
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceB(C6F5)3-catalyzed dehydrogenative cyclization of N-tosylhydrazones and anilines via a Lewis adduct: a combined experimental and computational investigation†
Murali Mohan
Guru‡
 ,
Sriman
De‡
,
Sayan
Dutta
,
Debasis
Koley
* and
Biplab
Maji
*
,
Sriman
De‡
,
Sayan
Dutta
,
Debasis
Koley
* and
Biplab
Maji
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur-741246, India. E-mail: koley@iiserkol.ac.in; bm@iiserkol.ac.in
First published on 5th July 2019
Abstract
Tris(pentafluorophenyl)borane-catalyzed dehydrogenative-cyclization of N-tosylhydrazones with aromatic amines has been disclosed. This metal-free catalytic protocol is compatible with a range of functional groups to provide both symmetrical and unsymmetrical 3,4,5-triaryl-1,2,4-triazoles. Mechanistic experiments and density functional theory (DFT) studies suggest an initial Lewis adduct formation of N-tosylhydrazone with B(C6F5)3 followed by sequential intermolecular amination of the borane adduct with aniline, intramolecular cyclization and frustrated Lewis pair (FLP)-catalyzed dehydrogenation for the generation of substituted 1,2,4-triazoles.
Introduction
Tris(pentafluorophenyl)borane has recently emerged as a powerful Lewis acid catalyst.1–3 The strong Lewis acidity2,3 at the boron center allows us to establish a wide range of organic transformations via C–B,4–6 C–C,7–10 C–N,11,12 C–O13,14 and C–Si15–18 bond formations. Pioneered by Stephan and Erker, B(C6F5)3 has gained popularity in frustrated Lewis pair (FLP) chemistry which encompasses widespread applications in organic reactions.19–22 Indeed, numerous efforts have been devoted to the activation of alkenes and alkynes by FLPs for the formation of cyclic scaffolds.23–25 However, in many cases, the highly desirable catalytic reaction is obstructed by the initially formed stable borate adduct.26–28 Consequently, B(C6F5)3 catalyzed cyclization leading to important heterocyclic scaffolds is rare.29–32In this context, 1,2,4-triazoles are omnipresent heterocyclic motifs in numerous biologically active compounds33,34 and they also have widespread applications in organic light emitting diodes,35,36 organic photovoltaic cells, electroluminescent devices,37,38 bi-stable resistive memory devices,39 pesticides, and medicines.40 Given their applications, a number of strategies have been implemented for the synthesis of 1,2,4-triazoles.41–45 However, most of these methods are limited due to the use of a super-stoichiometric amount of reagents or oxidants, low chemo-selectivity, narrow functional group tolerance, multiple reaction steps, and the production of copious waste. And catalytic metal- and oxidant-free, step-economical processes for an efficient synthesis of substituted 1,2,4-triazoles are in demand.
Lewis acid–base adducts are potential intermediates in a multitude of transformations.46–48 Recently, Stephan and coworkers have reported that diphenyldiazomethane, obtained from the corresponding N-tosylhydrazone in the presence of a base, readily forms adducts with B(C6F5)3 (Scheme 1a).49,50 This has shifted the gear for metal-free N2 activation closer to reality. Conversely, the direct interaction of N-tosylhydrazones with B(C6F5)3 and their application in catalytic transformations have not been reported yet.
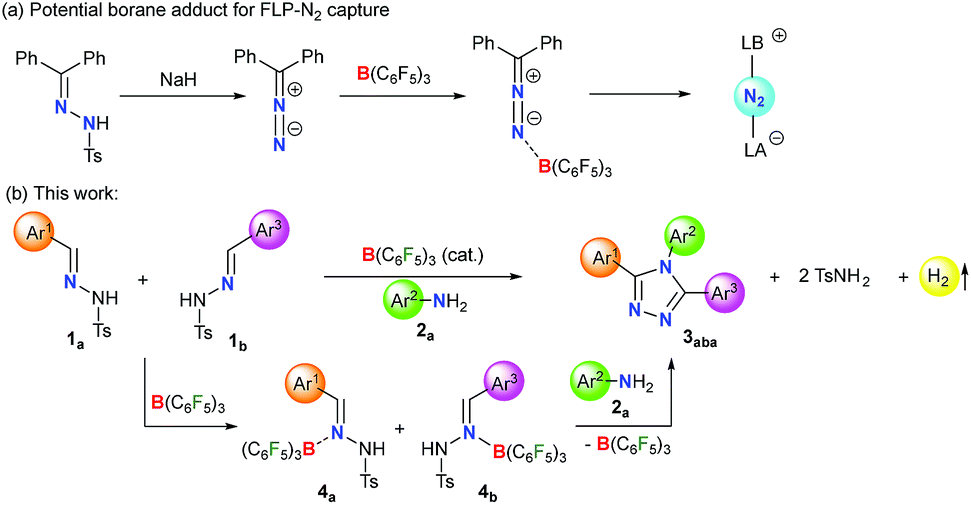 | ||
| Scheme 1 Applications of borane adducts in main group chemistry. | ||
On the other hand, environmentally benign acceptorless catalytic dehydrogenation which is highly challenging even for transition-metal complexes51,52 is rare under metal-free conditions.53–56 Very recently, we have presented a manganese-catalyzed acceptorless-dehydrogenative olefination of heteroarenes with primary alcohols.57 Herein, we report B(C6F5)3 catalyzed acceptorless-dehydrogenative-cyclization of N-tosylhydrazones 1 and anilines 2 to triaryl-1,2,4-triazoles 3via a borane adduct 4 followed by sequential C–N/N–N bond formation (Scheme 1b). Furthermore, extensive DFT calculations are performed not only to understand the mechanistic features of borane catalyzed triazole formation but also to assist future development of similar classes of reactions.
Results and discussion
Initially, to check the reactivity of hydrazone 1a with B(C6F5)3, a stoichiometric reaction in benzene at room temperature was carried out. It leads to the formation of a Lewis adduct 4a (δ11B – 2.5 ppm) after 1 h in 82% yield (Scheme 2). When 4a was reacted with an aromatic amine 2a at 80 °C in benzene, 3,4,5-triaryl-1,2,4-triazole 3aaa was formed in 85% NMR yield after 12 h along with an equimolar amount of TsNH2 and B(C6F5)3.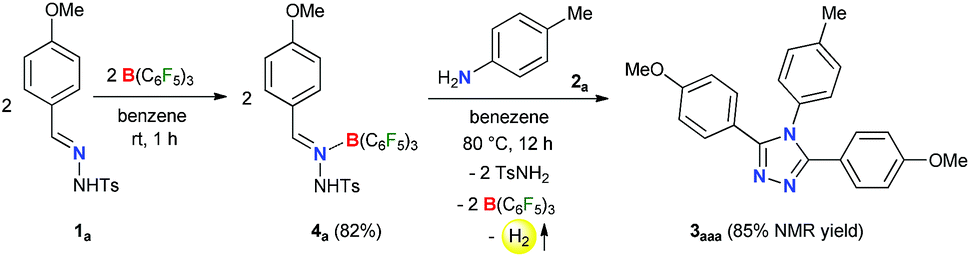 | ||
| Scheme 2 Borane adduct of N-tosylhydrazone 1a and its transformation. | ||
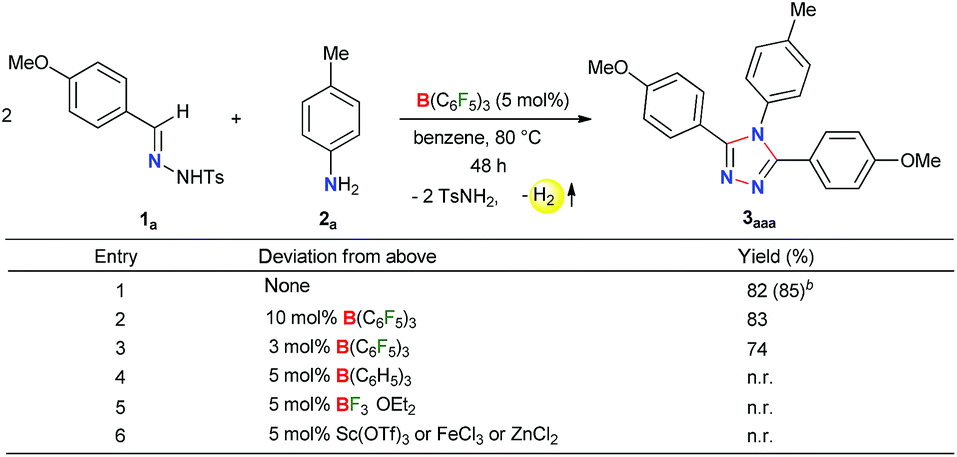
As B(C6F5)3 was finally released from the adduct 4a after its reaction with the amine 2a, it was posited that catalytic turnover should be possible under thermal conditions. Interestingly, when the reaction was implemented with 5 mol% B(C6F5)3 at 80 °C 1,2,4-triazole 3aaa was obtained in 82% isolated yield along with 68% of TsNH2 as a byproduct (Table 1, entry 1). While the increased catalyst loading did not significantly improve the yield (entry 2), a slight decrease in the product yield was observed with 3 mol% B(C6F5)3 (entry 3). The use of less Lewis acidic boranes such as BPh3 was unproductive (entry 4). In addition, less hindered boranes like BF3·OEt2 resulted in no detectable product formation (entry 5). Other Lewis acid catalysts like Sc(OTf)3, FeCl3 and ZnCl2 were also not effective for this reaction (entry 6, Schemes S9 and S10 in the ESI†).
