)silanes – a class of substances that is easily accessible by a one-step protocol in high yields. A combined experimental and theoretical study of effective, global and intrinsic Lewis acidity measures gives a consistent Lewis acidity trend forSi(catX)2of X =tBu < H ≪ F < Cl < Br. Their FIAs range between moderate strengths like that of PF5 (X = H) to extreme Lewis acidity like Al(OC(CF3)3)3 (X = Br) (Fig. 5, further reference FIA data can be found in ref. 4b). Thus, we herein identified and synthesized the Lewis acid Si(catBr)2, which constitutes a new record holder of reachable Lewis acidity within neutral silanes. Inspection of the solid-state structures reemphasized the inability of structural parameters to accurately reflect Lewis acidity. Finally, this contribution demonstrates a correlation between the Lewis acidity of the investigated bis(catecholato)silanes and their efficiency in halide abstraction reactions and catalytic applications. Although the presence of acetonitrile in the strongest acids (X = Cl, Br) might limit their compatibility with very reactive cations, applicability in manifold transformations can be conceived. | ||
| Fig. 5 Comparison of FIA of the Si(catX)2 class with other selected Lewis acids (all values obtained at DLPNO-CCSD(T)/aug-cc-pVQZ level of theory). | ||
The applied theoretical analyses provide a rationale for the origin of the FIA in bis(catecholato)silanes and deliver inspiration for future modifications within this class of Lewis acids. Contradicting the general notion that the inductive effects of the substituents X (F > Cl > Br) are decisive, it is rather the decreasing ability of X for π-backdonation into the catechol aromatic system (F > Cl > Br) that dominates the observed Lewis acidities. These insights might guide future design principles for even stronger Lewis acids.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank Prof. H.-J. Himmel for his support, the FCI and DFG for financial support and the BWFor/BWUniCluster for computational resources. D. H. thanks the FES for a PhD scholarship.References
- (a) H. Yamamoto, Lewis acids in organic synthesis, Wiley-VCH, Weinheim, 2002 Search PubMed; (b) A. Corma and H. García, Chem. Rev., 2003, 103, 4307–4366 CrossRef CAS; (c) J. Eames and M. Watkinson, in Metalloenzymes and electrophilic catalysis, John Wiley & Sons Ltd, 2007, vol. 1, pp. 508–519 Search PubMed; (d) H. Yamamoto and K. Ishihara, Acid catalysis in modern organic synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed; (e) E. Y.-X. Chen, in Frustrated Lewis Pairs II: Expanding the Scope, ed. G. Erker and D. W. Stephan, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 239–260 Search PubMed.
- (a) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- R. Maskey, M. Schädler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717–1720 CrossRef CAS PubMed.
- (a) L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659–7663 CrossRef PubMed; (b) L. Greb, Chem.–Eur. J., 2018, 24, 17881–17896 CrossRef CAS PubMed.
- L. A. Körte, J. Schwabedissen, M. Soffner, S. Blomeyer, C. G. Reuter, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2017, 56, 8578–8582 CrossRef PubMed.
- (a) T. Belgardt, J. Storre, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Inorg. Chem., 1995, 34, 3821–3822 CrossRef CAS; (b) G. S. Hair, A. H. Cowley, R. A. Jones, B. G. McBurnett and A. Voigt, J. Am. Chem. Soc., 1999, 121, 4922–4923 CrossRef CAS; (c) A. Wiesner, T. W. Gries, S. Steinhauer, H. Beckers and S. Riedel, Angew. Chem., Int. Ed., 2017, 56, 8263–8266 CrossRef CAS PubMed; (d) J. F. Kogel, D. A. Sorokin, A. Khvorost, M. Scott, K. Harms, D. Himmel, I. Krossing and J. Sundermeyer, Chem. Sci., 2018, 9, 245–253 RSC.
- (a) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed; (b) B. Pan and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 9564–9567 CrossRef CAS PubMed.
- (a) A. D. Dilman and S. L. Ioffe, Chem. Rev., 2003, 103, 733–772 CrossRef CAS PubMed; (b) L. Ratjen, M. van Gemmeren, F. Pesciaioli and B. List, Angew. Chem., Int. Ed., 2014, 53, 8765–8769 CrossRef CAS PubMed; (c) D. Höfler, M. v. Gemmeren, P. Wedemann, K. Kaupmees, I. Leito, M. Leutzsch, J. B. Lingnau and B. List, Angew. Chem., Int. Ed., 2017, 56, 1411–1415 CrossRef PubMed.