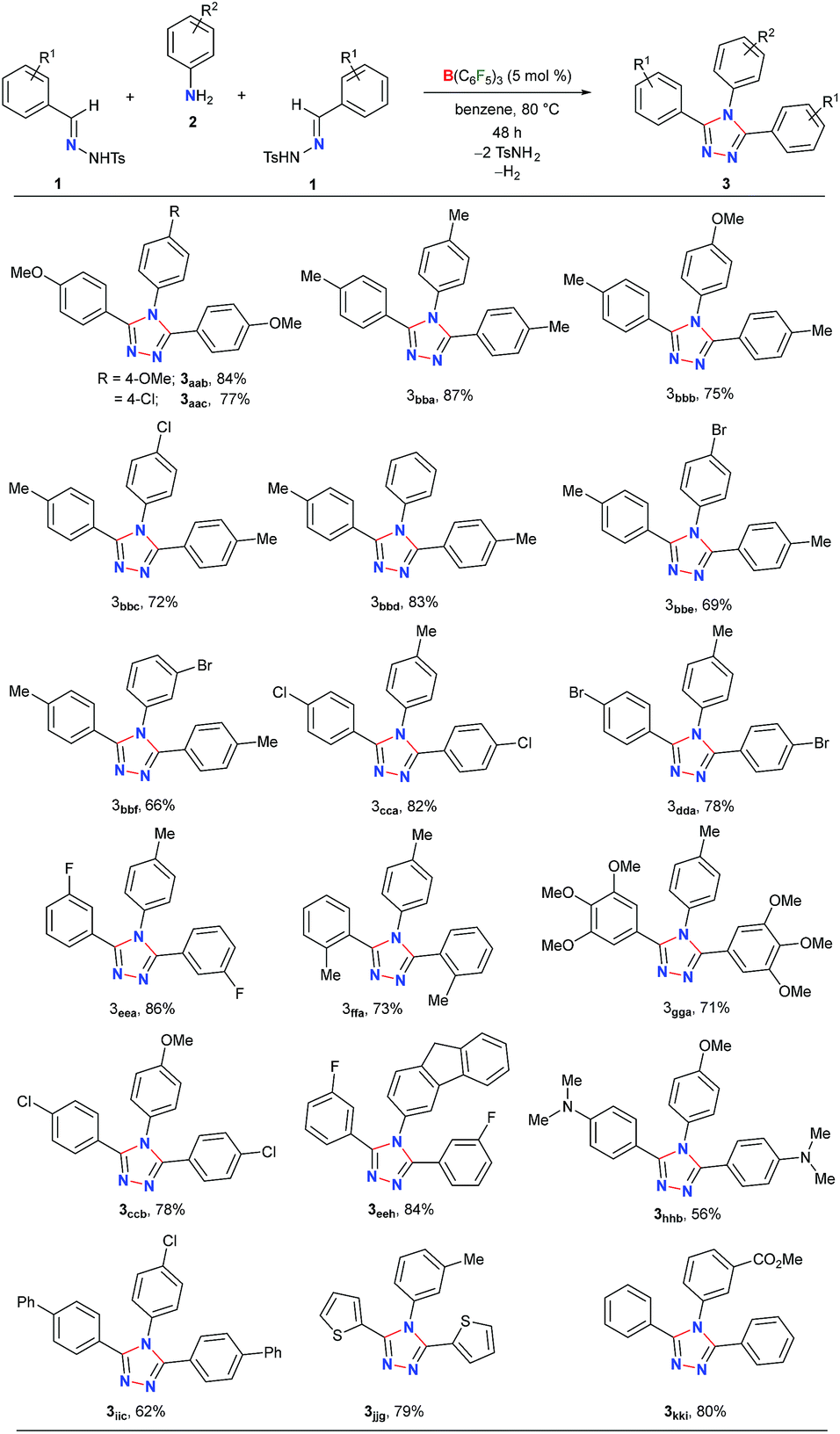
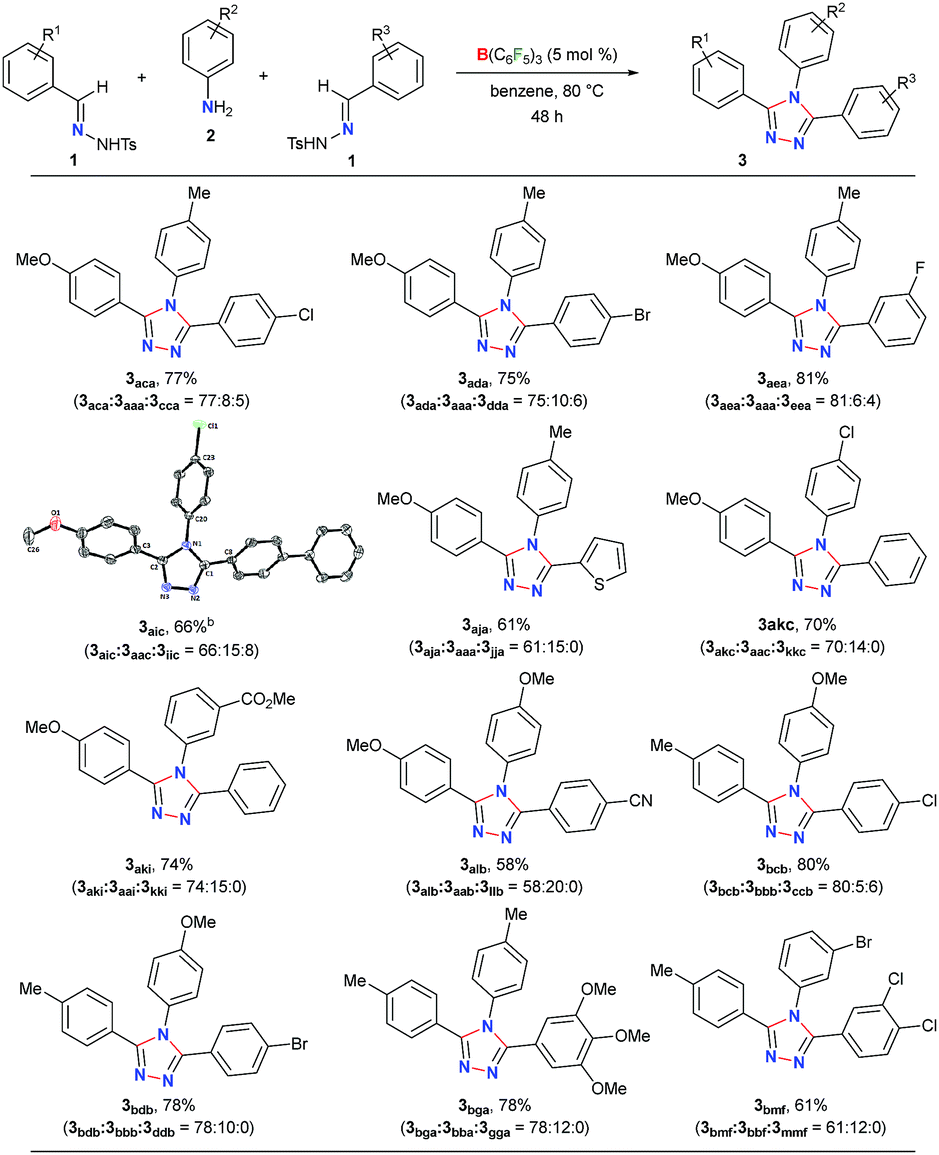
Next, the scope of this metal-free protocol was explored. A series of hydrazones 1 could be employed with various anilines 2 to afford symmetrical 3,4,5-triaryl-1,2,4-triazoles 3 in good to excellent yields (Table 2). The reaction of 1a and 1b with anilines 2a–f bearing OMe-, Me-, H-, Cl- and Br-substituents at the para- or meta-position of the aryl ring afforded the triazoles 3aab–3bbf in 66–87% yields. Similarly, the hydrazones 1c–g having substituents with different steric and electronic properties at the ortho-, meta-, and para-position on the aromatic rings could readily be cyclized with 2a to produce 3cca–3gga in 71–86% yields. Electronically biased aryl rings could be installed smoothly to obtain 3ccb, 3hhb, and 3iic in 56–78% yields. Likewise, thiophene- and fluorine-containing 1,2,4-triazoles 3eeh and 3jjg could be synthesized in 79% and 84% yields, respectively. Notably, an ester-group could also be tolerated under the reaction conditions to furnish 3kki in 80% yield. On the other hand, substrates containing amides, nitro groups, olefins, primary alcohols, primary amino groups, N-tosylhydrazone of aliphatic aldehydes, and N-methanesulfonyl hydrazone were found to be unsuitable as either the starting materials remained intact or a complex mixture of products was formed (Schemes S1–S5 in the ESI†).
To further demonstrate the applicability of our protocol, we examined the possibility of obtaining unsymmetrical 1,2,4-triazoles by employing two different hydrazones (Table 3). In fact, the synthesis of unsymmetrically substituted 1,2,4-triazoles is considered to be highly challenging and to the best of our knowledge, their single step synthesis from readily accessible starting materials is less explored.41–45 Gratifyingly, when 2a was reacted with an equimolar mixture of 1a and 1c the unsymmetrical 1,2,4-triazole 3aca was obtained as a major product in 77% yield and symmetrical triazoles were only obtained in minor amounts. The products could be purified via column chromatography on silica-gel using an ethyl acetate/hexane mixture as the eluent. Similar reactivity and selectivity were also observed for the synthesis of 3ada and 3aea. Likewise, biphenyl-, haloaromatic- and heteroaromatic-rings could be installed without any difficulty to give the triazoles 3aic, 3aja, 3akc, 3bcb and 3bdb in 61–80% yields. Colorless crystals of 3aic, grown from a saturated benzene solution stored at room temperature, were suitable for single-crystal X-ray analysis and clearly confirmed the structure of the product (Fig. S3 in the ESI†).58 It is noteworthy that the reaction is highly chemoselective as the reactive carbomethoxy- and cyano-groups remain unaffected under the present reaction conditions to provide 3aki and 3alb in 74% and 58% yields, respectively. The steric hindrance was found to exert a negligible influence on the reaction to give the desired product 3bga and 3bmf in good yields.
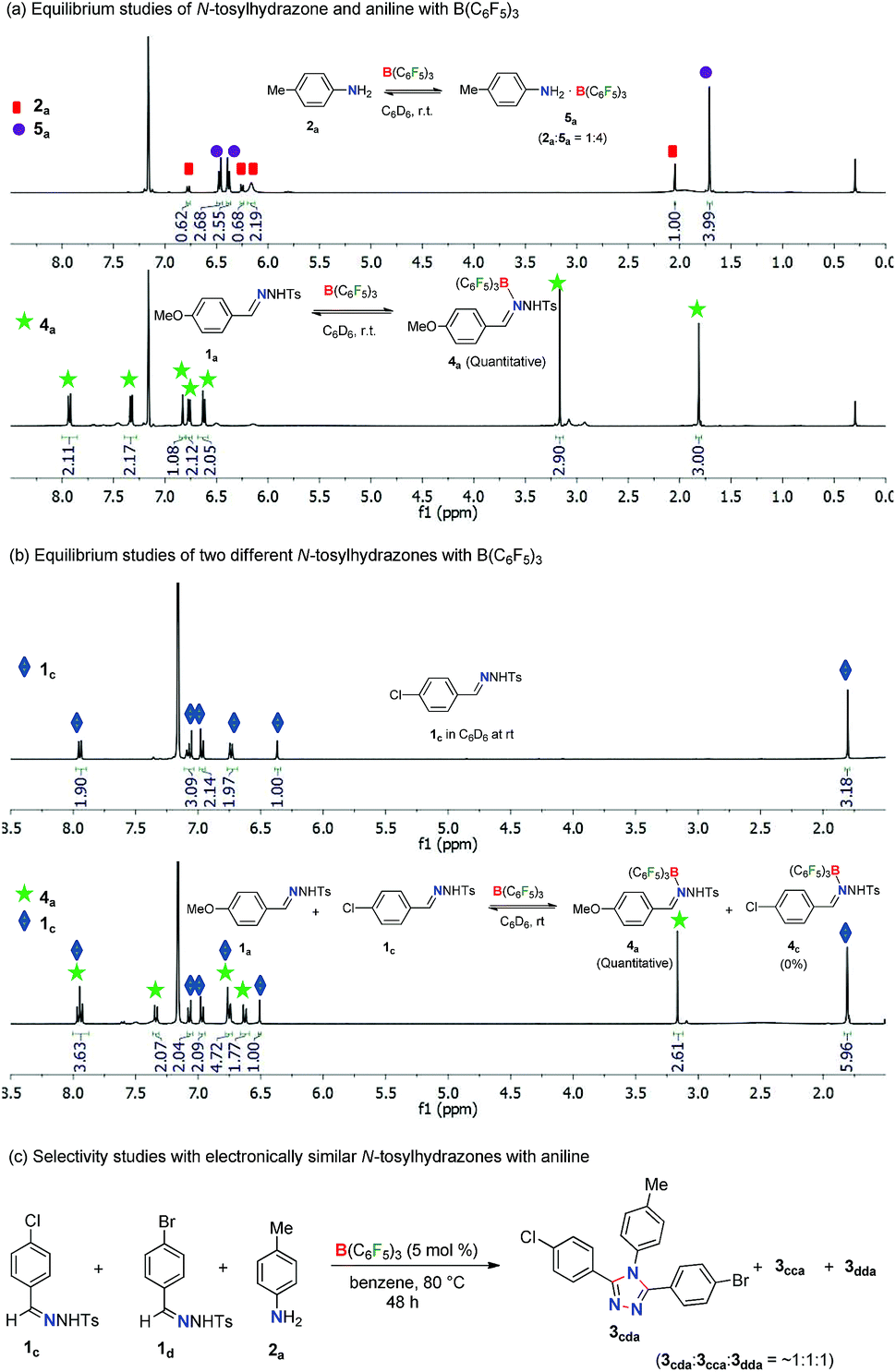
In order to obtain insight into the reaction mechanism, several equilibrium studies were performed initially with N-tosylhydrazones and anilines in the presence of Lewis acids (Scheme 3). At the onset, the relative Lewis basicity of aniline and N-tosylhydrazone towards B(C6F5)3 was analyzed (Scheme 3a). Thus, in a stoichiometric reaction of 2a and B(C6F5)3 at room temperature in C6D6, the ratio of 2a and 5a (B(C6F5)3 adduct of 2a) was found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (2a:5a = 1:4). Similarly, the stoichiometric mixture of 1a and B(C6F5)3 in C6D6 at room temperature afforded quantitative formation of 4a (B(C6F5)3 adduct 1a) where the ration was 1a:4a = 0:100. This confirms the tendency of N-tosylhydrazone 1 to form a stronger Lewis acid–base adduct with B(C6F5)3 in comparison to aniline 2. Moreover, a competitive equilibrium study was performed with two electronically biased N-tosylhydrazones (1avs.1c) with B(C6F5)3 (Scheme 3b). Accordingly, a stoichiometric (1:1) mixture of 1a and 1c was treated with 1 equivalent of B(C6F5)3 in C6D6 at room temperature. This selectively afforded the Lewis acid–base adduct 4a whereas 1c remained unreacted (4a:1c = 1:1). Thus, N-tosylhydrazones having electron rich arenes will form strong Lewis acid–base adducts than hydrazones having electron deficient arenes. This is possibly a crucial factor for the selectivity observed during the synthesis of unsymmetrical 1,2,4-triazoles as shown in Table 3. Along this direction, in fact, the reaction of aniline 2a with a mixture of 1c and 1d having electronically similar substituents (Cl and Br) on arenes in the presence of the B(C6F5)3 catalyst provided a mixture of two symmetrical and unsymmetrical 1,2,4-triazoles (Scheme 3c) as both the hydrazones have similar probabilities for the formation of Lewis acid–base adducts with B(C6F5)3. Thus, electronically biased hydrazones are good candidates for better selectivity of unsymmetrical triazoles.
:4 (2a:5a = 1:4). Similarly, the stoichiometric mixture of 1a and B(C6F5)3 in C6D6 at room temperature afforded quantitative formation of 4a (B(C6F5)3 adduct 1a) where the ration was 1a:4a = 0:100. This confirms the tendency of N-tosylhydrazone 1 to form a stronger Lewis acid–base adduct with B(C6F5)3 in comparison to aniline 2. Moreover, a competitive equilibrium study was performed with two electronically biased N-tosylhydrazones (1avs.1c) with B(C6F5)3 (Scheme 3b). Accordingly, a stoichiometric (1:1) mixture of 1a and 1c was treated with 1 equivalent of B(C6F5)3 in C6D6 at room temperature. This selectively afforded the Lewis acid–base adduct 4a whereas 1c remained unreacted (4a:1c = 1:1). Thus, N-tosylhydrazones having electron rich arenes will form strong Lewis acid–base adducts than hydrazones having electron deficient arenes. This is possibly a crucial factor for the selectivity observed during the synthesis of unsymmetrical 1,2,4-triazoles as shown in Table 3. Along this direction, in fact, the reaction of aniline 2a with a mixture of 1c and 1d having electronically similar substituents (Cl and Br) on arenes in the presence of the B(C6F5)3 catalyst provided a mixture of two symmetrical and unsymmetrical 1,2,4-triazoles (Scheme 3c) as both the hydrazones have similar probabilities for the formation of Lewis acid–base adducts with B(C6F5)3. Thus, electronically biased hydrazones are good candidates for better selectivity of unsymmetrical triazoles.
 | ||
| Scheme 3 Equilibrium and selectivity studies for 1,2,4-triazole formation. | ||
In the case of competitive equilibrium studies of two different anilines with B(C6F5)3, a stoichiometric (1:1:1) mixture of 2a, 2c and B(C6F5)3 in C6D6 at room temperature gave their corresponding Lewis acid–base adduct 5a and 5c, respectively in an 8:1 ratio (Scheme S8 in the ESI†). This indicates that highly basic anilines will form stronger adducts with B(C6F5)3. It is also noteworthy that other Lewis acids like Sc(OTf)3 and BPh3 did not form any Lewis acid–base adducts with N-tosylhydrazones or anilines under similar reaction conditions (Schemes S9 and S10 in the ESI†).
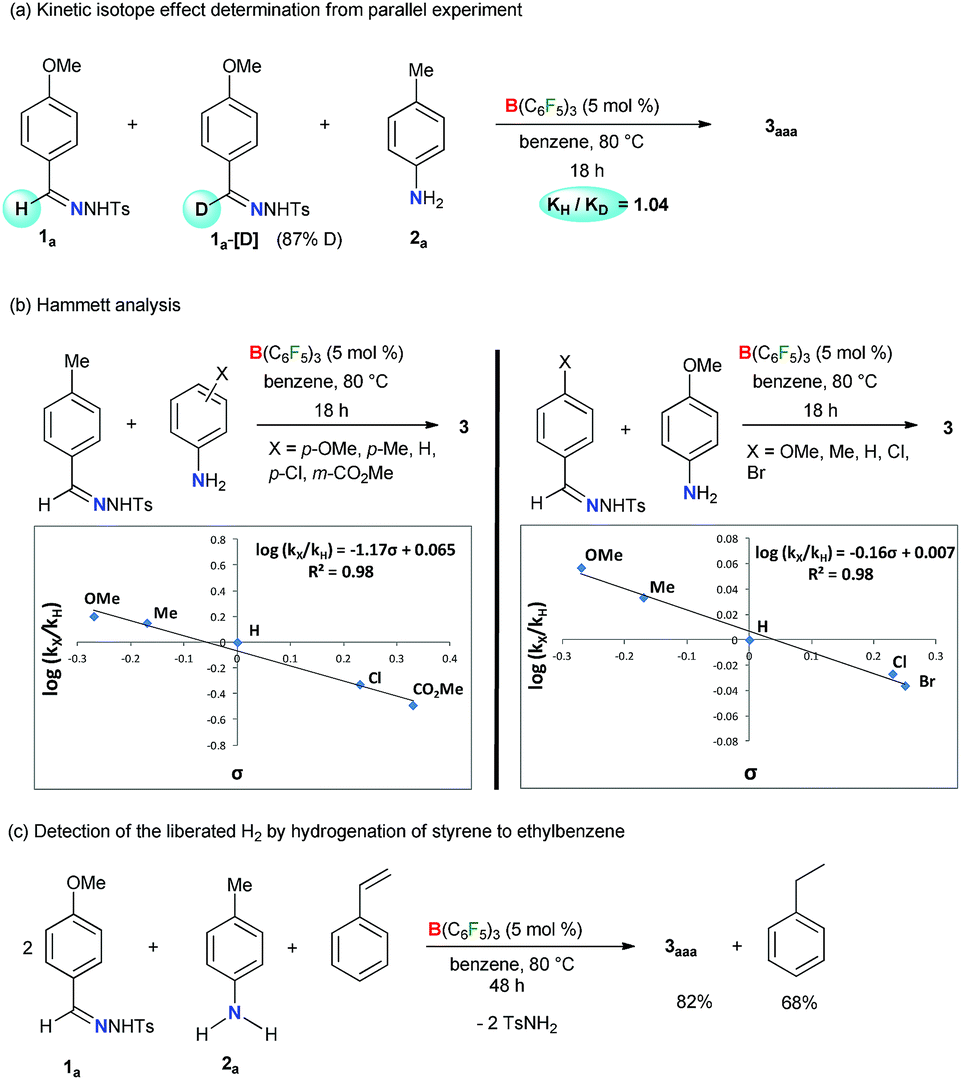
In addition, the kinetic isotope effect (KIE) of 1.0 measured from a parallel reaction of 2a with 1a and its deuterated analogue 1a-[D] suggests that the breakage of the imine C–H bond of 1a is not involved in the rate-determining step (Scheme 4a). Kinetic monitoring of the reaction suggests an exponential decay of both the reactants 1a and 2a, whereas sigmoidal increase of 3aaa was observed (Fig. S1 in the ESI†). The initial rate curve for triazole product formation possibly indicates that at the beginning, product formation is slower due to accumulation of reactive intermediates. Further kinetic studies revealed that the reaction is first order in the catalyst (Scheme S11 in the ESI†) and aniline 2a (Scheme S12 in the ESI†), and zero order in the N-tosylhydrazone 1a (Scheme S13 in the ESI†). In addition, the electronic influence of the aryl-substituents on both the reactants was investigated by a Hammett correlation study (Scheme 4b). By varying different electronic groups on the aryl-ring of 2 a small ρ = −1.17 was obtained. This plausibly indicates a weak resonance interaction involving a positive-charge at the N center of aniline in the rate-determining-step. On the other hand, a negligible substituent effect (ρ = −0.16) was determined for N-tosylhydrazones. Meanwhile, the evolution of H2 gas as a byproduct was confirmed by the transfer hydrogenation59,60 of styrene under the reaction conditions (Scheme 4c). A 2:1:1 mixture of 1a, 2a and styrene in the presence of the B(C6F5)3 catalyst afforded 82% of 3aaa along with 68% of ethylbenzene (Scheme 4c). Moreover, we performed a number of control experiments using amidines, azines, and imines as possible reaction intermediates albeit none of them proceeded to give 1,2,4-triazoles (Schemes S14–S16 in the ESI†).