- (a) A. G. Myers, S. E. Kephart and H. Chen, J. Am. Chem. Soc., 1992, 114, 7922–7923 CrossRef CAS; (b) K. Matsumoto, K. Oshima and K. Utimoto, J. Org. Chem., 1994, 59, 7152–7155 CrossRef CAS; (c) J. W. A. Kinnaird, P. Y. Ng, K. Kubota, X. Wang and J. L. Leighton, J. Am. Chem. Soc., 2002, 124, 7920–7921 CrossRef CAS PubMed; (d) X. Zhang, K. N. Houk and J. L. Leighton, Angew. Chem., Int. Ed., 2005, 44, 938–941 CrossRef CAS; (e) R. Hrdina, C. E. Müller, R. C. Wende, K. M. Lippert, M. Benassi, B. Spengler and P. R. Schreiner, J. Am. Chem. Soc., 2011, 133, 7624–7627 CrossRef CAS PubMed; (f) W. A. Chalifoux, S. K. Reznik and J. L. Leighton, Nature, 2012, 487, 86 CrossRef CAS PubMed.
- (a) S. E. Denmark and T. Wynn, J. Am. Chem. Soc., 2001, 123, 6199–6200 CrossRef CAS; (b) S. E. Denmark and G. L. Beutner, J. Am. Chem. Soc., 2003, 125, 7800–7801 CrossRef CAS PubMed; (c) S. E. Denmark, G. L. Beutner, T. Wynn and M. D. Eastgate, J. Am. Chem. Soc., 2005, 127, 3774–3789 CrossRef CAS PubMed; (d) A. V. Malkov, A. J. Liddon, P. Ramirez-Lopez, L. Bendova, D. Haigh and P. Kocovsky, Angew. Chem., Int. Ed., 2006, 45, 1432–1435 CrossRef CAS PubMed; (e) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS; (f) R. Hrdina, F. Opekar, J. Roithova and M. Kotora, Chem. Commun., 2009, 2314–2316 RSC.
- (a) H. J. Frohn, M. Giesen, A. Klose, A. Lewin and V. V. Bardin, J. Organomet. Chem., 1996, 506, 155–164 CrossRef CAS; (b) A. D. Dilman, V. V. Levin, M. Karni and Y. Apeloig, J. Org. Chem., 2006, 71, 7214–7223 CrossRef CAS PubMed; (c) A. D. Dilman, V. V. Levin, A. A. Korlyukov, P. A. Belyakov, M. I. Struchkova, M. Y. Antipin and V. A. Tartakovsky, J. Organomet. Chem., 2008, 693, 1005–1019 CrossRef CAS.
- U. Dettlaff-Weglikowska, E. Hey-Hawkins and H. G. von Schnering, Z. Naturforsch., B: J. Chem. Sci., 1991, 46, 609 CAS.
- (a) M. Wiesemann and B. Hoge, Chem.–Eur. J., 2018, 24, 16457–16471 CrossRef CAS; (b) S. Steinhauer, J. Bader, H.-G. Stammler, N. Ignat'ev and B. Hoge, Angew. Chem., Int. Ed., 2014, 53, 5206–5209 CAS; (c) N. Schwarze, B. Kurscheid, S. Steinhauer, B. Neumann, H. G. Stammler, N. Ignat'ev and B. Hoge, Chemistry, 2016, 22, 17460–17467 CrossRef CAS PubMed; (d) B. Waerder, M. Pieper, L. A. Körte, T. A. Kinder, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2015, 54, 13416–13419 CrossRef CAS PubMed.
- A. L. Liberman-Martin, R. G. Bergman and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 5328–5331 CrossRef CAS PubMed.