 | ||
| Scheme 4 Kinetic and mechanistic studies for the synthesis of 1,2,4-triazole. | ||
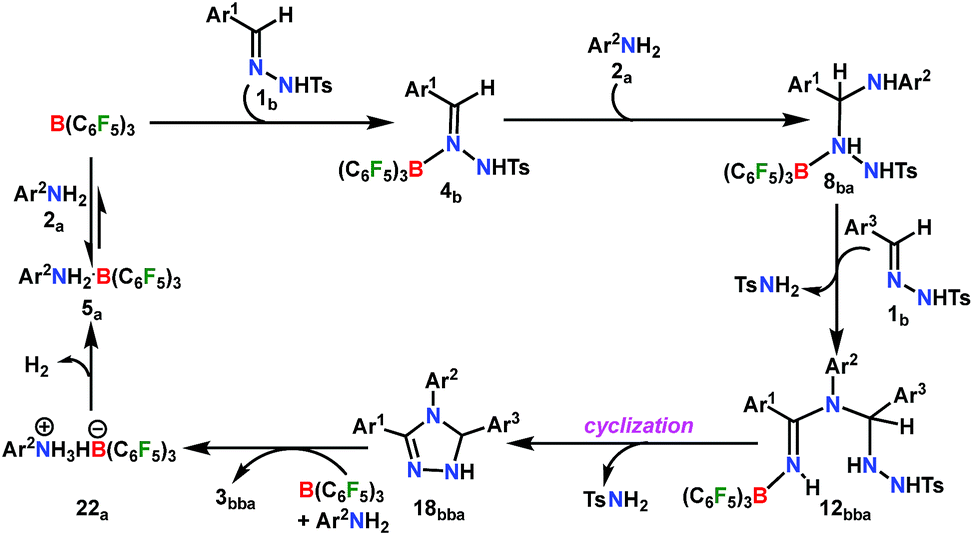
A plausible mechanism is proposed on the basis of the above experimental observations and previous literature reports (Scheme 5).54,55 To the best of our knowledge DFT calculations of N–N cyclization leading to 1,2,4 triazole fragments are obscure. Based on the proposed mechanism of the B(C6F5)3-catalyzed acceptorless-dehydrogenative-cyclization of N-tosylhydrazones with anilines (Scheme 5), we have performed DFT calculations to investigate the detailed reaction mechanism and to gain insight into the driving force for the formation of the 1,2,4-triazole moiety 3bba. Additionally, the calculations seek to address some pertinent questions regarding the studied system: (a) the specific role of B(C6F5)3 in the reaction, (b) the rate-limiting-step in the reaction, and (c) product distribution for unsymmetrical coupling.
 | ||
| Scheme 5 Plausible reaction mechanism. | ||
The reaction is initiated with the coordination of B(C6F5)3 to the sp2 nitrogen (N1) in N-tosylhydrazone (1b) which results in the formation of an isoenergetic encounter complex 4bp (Scheme 6). The approach of the nucleophilic N1 center in 1b towards the electron-deficient B center in B(C6F5)3 furnishes the slightly more stable Lewis adduct 4bvia a transition state [4p-4]b‡ with an activation barrier of 10.9 kcal mol−1. Despite the fact that the N2 center in 1b is significantly electron-rich (−0.646 e) compared to the N1 center (−0.242 e), as obtained by the natural population analysis (NPA), B(C6F5)3 gets coordinated to the N1 center. This is attributed to the fact that the lone pair orbital located on the N1 atom (HOMO-4) is significantly destabilized compared to the one on the N2 atom (HOMO-5) by 0.4 eV (Fig. S100 in the ESI†). Furthermore, coordination at the N2 center resulted in adduct 4b′, which is less stable than 4bp by 2.4 kcal mol−1 (Scheme S17 in the ESI†).
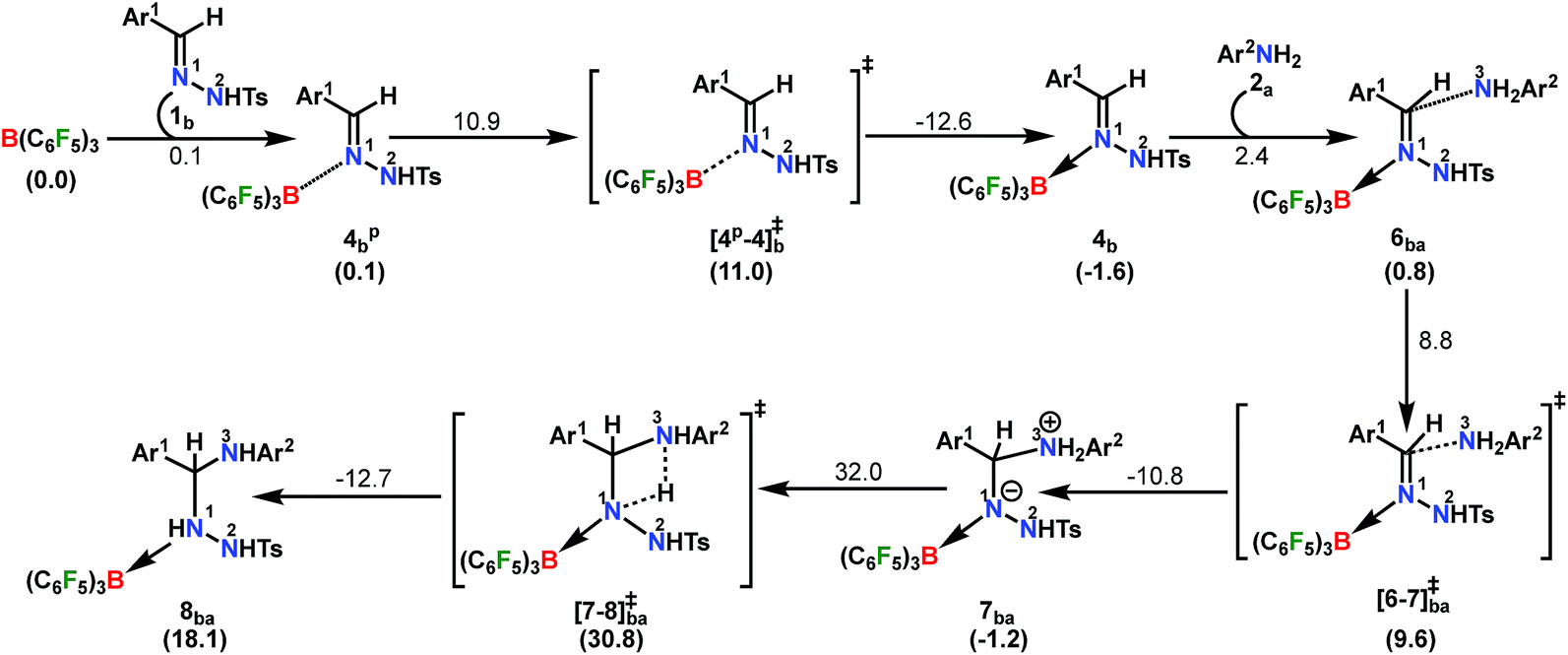 | ||
| Scheme 6 Part I: the reaction pathway for the formation of the intermediate 8ba. The energy values above the arrows denote the Gibbs free energy changes (ΔGLS) of the individual steps. The values within parentheses are the relative ΔGLS energies w.r.t the starting structures. All energy terms are in kcal mol−1. | ||
To cast light on the origin of the activation barrier and the bonding scenario in [4p-4]b‡, EDA-NOCV (energy decomposition analysis-natural orbital for chemical valence) analysis was performed, considering 1b and B(C6F5)3 as interacting fragments (Table S4 in the ESI†). Examination of the individual energy terms of the EDA reveals that the B–N1 bond has a higher electrostatic character (ΔEelstat: 39.8%) than the covalent character (ΔEorb: 33.5%). Importantly, the major contribution to the total covalent interaction (ΔEorb) originates from the donation of the lone pair on the N1 center in 1b to the vacant 2pz orbital of boron in the B(C6F5)3 fragment (Fig. S101 in the ESI†). We have calculated the associated eigenvalue of 0.49 e quantifying the amount of charge flow from donor → acceptor fragments. Additionally, the B(C6F5)3 fragment has the predominant contribution to the destabilizing distortion energy (ΔEdis). The calculated electron density [ρ(r)] of 0.112 at the (3, −1) bond critical point (BCP) of the B–N1 bond in 4b along with the respective Laplacian of +0.192 [∇2ρ(r)] suggests a donor–acceptor type interaction.61,62 Thereafter, the coordination of the substituted aniline (2a) to 4b affords the intermediate 6ba which finally leads to a slightly more stable Zwitterionic complex 7ba accompanied by a moderately low energy barrier of 8.8 kcal mol−1. The imaginary mode in [6-7]ba‡ portrays the formation of the C–N3 bond (1.849 Å) along with concomitant elongation of the C–N1 bond (1.383 Å). It is worthwhile to mention that the HOMO in 7ba represents the lone pair orbital located on the N1 atom (Fig. S100 in the ESI†). The subsequent proton transfer from N3 to the N1 center in 7ba furnishes the significantly less stable intermediate 8bavia a four-membered transition state [7-8]ba‡ (Scheme 6, Fig. 1a). The step 7ba → 8ba involving proton migration requires an activation barrier of 32.0 kcal mol−1 and thus becomes the rate-limiting step for the overall transformation.63 Indeed, this is supported by the experimental rate curve with a slower rate at the beginning of the reaction (vide supra, Fig. S1 in the ESI†). The single imaginary mode in [7-8]ba‡ depicts the synchronous breakage of N3–H (1.296 Å) and formation of N1–H (1.353 Å) bonds. In [7-8]ba‡, the B–N1 bond gets significantly elongated (1.602/1.676 Å in 7ba/[7-8]ba‡) and this weakening of the donor–acceptor bond is reflected in the reduced NPA charge on the B center (+0.467/+0.488 e in 7ba/[7-8]ba‡). It should be noted that both aniline and TsNH2 assisted alternative intermolecular proton transfer between the two nitrogen centers (N3 → N1) are less favorable than the intramolecular path reported in Scheme 6 (Scheme S18a and b in the ESI†).
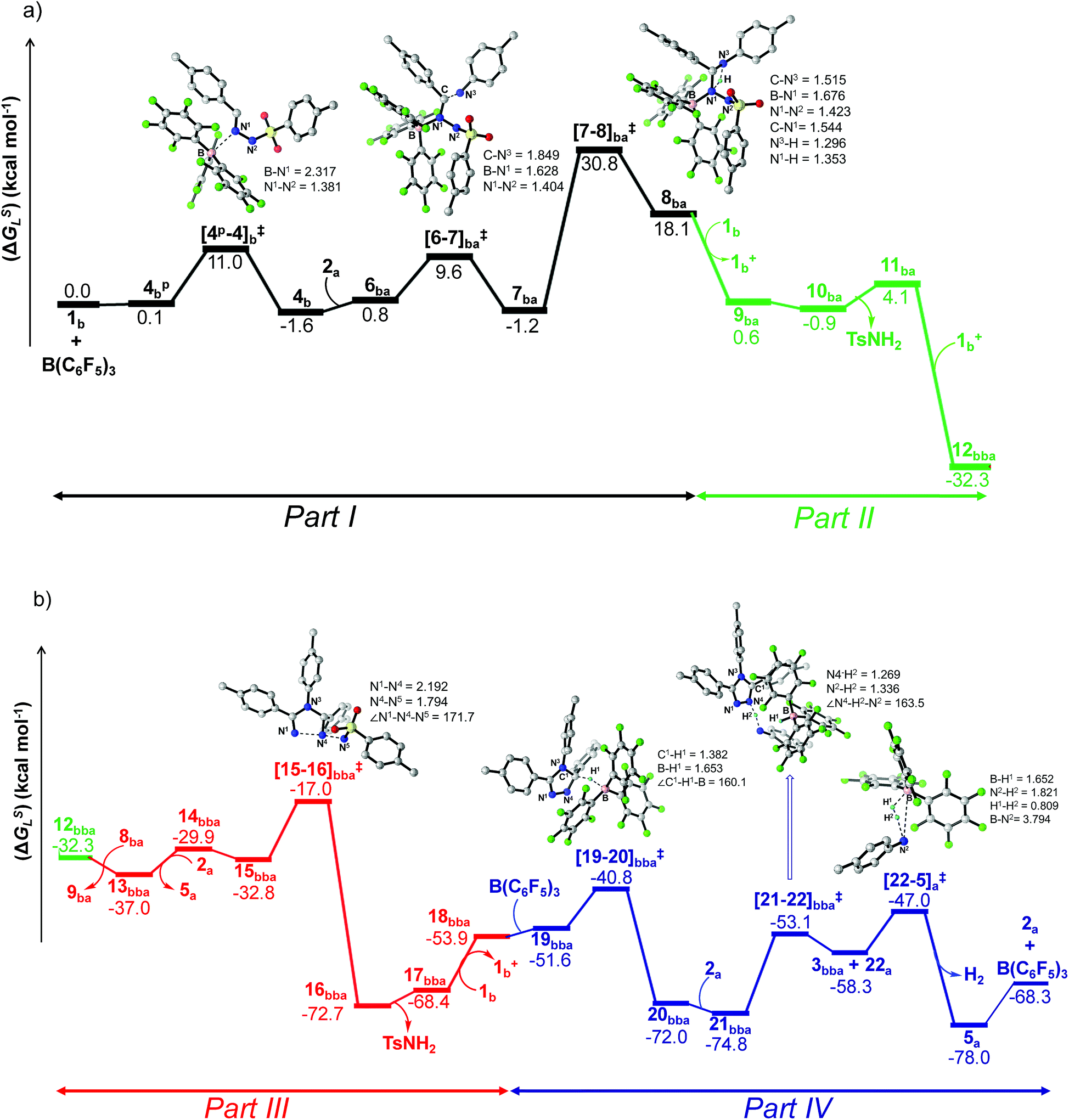 | ||
| Fig. 1 Energy profile and 3D figures of optimized transition states with selected geometrical parameters for 1,2,4-triazole product (3bba) formation at the B3LYP-D3/TZVP//B3LYP/SVP level. The black, green, red and blue colored pathways represent sections (a) Part I and Part II, and (b) Part III and Part IV, respectively (refer Schemes 6–9). Bond distances are in angstroms (Å) and bond angles are in degrees (°). | ||
From here on, the coupling of a second N-tosylhydrazone unit is required for the progress of the reaction. This is accomplished through an initial proton transfer from the C center in 8ba to N4 in 1b. Such a proton abstraction from the tertiary C atom is manifested with N1–N2 bond elongation. This intermolecular proton transfer is clearly favorable (−17.5 kcal mol−1), creating charged species 1b+ and 9ba respectively (Scheme 7), whereas the coordination of 1b instead of proton transfer is highly unfavorable (refer Scheme S18c in the ESI†). In accordance with the experimental findings, KIE measurements suggest the non-involvement of imine C–H bond cleavage in the rate-determining step (Scheme 4a). Though obvious, it is important to note that hydrogen abstraction from electronegative N centers in 8ba is undoubtedly difficult, leading to highly unstable intermediates (Scheme S18d in the ESI†). Close inspection of the structural parameters in 9ba indicates considerable elongation, rather than dissociation of the N1–N2 bond (1.450 Å/2.987 Å = 8ba/9ba) and generation of a partial double bond character in the C–N1 bond (1.568 Å/1.323 Å = 8ba/9ba). The N2 center of the -NHTS unit in 9ba shows significant hydrogen bonding interactions with the H2 atom connected to the N3 center, as evidenced by the N3–H2 (1.088 Å) and N2–H2 (1.622 Å) bond lengths. The dissociation of the TsNH2 fragment is quite evident from 10ba with a shorter N2–H2 distance (1.057 Å) and further elongated N1–N2 distance (3.634 Å). Complete removal of TsNH2 will generate a highly nucleophilic N3 center in 11ba which will immediately coordinate with the preformed cationic intermediate 1b+ to generate substantially stable 12bba (Scheme 7). The coupling of two oppositely charged species is further facilitated by the exothermicity of C–N3 bond formation.