- (a) C. L. Frye, J. Am. Chem. Soc., 1964, 86, 3170–3171 CrossRef CAS; (b) C. M. S. Yoder and J. J. Zuckerman, Inorg. Chem., 1967, 6, 163–164 CrossRef CAS; (c) F. P. Boer, J. J. Flynn and J. W. Turley, J. Am. Chem. Soc., 1968, 90, 6973–6977 CrossRef CAS; (d) J. J. Harland, R. O. Day, J. F. Vollano, A. C. Sau and R. R. Holmes, J. Am. Chem. Soc., 1981, 103, 5269–5270 CrossRef CAS; (e) R. R. Holmes, R. O. Day, J. J. Harland and J. M. Holmes, Organometallics, 1984, 3, 347–353 CrossRef CAS; (f) R. R. Holmes, R. O. Day, J. J. Harland, A. C. Sau and J. M. Holmes, Organometallics, 1984, 3, 341–347 CrossRef CAS; (g) R. R. Holmes, R. O. Day, V. Chandrasekhar, J. J. Harland and J. M. Holmes, Inorg. Chem., 1985, 24, 2016–2020 CrossRef CAS; (h) K. C. K. Swamy, V. Chandrasekhar, J. J. Harland, J. M. Holmes, R. O. Day and R. R. Holmes, J. Am. Chem. Soc., 1990, 112, 2341–2348 CrossRef CAS; (i) E. Hey-Hawkins, U. Dettlaff-Weglikowska, D. Thiery and H. G. von Schnering, Polyhedron, 1992, 11, 1789–1794 CrossRef CAS; (j) R. Tacke, J. Becht, A. Lopez-Mras and J. Sperlich, J. Organomet. Chem., 1993, 446, 1–8 CrossRef CAS; (k) J. H. Small, D. J. McCord, J. Greaves and K. J. Shea, J. Am. Chem. Soc., 1995, 117, 11588–11589 CrossRef CAS; (l) R. Tacke, M. Pülm and B. Wagner, in Adv. Organomet. Chem., ed. R. West and A. F. Hill, Academic Press, 1999, vol. 44, pp. 221–273 Search PubMed; (m) J. B. Lambert and S. R. Singer, J. Organomet. Chem., 2004, 689, 2293–2302 CrossRef CAS.
- (a) F. Carré, G. Cerveau, C. Chuit, R. J. P. Corriu and C. Réyé, Angew. Chem., Int. Ed., 1989, 28, 489–491 CrossRef; (b) S. P. Narula, R. Shankar, M. Kumar and Meenu, Inorg. Chem., 1994, 33, 2716–2718 CrossRef CAS; (c) F. E. Hahn, M. Keck and K. N. Raymond, Inorg. Chem., 1995, 34, 1402–1407 CrossRef CAS.
- (a) M. Kira, K. Sato and H. Sakurai, J. Org. Chem., 1987, 52, 948–949 CrossRef CAS; (b) M. Kira, K. Sato and H. Sakurai, J. Am. Chem. Soc., 1990, 112, 257–260 CrossRef CAS.
- (a) V. Corcé, L.-M. Chamoreau, E. Derat, J.-P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., 2015, 127, 11576–11580 CrossRef; (b) M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475–478 CrossRef CAS PubMed; (c) C. Leveque, L. Chenneberg, V. Corce, C. Ollivier and L. Fensterbank, Chem. Commun., 2016, 52, 9877–9880 RSC.
- (a) A. Rosenheim and O. Sorge, Ber. Dtsch. Chem. Ges., 1920, 53, 932–939 CrossRef; (b) A. Rosenheim, B. Raibmann and G. Schendel, Z. Anorg. Allg. Chem., 1931, 196, 160–176 CrossRef CAS; (c) A. Weiss, G. Reiff and A. Weiss, Z. Anorg. Allg. Chem., 1961, 311, 151–179 CrossRef CAS; (d) H. Bartels and H. Erlenmeyer, Helv. Chim. Acta, 1964, 47, 7–13 CrossRef CAS; (e) J. J. Flynn and F. P. Boer, J. Am. Chem. Soc., 1969, 91, 5756–5761 CrossRef CAS; (f) D. W. Barnum, Inorg. Chem., 1972, 11, 1424–1429 CrossRef CAS; (g) D. F. Evans, J. Parr and E. N. Coker, Polyhedron, 1990, 9, 813–823 CrossRef CAS; (h) J. V. Kingston, B. Vargheese and M. N. S. Rao, Main Group Chem., 2000, 3, 79–90 CrossRef CAS; (i) R. Tacke and O. Seiler, in Silicon Chemistry, ed. U. Schubert and P. Jutzi, Wiley-VCH, 2007 Search PubMed.
- (a) H. R. Allcock, T. A. Nugent and L. A. Smeltz, Synthesis and Reactivity in Inorganic and Metal-Organic Chemistry, 1972, vol. 2, pp. 97–104 Search PubMed; (b) A. K. Chekalov, A. I. Prokof'ev, N. N. Bubnov, S. P. Solodovnikov, A. A. Zhdanov and M. I. Kabachnik, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1981, 30, 2064–2071 CrossRef.
- H. Meyer and G. Nagorsen, Angew. Chem., Int. Ed., 1979, 18, 551–553 CrossRef.