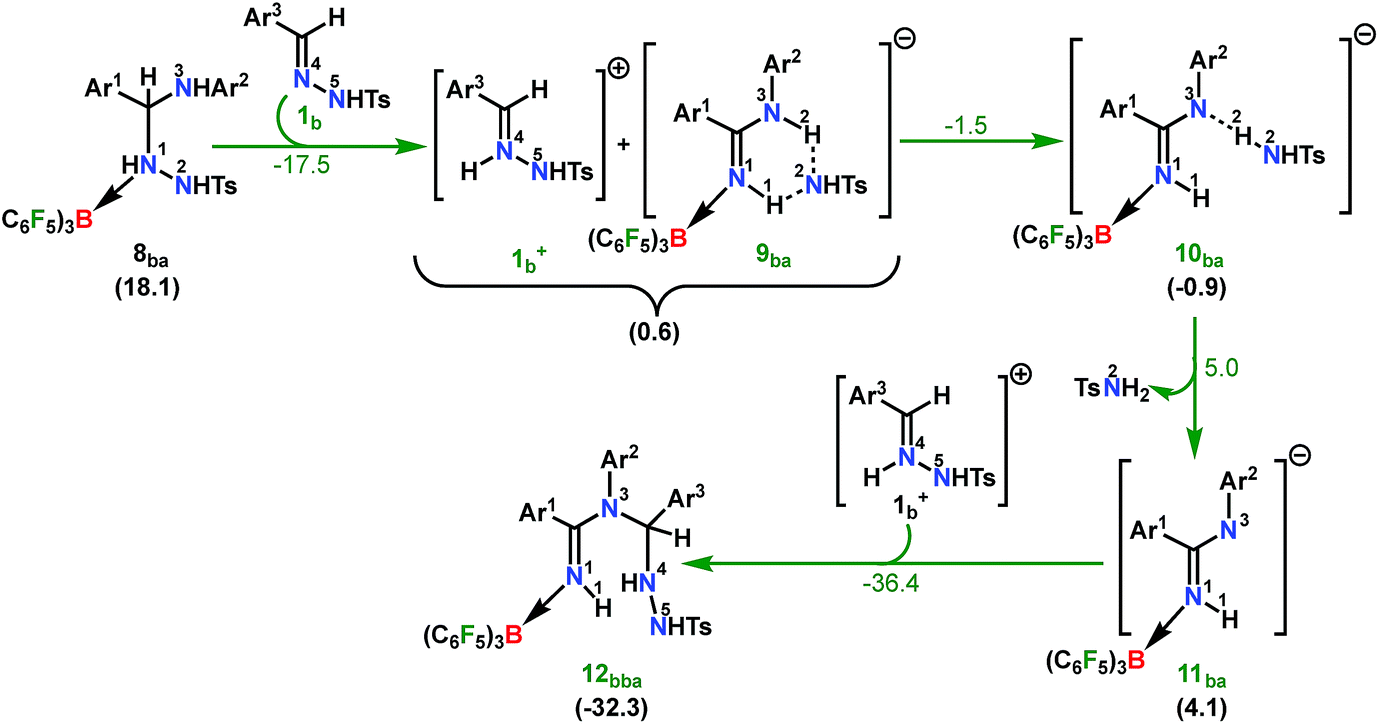 | ||
| Scheme 7 Part II: the reaction pathway for the formation of the intermediate 12bba. For other information refer the caption of Scheme 6. | ||
From 12bba the cyclization step is necessary to generate the triazole product 3bba (Scheme 5). Under these circumstances, it might be conceivable that the liberation of a second TsNH2 unit will facilitate N1–N4 bond formation. Thus, protonation at the N5 center is necessary, similar to the preceding step 8ba → 1b+ + 9ba in Part II (Scheme 7). Unlike in 8ba, the possibility of C–H abstraction in 12bba either as a proton or hydride transfer is unfeasible (refer Scheme S18e in the ESI†). However, the formation of cationic species 13bba after protonation along with anionic 9ba generation is possible, but it is less exothermic than the previous transfer (8ba → 1b+ + 9ba; Schemes 7 and 8). Unfortunately, the addition of protons to any other electronegative center resulted in either high energy intermediates or reaction dead ends (refer Scheme S18f in the ESI†). In order to enhance the nucleophilicity at the N1 center, B(C6F5)3 was uncoordinated in the presence of an aryl amine (2a) to generate notably unstable 14bba (Scheme 8). Subsequent rearrangement to isomeric 15bba provides adequate structural disposition for facile cyclization to proceed. We have explored numerous possibilities for N1–N4 bond formation. However, none of them gave promising alternatives; instead the activation barriers are too high or transition states could not be optimized after numerous attempts (Scheme S18g in the ESI†). The cyclization step involving the transition state [15-16]bba‡ requires an activation barrier of 15.8 kcal mol−1 and it witnesses a progressive removal of the TsNH2 unit.
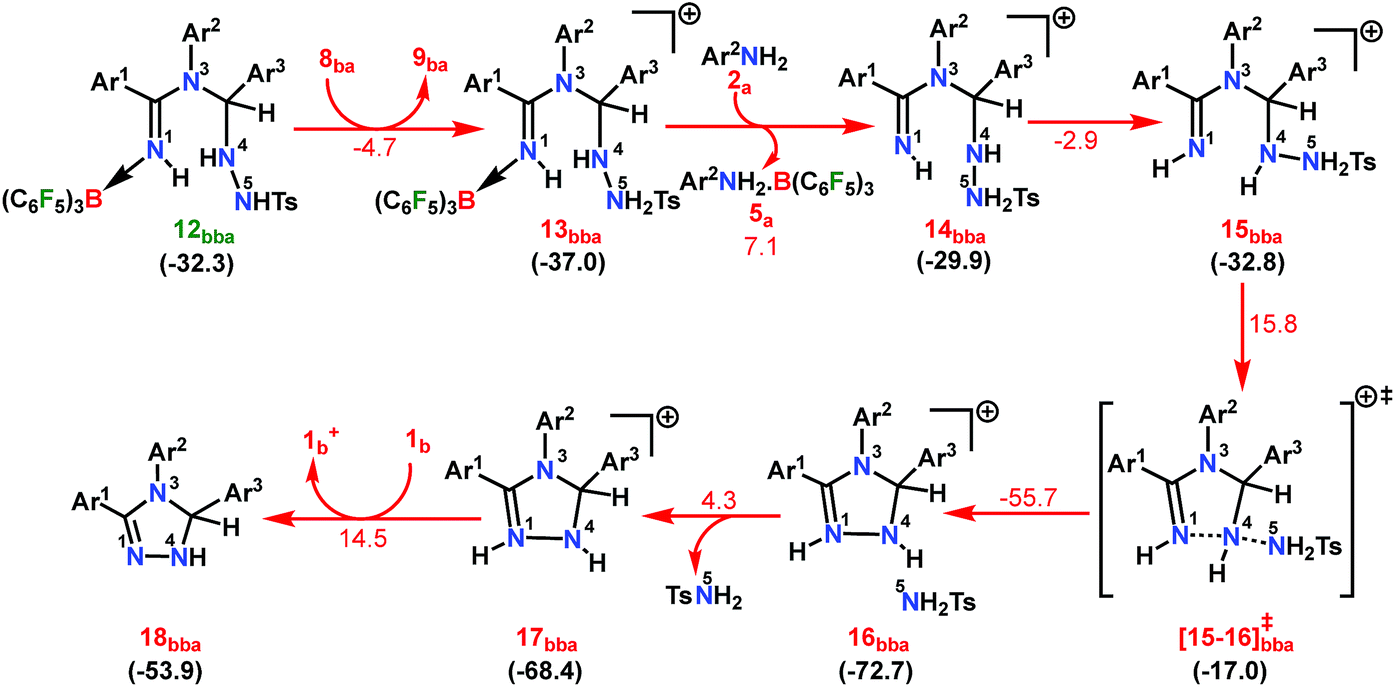 | ||
| Scheme 8 Part III: the reaction pathway for the formation of the intermediate 18bba. For other information refer the caption of Scheme 6. | ||
As expected, the transition vector in [15-16]bba‡ depicts the breaking of the N4-N5 bond (1.794 Å) with the concomitant formation of the N1-N4 bond (2.192 Å). Complete removal of TsNH2 affords the saturated triazole intermediate 17bba, which is 35.6 kcal mol−1 more stable than 15bba. Subsequent deprotonation at the N1 center by 1b will generate the significantly less stable intermediate 18bba.64 This step is facile with an activation barrier of only 1.6 kcal mol−1 (Scheme S19 in the ESI†). The protonated form 1b+ generated can have two fates: it may either participate in the preceding steps reported in Part II (Scheme 7) or can undergo an endergonic exchange of protons to a free amine (2a + 1b+ → 2a+ + 1b; 4.8 kcal mol−1). Generally, the intermediates formed after coupling of the second N-tosylhydrazone moiety are highly stable compared to the starting structures and the driving force for the subsequent reactions is the increasing exergonicity towards product formation (refer Fig. 1b). This statement is supported by high-resolution-mass-spectrometry studies which clearly detect a similar skeleton to 18bba (Fig. S2 in the ESI†). In order to address the positional effect of B(C6F5)3 in the cyclization step, we calculated two isomers in which it coordinates to other N centers (N4 and N5) in 12bba. The resulting intermediates 35bba and 36bba are unstable and did not provide a low energy route to the cyclization step (Scheme S18h in the ESI†). Furthermore, in the absence of B(C6F5)3 the cyclization step leading to 18bba encounters a high transition barrier (38.2 kcal mol−1; Scheme S18i in the ESI†) and thus underscores the significance of B(C6F5)3 in this current transformation.
In the next step, 18bba undergoes dehydrogenative aromatization54,55 to furnish triazole 3bba and an ion pair 22a through B(C6F5)3 mediated hydride abstraction (19bba → 20bba) followed by proton abstraction involving substituted anilines (21bba → 3bba + 22a; Scheme 9). We have calculated intrinsic activation barriers of 13.1 and 21.7 kcal mol−1 for the hydride and proton abstraction steps, respectively. Thereafter, two hydrogen atoms in the ion pair 22a produce a H2 molecule via the four-membered transition state [22-5]a‡ (Scheme 9, Fig. 1b). Liberation of H2 along with the formation of the frustrated Lewis acid–base pair (FLP) adduct 5a is facile with a barrier of only 11.3 kcal mol−1 (Scheme 9). The evolution of H2 was also confirmed by the transfer hydrogenation of styrene (vide supra, Scheme 4c). Finally, maintaining an endoergic equilibrium, the FLP adduct regenerates B(C6F5)3 and substrate 2a.65 In sum, the computational results do have concurrence with the experimental findings, particularly in understanding the dual role of B(C6F5)3 in activating the N-tosylhydrazone towards nucleophilic attack and acceptor-less liberation of H2 with the formation of a FLP (Scheme 9). Additionally, the rate determining step involving intramolecular proton transfer (7ba → 8ba; Δ‡GLS = 32.0 kcal mol−1) can be surmounted at a reaction temperature of 80 °C.66 Optimized geometries of the transition states with selected geometrical parameters along with the energy profiles are shown in Fig. 1.
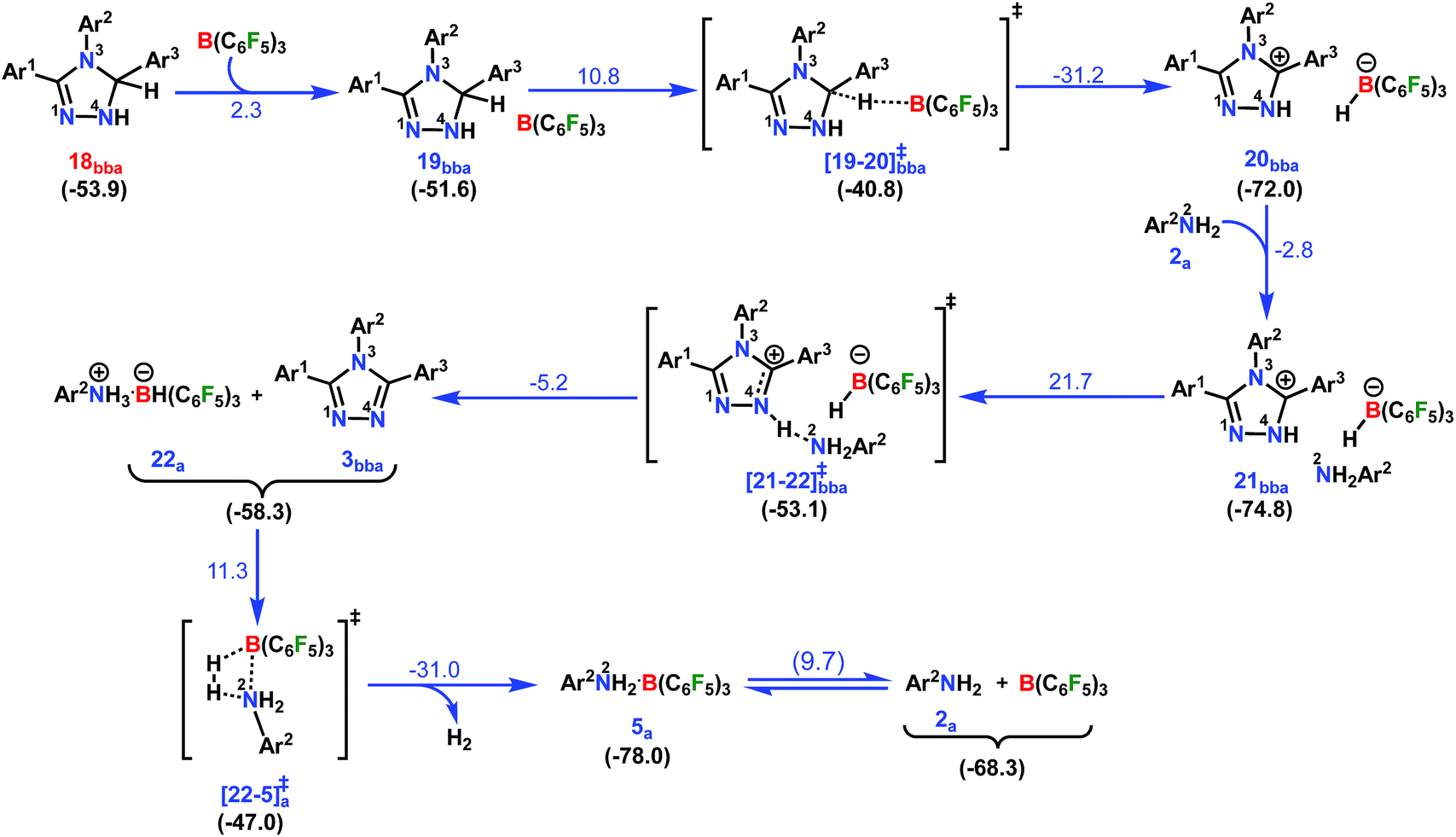 | ||
| Scheme 9 Part IV: the reaction pathway for the formation of the desired product 1,2,4-triazole complex (3bba) and the hydrogen evolution step. For other information refer the caption of Scheme 6. | ||
For unsymmetrical systems
An equimolar mixture of 1a and 1c in the presence of 2a afforded the unsymmetrical triazole 3aca as the major product (77%) compared to the symmetrical counterpart (vide supra; Table 3). To provide reasonable justification for this observation we decided to compare the relative propensity of Lewis acid–base adduct formation of N-tosylhydrazones 1a and 1c with B(C6F5)3 (B(C6F5)3 → 4a/cp → 4a/c). As expected, the formation of 4a is more facile than 4c by ca. 3.0 kcal mol−1 (Fig. S102 in the ESI†). This is in accordance with the experimentally observed equilibrium ratio in Scheme 3b. Eventually the activation barrier for B(C6F5)3 coordination is favorable for the –OMe substituent by 3.6 kcal mol−1 (ΔΔ‡GLS; Fig. S102 in the ESI†). What is more interesting is that the overall energy span for Part I is substantially higher for the –Cl substituent than –OMe (42.5 kcal mol−1vs. 33.3 kcal mol−1), clearly indicating the preference for 1a to undergo B(C6F5)3 assisted intra-molecular proton transfer in a facile manner. Therefore, when 8aa couples with another hydrazone unit, the preferred choice will be the chloro-substituted analogue 1c as most of the 1a will be available in the adduct form 4a. The combination of 8aa with 1c will lead directly to 12aca in a favorable fashion with an exothermicity of −50.6 kcal mol−1 (Fig. S103 in the ESI†). From 12aca, the generation of 16aca requires a barrier of 20.4 kcal mol−1 which is almost similar to the value obtained in the previous case (20.0 kcal mol−1: 12bba → 16bba: Fig. 1b). This barrier is 1.1 kcal mol−1 lower than the symmetrical case (Fig. S103 in the ESI†) further supporting the preference for unsymmetrical triazole (3aca) formation (Fig. S103 in the ESI†). After the formation of the triazole ring in 16aca, which is 71.5 kcal mol−1 more stable than the starting materials, the subsequent B(C6F5)3 assisted dehydrogenation follows an analogous mechanism as outlined before (Scheme 9; Fig. 1b).67Conclusions
In summary, we have demonstrated B(C6F5)3 catalyzed metal-free, one-pot, dehydrogenative-cyclization of hydrazones with anilines to furnish both symmetrical and unsymmetrical 3,4,5-triaryl-1,2,4-triazoles. The isolation of the N-tosylhydrazone-borane adduct is also reported for the first time. Mechanistic experiments and DFT calculations suggest that the B(C6F5)3 catalyst serves a dual role: the activation of the hydrazone for the nucleophilic attack and the formation of an FLP for dehydrogenation. Calculations also reveal that the rate-determining step involves intra-molecular hydrogen transfer between the N-centers after aniline gets bonded to the N-Tosylhydrazone unit. The chemo-selective, step-economical, oxidant-free and mild reaction protocol could give a potential platform for the increasing focus on main-group catalyzed chemical transformation without using transition metal.Computational methods
All computations are performed using Gaussian 0968 and ADF 2018.10369 quantum codes. Geometry optimizations of the saddle points without any symmetry constraints are carried out using the B3LYP hybrid functional70 in conjunction with the SVP basis set71 in the Gaussian 09 program package. Harmonic force constants are computed at the optimized geometries to characterize the nature of the stationary points as minima (Nimg = 0) or transition states (Nimg = 1). Transition states are located by using the linear synchronous transit (LST)72 scan method in which the reaction coordinate was kept fixed at different distances while all other degrees of freedom are relaxed. After the linear transit search, the transition states are optimized by using the default Berny algorithm implemented in the Gaussian 09 code. All transition states are validated by intrinsic reaction coordinate (IRC) calculations. In addition, single point calculations were performed on the B3LYP/SVP optimized structures using the dispersion corrected hybrid functional B3LYP-D373 in conjunction with a large basis set (triple-ζ quality split valence plus polarization, TZVP).74 The effect of solvation (benzene, dielectric constant ε = 2.27) was assessed by a self-consistent reaction field (SCRF) approach, using the conductor-like polarizable continuum model (CPCM).75 Tight wave function convergence criteria and an “ultrafine” (99950) grid were used for the single point calculations. Natural bond orbital (NBO)76 analysis was performed at the B3LYP-D3/TZVP//B3LYP/SVP level using the NBO Version 3.1 program. QTAIM (quantum theory of atoms in molecules) calculations are also performed to characterize the electron distribution around some selected bonds in the chemical species applying Bader's AIM (atoms-in-molecule) theory.77 Furthermore, to gain insight into the bonding scenario in the transition state [4p-4]b‡, EDA (energy decomposition analysis) calculations in conjunction with the NOCV (natural orbital for chemical valence)78 method are undertaken using the ADF 2018.103 package. Implementation and application of the EDA method, which was originally developed by Morokuma79 and later modified by Ziegler and Rauk,80 can be found elsewhere.81–85 The figures provided in the manuscript are generated using ChemDraw Ultra 12.0 and CYLview86 visualization software.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the IISER Kolkata (Start-up grant) for financial support. M. M. G. thanks the SERB (PDF/2017/000028) for the NPDF fellowship. S. De and S. Dutta thank the UGC and CSIR, respectively for senior research fellowships. D. K. acknowledges the funding from the bilateral DST-DFG (INT/FRG/DFG/P-05/2017) scheme and the IISER Kolkata for computational facility. The authors thank Dr. S. Lakhdar (CNRS–ENSI Caen), and Prof. Dr. H. Mayr (LMU Munich) for helpful discussions. Dedicated to Professor Dr. Vinod K. Singh on the occasion of his 60th birthday.Notes and references
- W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC.
- (a) K. Ishihara, N. Hananki and H. Yamamoto, Synlett, 1993, 577–579 CrossRef CAS; (b) K. Ishihara, M. Funahashi, M. Miyata and H. Yamamoto, Synlett, 1994, 963–964 CrossRef CAS; (c) K. Ishihara, N. Hanaki, M. Funahashi, M. Miyata and H. Yamamoto, Bull. Chem. Soc. Jpn., 1995, 68, 1721–1730 CrossRef CAS; (d) G. Erker, Dalton Trans., 2005, 1883–1890 RSC.
- J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 CrossRef CAS.
- R. L. Melen, L. C. Wilkins, B. M. Kariuki, H. Wadepohl, L. H. Gade, A. S. K. Hashmi, D. W. Stephan and M. M. Hansmann, Organometallics, 2015, 34, 4127–4137 CrossRef CAS.
- G. Kehr and G. Erker, Chem. Sci., 2016, 7, 56–65 RSC.
- J. S. McGough, S. M. Butler, I. A. Cade and M. J. Ingleson, Chem. Sci., 2016, 7, 3384–3389 RSC.
- S. Tamke, Z.-W. Qu, N. A. Sitte, U. Floerke, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 4336–4339 CrossRef CAS.
- M. Shang, M. Cao, Q. Wang and M. Wasa, Angew. Chem., Int. Ed., 2017, 56, 13338–13341 CrossRef CAS.
- C. K. Hazra, J. Jeong, H. Kim, M.-H. Baik, S. Park and S. Chang, Angew. Chem., Int. Ed., 2018, 57, 2692–2696 CrossRef CAS.
- M. Shang, J. Z. Chan, M. Cao, Y. Chang, Q. Wang, B. Cook, S. Torker and M. Wasa, J. Am. Chem. Soc., 2018, 140, 10593–10601 CrossRef CAS.
- M. Shang, X. Wang, S. M. Koo, J. Youn, J. Z. Chan, W. Yao, B. T. Hastings and M. Wasa, J. Am. Chem. Soc., 2017, 139, 95–98 CrossRef CAS.
- Y. Hoshimoto, T. Kinoshita, S. Hazra, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2018, 140, 7292–7300 CrossRef CAS.