- (a) S. Robert and K. Wilhelm, Z. Anorg. Allg. Chem., 1951, 266, 185–192 CrossRef; (b) J. Mihailo, Z. Anorg. Allg. Chem., 1957, 288, 324–332 CrossRef; (c) J. J. Zuckerman, J. Chem. Soc., 1962, 873–876 RSC.
- W. Hönle, U. Dettlaff-Weglikowska, L. Walz and H. G. von Schnering, Angew. Chem., Int. Ed., 1989, 28, 623–624 CrossRef.
- C. Y. Wong and J. D. Woollins, Coord. Chem. Rev., 1994, 130, 175–241 CrossRef CAS.
- (a) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS; (b) M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- E. L. Myers, C. P. Butts and V. K. Aggarwal, Chem. Commun., 2006, 4434–4436 RSC.
- H. C. Brown and Y. Okamoto, J. Am. Chem. Soc., 1958, 80, 4979–4987 CrossRef CAS.
- D. F. Evans, A. M. Z. Slawin, D. J. Williams, C. Y. Wong and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1992, 2383–2387 RSC.
- S. Steinhauer, T. Böttcher, N. Schwarze, B. Neumann, H.-G. Stammler and B. Hoge, Angew. Chem., Int. Ed., 2014, 53, 13269–13272 CrossRef CAS PubMed.
- (a) V. Jonas, G. Frenking and M. T. Reetz, J. Am. Chem. Soc., 1994, 116, 8741–8753 CrossRef CAS; (b) K. R. Leopold, M. Canagaratna and J. A. Phillips, Acc. Chem. Res., 1997, 30, 57–64 CrossRef CAS; (c) D. G. Anderson, A. J. Blake, S. Cradock, E. A. V. Ebsworth, D. W. H. Rankin and A. J. Welch, Angew. Chem., Int. Ed., 1986, 25, 107–108 CrossRef.
- T. Bottcher, S. Steinhauer, B. Neumann, H. G. Stammler, G. V. Roschenthaler and B. Hoge, Chem. Commun., 2014, 50, 6204–6206 RSC.
- E. P. A. Couzijn, J. C. Slootweg, A. W. Ehlers and K. Lammertsma, J. Am. Chem. Soc., 2010, 132, 18127–18140 CrossRef CAS PubMed.
- R. Tacke, J. Heermann, M. Pülm and E. Gottfried, Monatsh. Chem., 1999, 130, 99–107 CAS.
- R. Tacke, M. Pülm, I. Richter, B. Wagner and R. Willeke, Z. Anorg. Allg. Chem., 1999, 625, 2169–2177 CrossRef CAS.
- (a) C. B. Caputo and D. W. Stephan, Organometallics, 2012, 31, 27–30 CrossRef CAS; (b) S. S. Chitnis, F. Krischer and D. W. Stephan, Chem.–Eur. J., 2018, 24, 6543–6546 CrossRef CAS PubMed.
- (a) K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS; (b) L. Zhao, M. Hermann, W. H. E. Schwarz and G. Frenking, Nat. Rev. Chem., 2019, 3, 48–63 CrossRef CAS.
- (a) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS; (b) E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CAS.
- (a) R. F. W. Bader and H. Essén, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS; (b) D. Stalke, Chem.–Eur. J., 2011, 17, 9264–9278 CrossRef CAS PubMed.
- C. Outeiral, M. A. Vincent, A. Martin Pendas and P. L. A. Popelier, Chem. Sci., 2018, 9, 5517–5529 RSC.
- M. Fugel, M. F. Hesse, R. Pal, J. Beckmann, D. Jayatilaka, M. J. Turner, A. Karton, P. Bultinck, G. S. Chandler and S. Grabowsky, Chemistry, 2018, 24, 15275–15286 CrossRef CAS PubMed.
- (a) R. G. Parr, L. v. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS; (b) P. K. Chattaraj, U. Sarkar and D. R. Roy, Chem. Rev., 2006, 106, 2065–2091 CrossRef CAS PubMed; (c) A. R. Jupp, T. C. Johnstone and D. W. Stephan, Dalton Trans., 2018, 47, 7029–7035 RSC; (d) A. R. Jupp, T. C. Johnstone and D. W. Stephan, Inorg. Chem., 2018, 57, 14764–14771 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic and computational details. CCDC 1905749–1905760. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02167a |
| This journal is © The Royal Society of Chemistry 2019 |