- D. J. Faizi, A. Issaian, A. J. Davis and S. A. Blum, J. Am. Chem. Soc., 2016, 138, 2126–2129 CrossRef CAS PubMed.
- H. H. San, S.-J. Wang, M. Jiang and X.-Y. Tang, Org. Lett., 2018, 20, 4672–4676 CrossRef CAS PubMed.
- C. K. Hazra, N. Gandhamsetty, S. Park and S. Chang, Nat. Commun., 2016, 7, 13431 CrossRef CAS PubMed.
- P. Smirnov and M. Oestreich, Organometallics, 2016, 35, 2433–2434 CrossRef CAS.
- Y. Han, S. Zhang, J. He and Y. Zhang, J. Am. Chem. Soc., 2017, 139, 7399–7407 CrossRef CAS.
- Y. Ma, L. Zhang, Y. Luo, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2017, 139, 12434–12437 CrossRef CAS.
- K. Chernichenko, A. Madarasz, I. Papai, M. Nieger, M. Leskelae and T. Repo, Nat. Chem., 2013, 5, 718–723 CrossRef CAS.
- M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513–516 CrossRef.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS.
- W. Meng, X. Feng and H. Du, Acc. Chem. Res., 2018, 51, 191–201 CrossRef CAS PubMed.
- A. Fukazawa, H. Yamada and S. Yamaguchi, Angew. Chem., Int. Ed., 2008, 47, 5582–5585 CrossRef CAS.
- C. M. Moemming, G. Kehr, B. Wibbeling, R. Froehlich, B. Schirmer, S. Grimme and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 2414–2417 CrossRef CAS.
- X. Zhao and D. W. Stephan, Chem. Sci., 2012, 3, 2123–2132 RSC.
- M. M. Hansmann, R. L. Melen, F. Rominger, A. S. K. Hashmi and D. W. Stephan, Chem. Commun., 2014, 50, 7243–7245 RSC.
- M. M. Hansmann, R. L. Melen, M. Rudolph, F. Rominger, H. Wadepohl, D. W. Stephan and A. S. K. Hashmi, J. Am. Chem. Soc., 2015, 137, 15469–15477 CrossRef CAS.
- L. C. Wilkins, J. R. Lawson, P. Wieneke, F. Rominger, A. S. K. Hashmi, M. M. Hansmann and R. L. Melen, Chem.–Eur. J., 2016, 22, 14618–14624 CrossRef CAS.
- T. Mahdi and D. W. Stephan, Chem.–Eur. J., 2015, 21, 11134–11142 CrossRef CAS.
- V. Fasano, J. E. Radcliffe and M. J. Ingleson, Organometallics, 2017, 36, 1623–1629 CrossRef CAS.
- Y. Soltani, L. C. Wilkins and R. L. Melen, Angew. Chem., Int. Ed., 2017, 56, 11995–11999 CrossRef CAS.
- K. Yuan and S. Wang, Org. Lett., 2017, 19, 1462–1465 CrossRef CAS.
- G. M. Brittenham, D. G. Nathan, N. F. Olivieri, M. J. Pippard and D. J. Weatherall, Lancet, 2003, 361, 183–184 CrossRef CAS.
- R. Tokala, S. Bale, I. P. Janrao, A. Vennela, N. P. Kumar, K. R. Senwar, C. Godugu and N. Shankaraiah, Bioorg. Med. Chem. Lett., 2018, 28, 1919–1924 CrossRef CAS.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- W. Song, L. Shi, L. Gao, P. Hu, H. Mu, Z. Xia, J. Huang and J. Su, ACS Appl. Mater. Interfaces, 2018, 10, 5714–5722 CrossRef CAS.
- H. Oka, R. V. Joshi, J. Tanabe, S. Lahiri, D. Vashi and P. Ghogale, WO Pat., WO2006114377A1, 2006.
- H. Oka, R. V. Joshi, J. Tanabe, S. Lahiri, D. Vashi and P. Ghogale, US Pat., US9113536B2, 2015.
- B. K. Barman, M. M. Guru, G. K. Panda, B. Maji and R. K. Vijayaraghavan, Chem. Commun., 2019, 55, 4643–4646 RSC.
- Y. Naito, F. Akahoshi, S. Takeda, T. Okada, M. Kajii, H. Nishimura, M. Sugiura, C. Fukaya and Y. Kagitani, J. Med. Chem., 1996, 39, 3019–3029 CrossRef CAS.
- A. Moulin, M. Bibian, A.-L. Blayo, S. El Habnouni, J. Martinez and J.-A. Fehrentz, Chem. Rev., 2010, 110, 1809–1827 CrossRef CAS.
- S. Ueda and H. Nagasawa, J. Am. Chem. Soc., 2009, 131, 15080–15081 CrossRef CAS.
- S. T. Staben and N. Blaquiere, Angew. Chem., Int. Ed., 2010, 49, 325–328 CrossRef CAS.
- H. Wang, Y. Ren, K. Wang, Y. Man, Y. Xiang, N. Li and B. Tang, Chem. Commun., 2017, 53, 9644–9647 RSC.
- Z. Ye, M. Ding, Y. Wu, Y. Li, W. Hua and F. Zhang, Green Chem., 2018, 20, 1732–1737 RSC.
- C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2013, 52, 4256–4259 CrossRef CAS.
- K. L. Bamford, L. E. Longobardi, L. Liu, S. Grimme and D. W. Stephan, Dalton Trans., 2017, 46, 5308–5319 RSC.
- T. C. Johnstone, G. N. J. H. Wee and D. W. Stephan, Angew. Chem., Int. Ed., 2018, 57, 5881–5884 CrossRef CAS.
- C. Tang, Q. Liang, A. R. Jupp, T. C. Johnstone, R. C. Neu, D. Song, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 16588–16592 CrossRef CAS.
- R. L. Melen, Angew. Chem., Int. Ed., 2018, 57, 880–882 CrossRef CAS.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 249 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed.
- Z. Lu, L. Schweighauser, H. Hausmann and H. A. Wegner, Angew. Chem., Int. Ed., 2015, 54, 15556–15559 CrossRef CAS.
- M. Kojima and M. Kanai, Angew. Chem., Int. Ed., 2016, 55, 12224–12227 CrossRef CAS.
- (a) A. F. G. Maier, S. Tussing, T. Schneider, U. Floerke, Z.-W. Qu, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 12219–12223 CrossRef CAS; (b) Z.-J. Cai, X.-M. Lu, Y. Zi, C. Yang, L.-J. Shen, J. Li, S.-Y. Wang and S.-J. Ji, Org. Lett., 2014, 16, 5108–5111 CrossRef CAS; (c) Z. Chen, Q. Yan, Z. Liu and Y. Zhang, Chem.–Eur. J., 2014, 20, 17635–17639 CrossRef CAS; (d) J.-P. Wan, D. Hu, Y. Liu and S. Sheng, ChemCatChem, 2015, 7, 901–903 CrossRef CAS; (e) J.-P. Wan, S. Cao and Y. Liu, Org. Lett., 2016, 18, 6034–6037 CrossRef CAS; (f) S. Panda, P. Maity and D. Manna, Org. Lett., 2017, 19, 1534–1537 CrossRef CAS.
- Z. Mo, A. Rit, J. Campos, E. L. Kolychev and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 3306–3309 CrossRef CAS.
- M. K. Barman, S. Waiba and B. Maji, Angew. Chem., Int. Ed., 2018, 57, 9126–9130 CrossRef CAS.
- CCDC 1866759 contains the supplementary crystallographic data for this paper.
- S. Keess and M. Oestreich, Chem. Sci., 2017, 8, 4688–4695 RSC.
- Q. Wang, J. Chen, X. Feng and H. Du, Org. Biomol. Chem., 2018, 16, 1448–1451 RSC.
- T. Mondal, S. Dutta, S. De, D. Thirumalai and D. Koley, J. Phys. Chem. A, 2019, 123, 565–581 CrossRef CAS.
- S. Dutta, B. Maity, D. Thirumalai and D. Koley, Inorg. Chem., 2018, 57, 3993–4008 CrossRef CAS.
- (a) D. Roy and R. B. Sunoj, Org. Lett., 2007, 9, 4873–4876 CrossRef CAS PubMed; (b) K. Ando, J. Org. Chem., 2010, 75, 8516–8521 CrossRef CAS; (c) R. Sharma, M. Thorley, J. P. McNamara, C. I. F. Watt and N. A. Burton, Phys. Chem. Chem. Phys., 2008, 10, 2475–2487 RSC.
- Deprotonation from the N4 center will afford highly unstable intermediates (ΔGLS = 37.6 kcal mol−1) compared to 17bba.
- L. Greb, S. Tamke and J. Paradies, Chem. Commun., 2014, 50, 2318–2320 RSC.
- Y. Ma, S.-J. Lou, G. Luo, G. Zhan, M. Nishiura, Y. Luo and Z. Hou, Angew. Chem., Int. Ed., 2018, 57, 15222–15226 CrossRef CAS PubMed.
- The energy span in Part IV (18aca → 20aca) is reduced by ca. 4.0 kcal mol−1 when intermediates for the step 17aca → 18aca were optimized in benzene continuum (B3LYP/SVP(CPCM) level).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS; (b) GUI 2013, SCM, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. Schäfer, H. Horn and R. J. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- T. A. Helgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225–232 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, J. Chem. Phys., 2010, 132, 154104–154122 CrossRef.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- (a) E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, University of Wisconsin, Madison, WI, 2001 Search PubMed; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- (a) M. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576–6580 CrossRef CAS; (b) M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS.
- (a) K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS; (b) K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- (a) T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1755–1759 CrossRef CAS; (b) T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- T. Mondal, S. Dutta, S. De and D. Koley, J. Org. Chem., 2019, 84, 257–272 CrossRef CAS.
- S. Dutta, T. Mondal, S. De, K. Rudra and D. Koley, Inorg. Chim. Acta, 2019, 485, 162–172 CrossRef CAS.
- G. Frenking, K. Wichmann, N. Fröhlich, C. Loschen, M. Lein, J. Frunzke and V. M. Rayon, Coord. Chem. Rev., 2003, 238–239, 55–82 CrossRef CAS.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS.
- T. Mondal, S. De and D. Koley, Inorg. Chem., 2017, 56, 10633–10643 CrossRef CAS.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, see http://www.cylview.org Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1866759. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02492a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |