
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst shuttling enabled by a thermoresponsive polymeric ligand: facilitating efficient cross-couplings with continuously recyclable ppm levels of palladium†
Erfei
Wang
and
Mao
Chen
 *
*
State Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200433, China. E-mail: chenmao@fudan.edu.cn; Web: http://chenmaofudan.wixsite.com/polymao
First published on 23rd July 2019
Abstract
A polymeric monophosphine ligand WePhos has been synthesized and complexed with palladium(II) acetate [Pd(OAc)2] to generate a thermoresponsive pre-catalyst that can shuttle between water and organic phases, with the change being regulated by temperature. The structure of the polymeric ligand was confirmed with matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry and size-exclusion chromatography (SEC) analysis, as well as nuclear magnetic resonance (NMR) measurements. This polymeric metal complex enables highly efficient Pd-catalyzed cross-couplings and tandem reactions using 50 to 500 ppm palladium, and this can facilitate reactions that are tolerant to a broad spectrum of (hetero)aryl substrates and functional groups, as demonstrated with 73 examples with up to 99% isolated yields. Notably, 97% Pd remained in the aqueous phase after 10 runs of catalyst recycling experiments, as determined via inductively coupled plasma-atomic emission spectrometry (ICP-AES) measurements, indicating highly efficient catalyst transfer. Furthermore, a continuous catalyst recycling approach has been successfully developed based on flow chemistry in combination with the catalyst shuttling behavior, allowing Suzuki–Miyaura couplings to be conducted at gram-scales with as little as 10 ppm Pd loading. Given the significance of transition-metal catalyzed cross-coupling and increasing interest in sustainable chemistry, this work is an important step towards the development of a responsive catalyst, in addition to having high activity, by tuning the structures of the ligands using polymer science.
Introduction
Transition-metal catalyzed cross-coupling reactions have become indispensable tools in organic synthesis. The palladium (Pd)-catalyzed Suzuki–Miyaura (SM) coupling is one of the most heavily used reactions for C–C bond formation,1–3 and has revolutionized synthetic approaches for pharmaceuticals,4 agrochemicals,5 polymer materials6 and so on.7 Notwithstanding its extraordinary advances, the cost of Pd and its separation, as well as the sustainability of this chemistry are almost always concerns when using SM coupling, especially in process research and development; these are also concerns with other precious metal catalyzed reactions.8–10 Consequently, methods featuring low Pd loading,11–16 recyclable catalysts17,18 and aqueous conditions19–21 have received increasing attention. For example, Lipshutz and co-workers disclosed that the ppm levels of Pd contained in the iron(III) chloride (FeCl3) salt catalyzed aqueous SM coupling with a dialkylphosphine ligand.11 They also reported that SM couplings could be facilitated using amphiphilic molecules in water (H2O).12,22,23 Based on previous achievements of using electron-rich phosphine ligands to promote SM couplings,24–31 Carrow and co-workers revealed that a tri(adamantyl)phosphine–Pd complex catalyzed a process with low Pd loadings in an organic solvent.13 Whereas polymeric materials loaded with Pd are easily recyclable for homo/heterogeneous SM couplings,32–42 in comparison to small molecule catalysts, polymeric catalysts generally exhibit: (a) limited functional group tolerance, (b) decreased reactivity toward deactivated aryl chlorides, and (c) unsatisfactory compatibility with heterocycles.43 Furthermore, tandem/iterative catalysis based on chemoselective SM coupling44–46 has rarely been realized with polymeric catalysts.In this research, a novel polymeric pre-catalyst was synthesized, which was composed of methoxy poly(ethylene glycol) (PEG) linked dicyclohexylphosphine ligand (WePhos) and Pd, which enables a general, efficient and low Pd loading SM coupling via a rapid catalyst transfer between aqueous and toluene phases (Scheme 1). This polymeric complex catalyzed reaction is tolerant of various functional groups as well as (hetero)aromatic rings, and is realized in tandem coupling with high chemoselectivity. Furthermore, taking advantage of the thermoresponsive catalyst shuttling, a continuous catalyst-recycling approach based on flow chemistry was developed to streamline scalable SM couplings using down to 10 ppm of Pd.
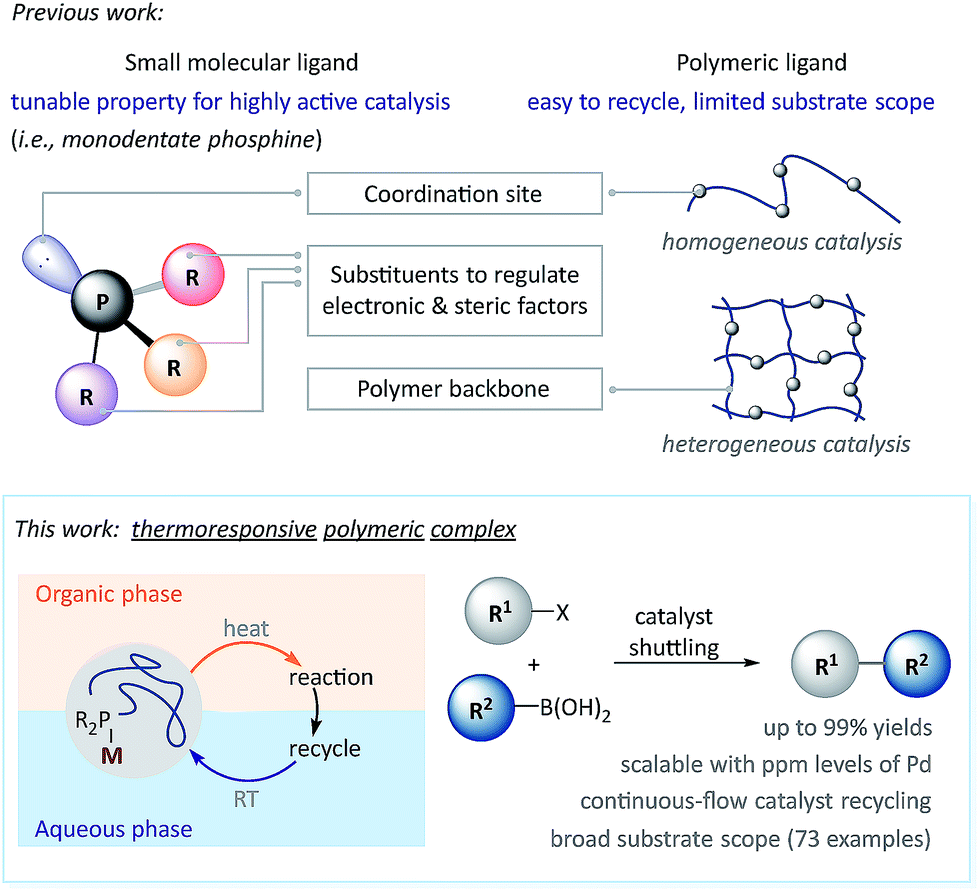 | ||
| Scheme 1 Ligand design in transition-metal catalyzed cross-coupling. | ||
The lower critical solution temperature (LCST) behaviour of PEG is known in polymer science,47–50 with PEGylated materials exhibiting dramatically decreased solubility in water upon heating above the LCST. It was thought that by introducing a PEGylated phosphine ligand, the corresponding Pd complex would display a thermoresponsive solubility in a biphasic system, thereby promoting a reaction in the organic phase and facilitating catalyst recycling simply by phase separation.
Results and discussion
At the beginning of the investigation, to confirm and visualize the shuttling effect, a yellow polymeric compound 1 was synthesized with PEG5000 (number average molecular weight, Mn ∼ 5000 g mol−1) and carboxyferrocene (Fig. 1). When 1 is dissolved in a mixture of water and toluene at 25 °C, only the water phase shows yellow coloration, however, upon heating to 90 °C, the toluene phase turns yellow within 30 s. The whole process is rapid and completely reversible, clearly exhibiting the thermoresponsive shuttling of a PEG-supported compound between water and organic phases. | ||
| Fig. 1 Optical images of the migration behavior of compound 1 between water and toluene phases in response to temperature. | ||
Subsequently, the new ligand WePhos50005a was synthesized using a synthetic route illustrated in Scheme 2. Starting from 4-bromophenol 2, a high overall yield of 83% was obtained for ligand 5a. This compound was air stable, and was characterized using matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry. As shown in Fig. 2a, a single set of peaks is observed in the MALDI-TOF mass spectrum, and each peak is separated by the molar mass of a single repeating unit in PEG (m/z = 44.05, Fig. 2b). The absolute m/z value was consistent with the calculated molecular weight of 5a. The y-intercept of the best-fit trend line of m/z versus the number of repeating units indicated the molecular weight of the chain-end group of 5a (Fig. 2c), which was consistent with the expected value. The proton-nuclear magnetic resonance (1H-NMR) analysis51 also confirmed the chemical structure of WePhos5000. Mixing 5a with palladium(II) acetate [Pd(OAc)2] in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, quantitatively generated pre-catalyst 6a. A peak shift from 1.24 to 45.21 ppm in the 31P-NMR spectra revealed the successful coordination of phosphine to the PdII center (Fig. 2d).52,53
:1 ratio, quantitatively generated pre-catalyst 6a. A peak shift from 1.24 to 45.21 ppm in the 31P-NMR spectra revealed the successful coordination of phosphine to the PdII center (Fig. 2d).52,53
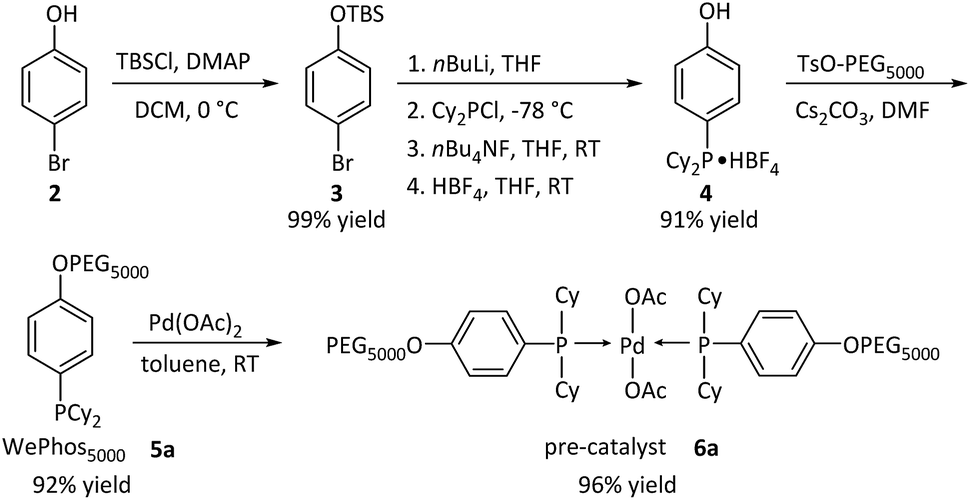 | ||
| Scheme 2 Synthetic pathway of WePhos50005a and pre-catalyst 6a; Cy = cyclohexyl group, AcO = acetate group. | ||
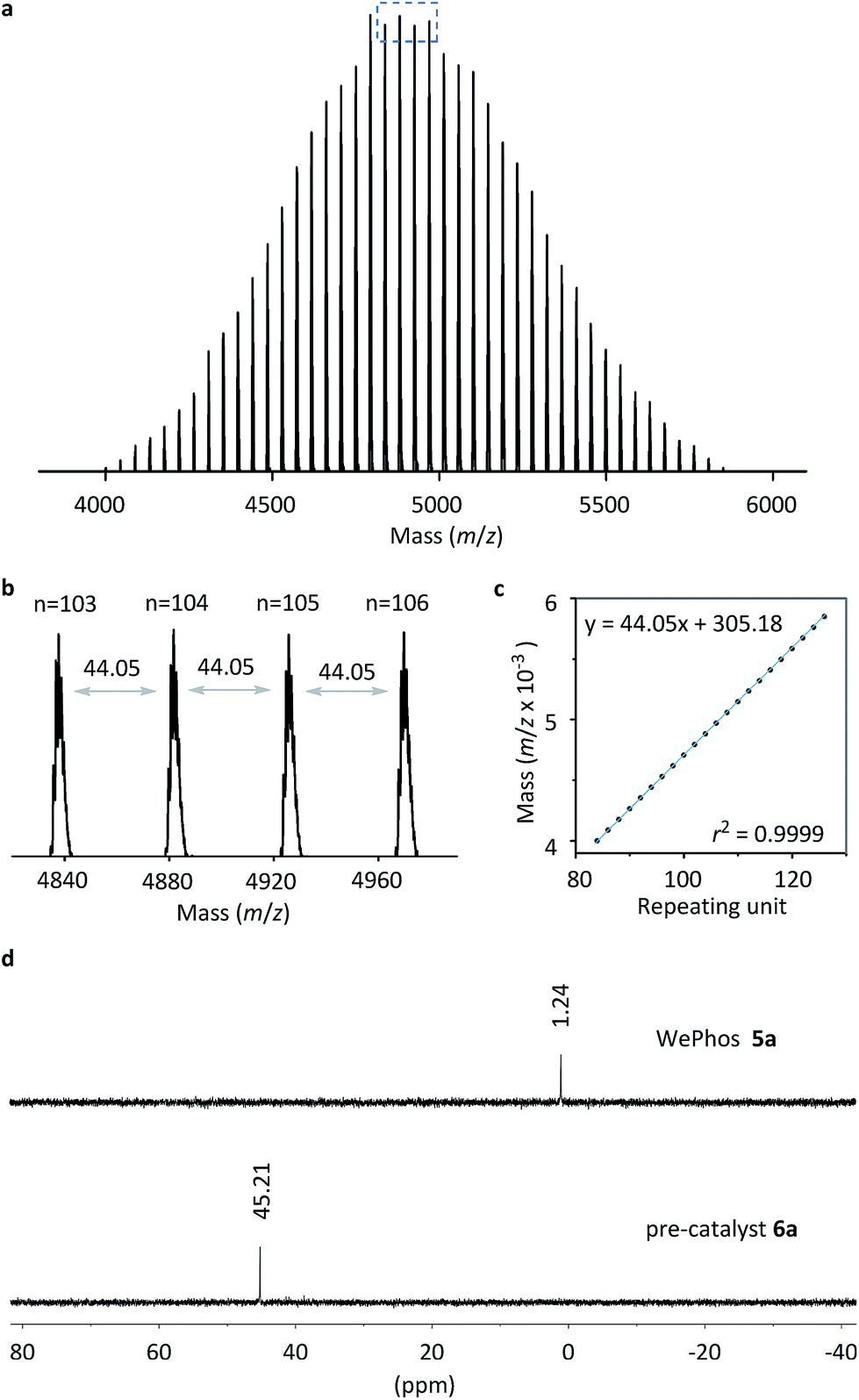 | ||
| Fig. 2 (a) MALDI-TOF mass spectrum of WePhos50005a. (b) Magnified MALDI-TOF mass spectrum of the blue box in (a). (c) m/z versus the number of repeating units for the MALDI-TOF mass spectrum of 5a. (d) 31P-NMR spectra of 5a and 6a. | ||
Using the same synthetic method, a series of WePhos ligands were prepared using PEGs of different molecular weights (Mn ∼ 750 g mol−1 for 5b, Mn ∼ 2000 g mol−1 for 5c, Mn ∼ 10000 g mol−1 for 5d). When these ligands were characterized using size-exclusion chromatography (SEC) (Fig. 3), the SEC profiles showed clear shifts from high to low retention times with no low molar mass tailing (molecular weight distribution, weight average molecular weight/number average molecular weight (Mw/Mn) = 1.05–1.09), which indicated the successful preparation of the polymeric ligands with a precise structure.
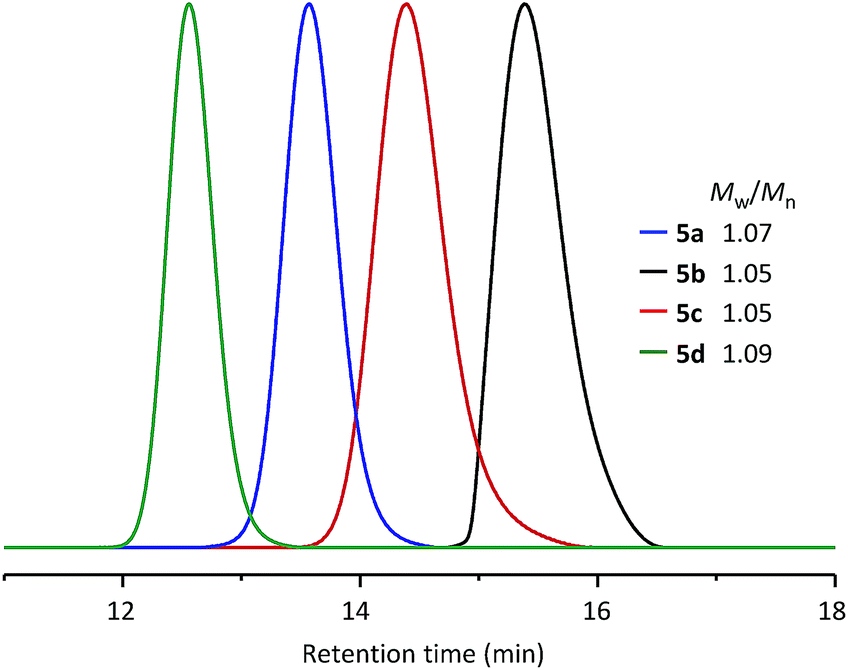 | ||
| Fig. 3 SEC results for WePhos ligands of different molecular weights. | ||
Next, Pd-complexes of ligands from 5a to 5d were used in the SM cross-coupling of bromobenzene and 4-methoxybenzeneboronic acid with potassium carbonate (K2CO3) as the base in water/toluene (v/v = 4/1) at 90 °C. As summarized in Fig. 4a, when the pre-catalyst 6a was used, the reaction gave the highest yield, in a reaction time of 20 min, determined using gas chromatography (GC, yield > 99%).54 After cooling down the reaction mixture with 6a to room temperature (RT), the metal-complex catalyst was transferred to the aqueous layer and the 4-methoxybiphenyl product was left in the toluene. Using a simple phase separation, the aqueous layer was collected and reused in the next SM cross-coupling reaction. The water phase containing the catalyst was recycled 10 times in this way and there was no clear decrease in GC yields (Fig. 4b). The final aqueous phase was analysed using inductively coupled plasma-atomic emission spectrometry (ICP-AES), which indicated that 97% of the Pd remained in the aqueous phase, which highlighted the robust catalyst shuttling facilitated by the thermoresponsive polymeric ligand.
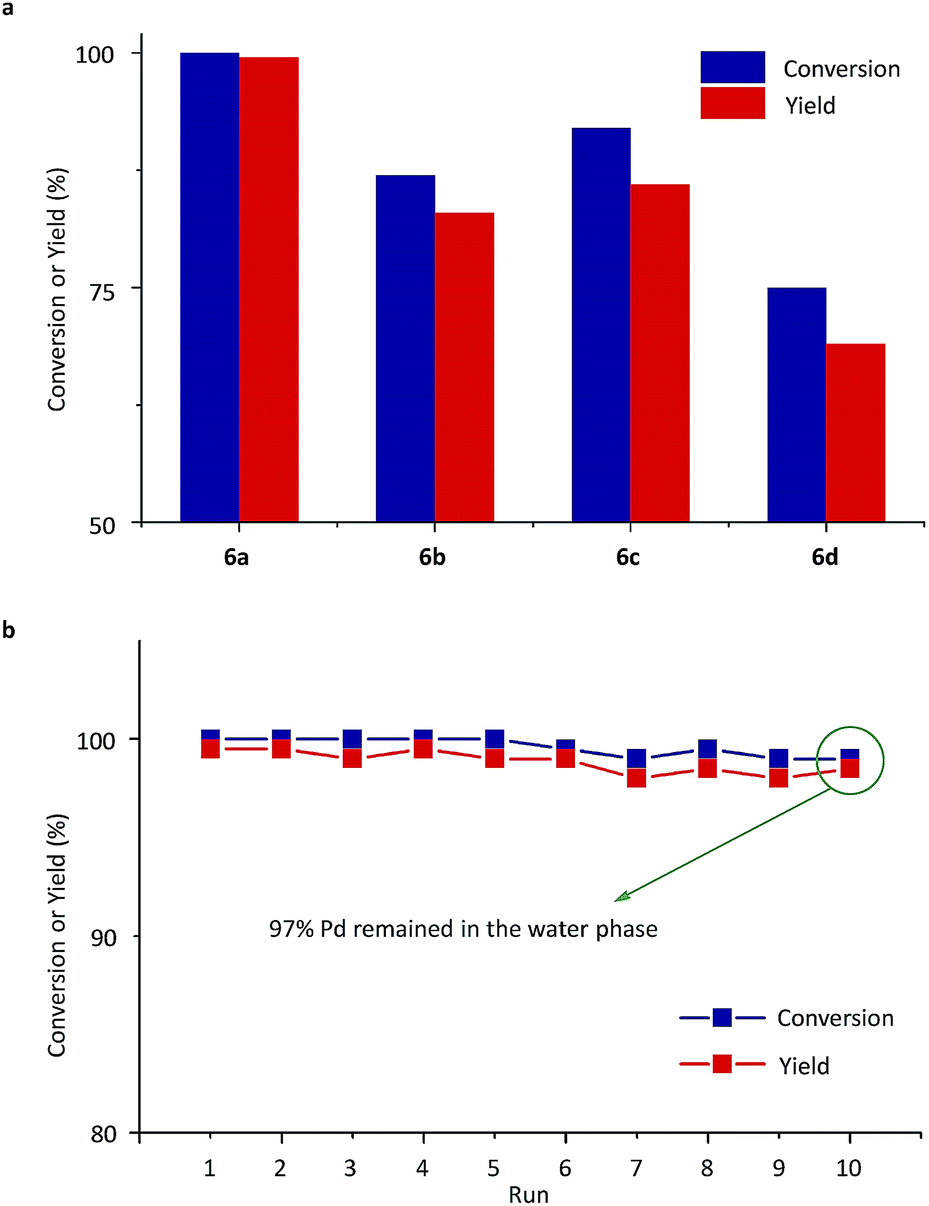 | ||
| Fig. 4 (a) Conversions and yields of SM cross-couplings between bromobenzene and 4-methoxybenzeneboronic acid with pre-catalysts 6a, 6b, 6c and 6d. (b) Conversions and yields of target product (4-methoxybiphenyl) for the catalyst recycling experiments with 6a. Conversions and yields were determined using GC analysis. | ||
With the optimized reaction conditions resolved, the substrate scope of the (hetero)aryl bromides was investigated (Scheme 3). All the substrates underwent complete conversion in 30 min to 2 h, giving products with good to excellent isolated yields with 50 to 500 ppm of catalyst. The SM reactions catalyzed with polymeric complexes could be successfully carried out with para-, meta-, and ortho-substituted aryl compounds and fused aryl compounds. Electron-deficient, electron-neutral and electron-rich aryl halides were all suitable substrates. The reaction conditions were compatible with a broad scope of functional groups including ester (9), amide (10), aldehyde (11), acetyl (12–14), and nitrile (14), as well as unprotected hydroxyl (13–18), amino (19) and carboxylic acid (20) groups. As is already known, heterocycles are core units of many important pharmaceuticals, organic materials and natural products.55 However, heterocycles are likely to coordinate with transition metals, thus representing a challenging class of starting materials for metal-catalyzed reactions with low catalyst dosages.56 With this method, a variety of heterocyclic compounds including pyridine (24–28), quinoline (29), carbazole (27), dibenzofuran (28), indole (30), pyrimidine (31–32), pyrazine (33), pyrazole (34), imidazo[1,2-a]pyrazine (35), thiophene (36–40), benzothiophene (41), and furan (42), could be used to generate corresponding products, representing the broadest substrate scope obtained with a polymeric catalyst as far as is known.
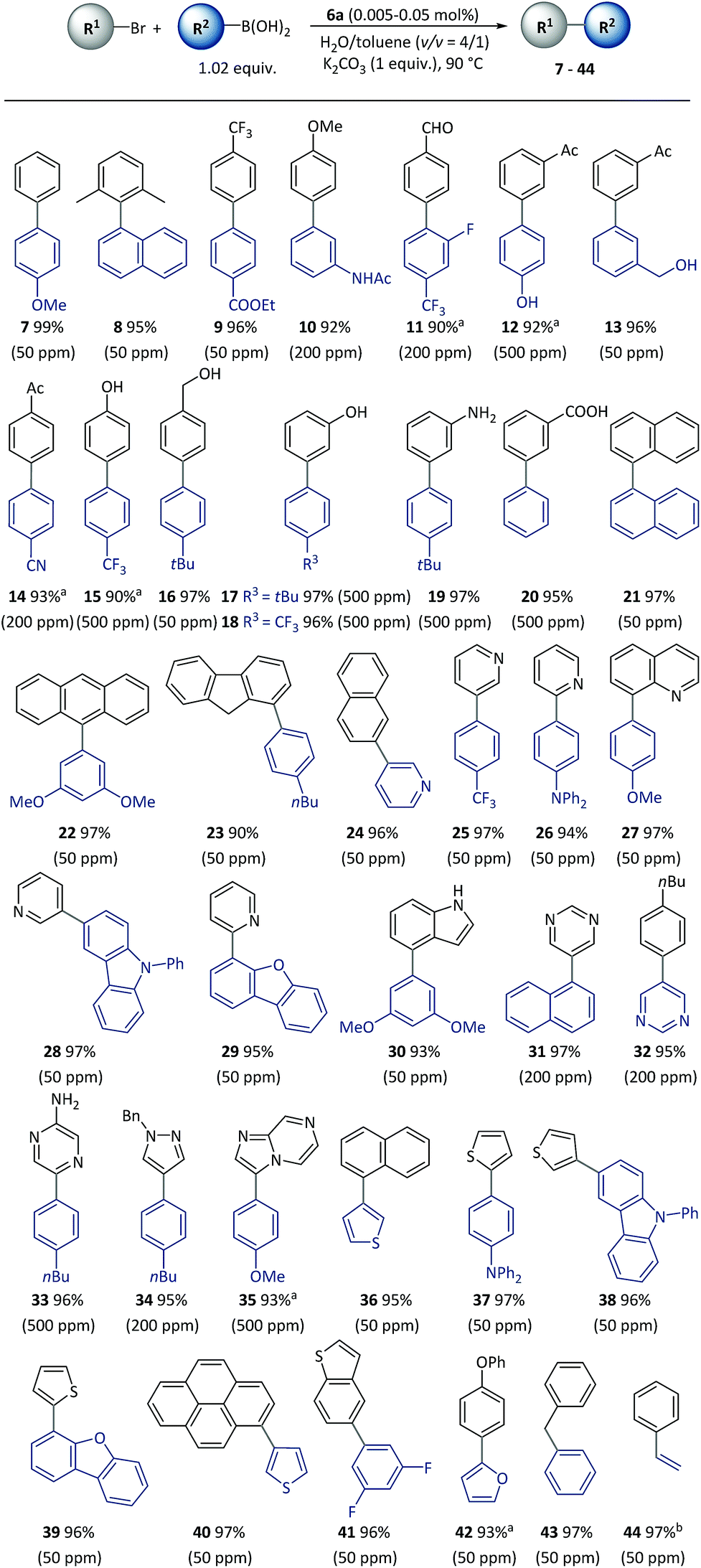 | ||
| Scheme 3 Cross-coupling of (hetero)aryl bromides using pre-catalyst 6a. Conditions: R1-Br (0.5 mmol), R2-B(OH)2 (0.51 mmol), 6a (50–500 ppm), K2CO3 (0.5 mmol), H2O (0.8 mL), toluene (0.2 mL), 90 °C, (30 min). Catalyst loadings are listed in parentheses. Isolated yields are based on aryl bromides. a2 h. bR2-BPin (boronic acid pinacol ester) was used instead of R2-B(OH)2. | ||
Furthermore, the SM cross-coupling using pre-catalyst 6a was found to be applicable to (hetero)aryl chlorides with a wide substrate scope as shown in Scheme 4. Aryl chlorides with a substituent group at different positions could effectively react with aryl boronic acids (45–48) to give biaryl compounds with yields of 94–98%. Functional groups [for example, acetyl (47), unprotected amino (48) and hydroxyl (49)] and heterocycles including pyridine (51–52), pyrimidine (53), thiophene (54–58) and dibenzofuran (58) underwent a smooth reaction with a low (50–500 ppm) catalyst loading, and there were complete conversions in 2–5 h.
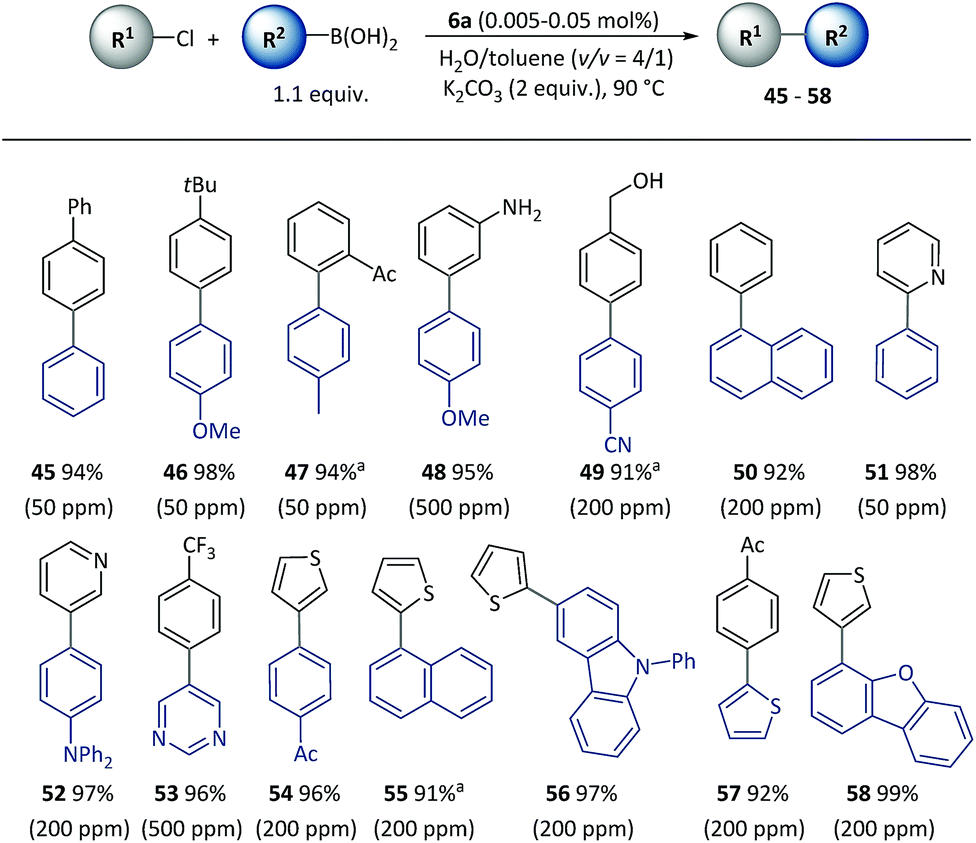 | ||
| Scheme 4 Cross-coupling of (hetero)aryl chlorides with pre-catalyst 6a. Conditions: R1-X (0.5 mmol), R2-B(OH)2 (0.55 mmol), 6a (50–500 ppm), K2CO3 (1.0 mmol), H2O (0.8 mL), toluene (0.2 mL), 90 °C, 2 h. Catalyst loadings are listed in parentheses. Isolated yields are based on aryl chlorides. a5 h. | ||
Given the different reactivities of C–Br and C–Cl bonds demonstrated in Pd catalyzed cross-coupling with many small molecule catalysts, the chemical selectivity of polymeric catalyst 6a was next evaluated, as shown in Scheme 5. Excellent selectivity was observed in all the cases examined. A variety of functionalized bi(hetero)aryl were produced with intact aryl chlorides groups (59–65) at above 90% isolated yields, which could be useful in further metal-catalyzed transformations. It is important to note that the chemoselectivity was not affected by steric or electronic factors with use of the polymeric catalyst.
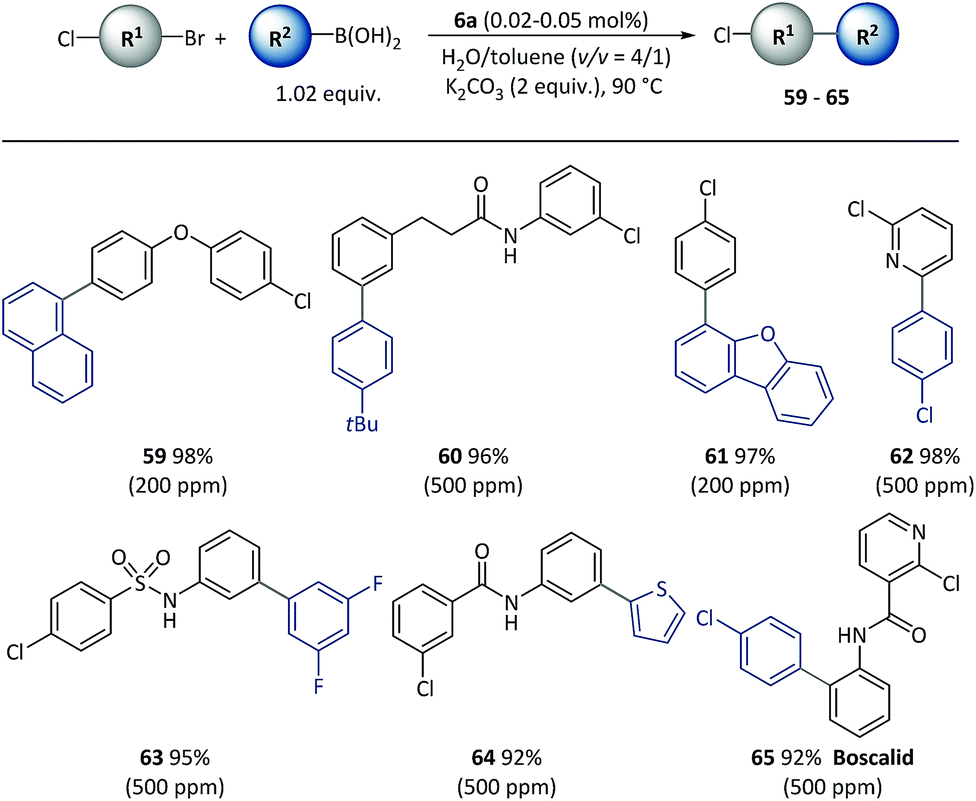 | ||
| Scheme 5 The chemoselective cross-coupling of conjunctive dihalide compounds using pre-catalyst 6a. Conditions: Cl-R1-Br (0.5 mmol), R2-B(OH)2 (0.51 mmol), 6a (50–500 ppm), K2CO3 (1.0 mmol), H2O (0.8 mL), toluene (0.2 mL), 90 °C, 2 h. Catalyst loadings are listed in parentheses. Isolated yields are based on conjunctive dihalide compounds. | ||
Tandem SM coupling based on chemoselective reaction facilitated modular and rapid synthesis of diversified molecules for applications such as material screening and drug discovery.44–46,57–59 Next, the polymeric catalyst was used in tandem SM coupling based on the different reactivities of electrophiles (Scheme 6). In these reactions, after the first step arylation via C–Br bond cleavage, boronic acids were subsequently added into the reaction mixtures without additional catalyst and base. Excellent yields (91–96%) were obtained for all examples after two-step syntheses in the presence of heterocycles [for example, thiophene (66), pyrimidine (67), pyridine (68) and furan (69)] and functional groups [for example, sulfonamide (69), amide (70) and ester (71, 72)], demonstrating the high efficiency achieved with this method.
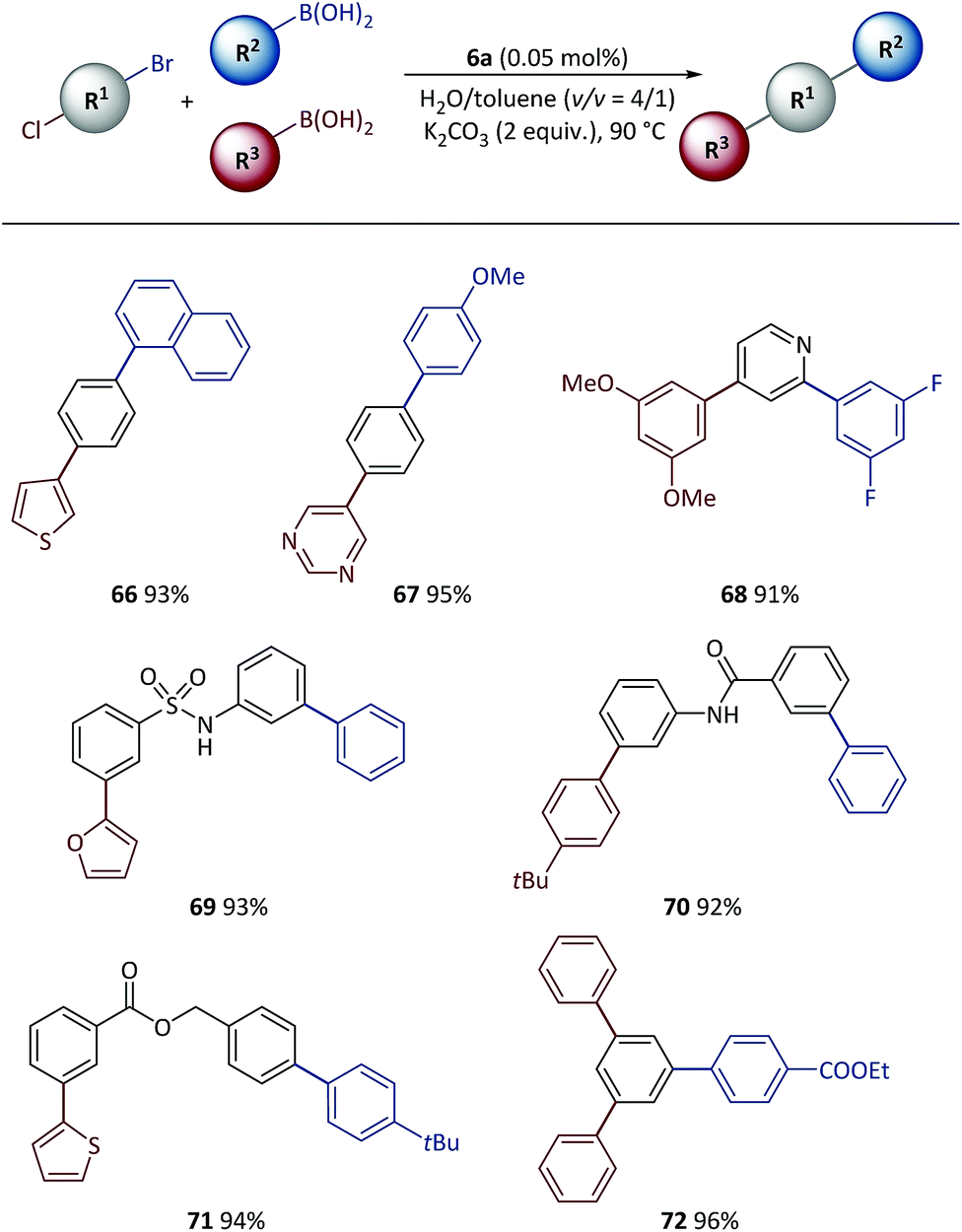 | ||
| Scheme 6 Tandem SM cross-coupling with pre-catalyst 6a. Conditions: R1-X (0.5 mmol), R2-B(OH)2 (0.51 mmol), R3-B(OH)2 (0.55 mmol), 6a (500 ppm), K2CO3 (1.0 mmol), H2O (0.8 mL), toluene (0.2 mL), 90 °C. Isolated yields based on conjunctive dihalide compounds. | ||
On the basis of the thermoresponsive catalyst migration, it was anticipated that a continuous-flow synthetic strategy60–67 would be practical for scaling-up the SM couplings at ultralow Pd loadings by continuous recycling of catalyst. To realize this idea, a continuous-flow setup was assembled (Fig. 5 and Scheme 7) to synthesize 73, a typical unit with aggregation-induced emission (AIE) behavior, which has been used in high-tech applications such as optoelectronic materials and biomedical sensors.68
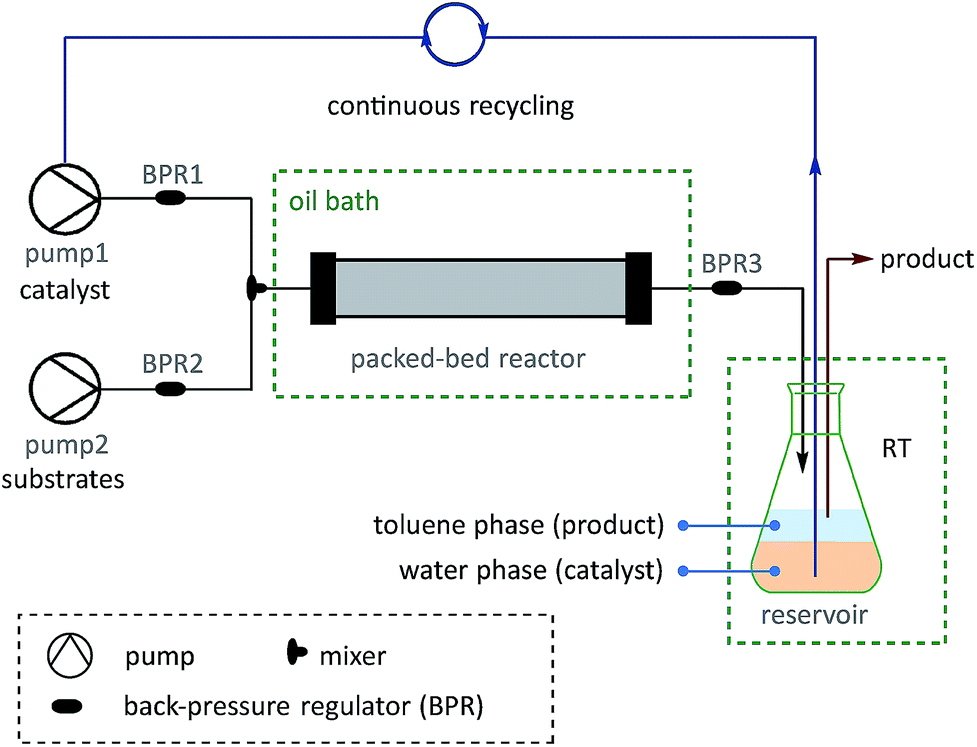 | ||
| Fig. 5 Set-up for continuous catalyst-recycling synthesis based on flow chemistry. | ||
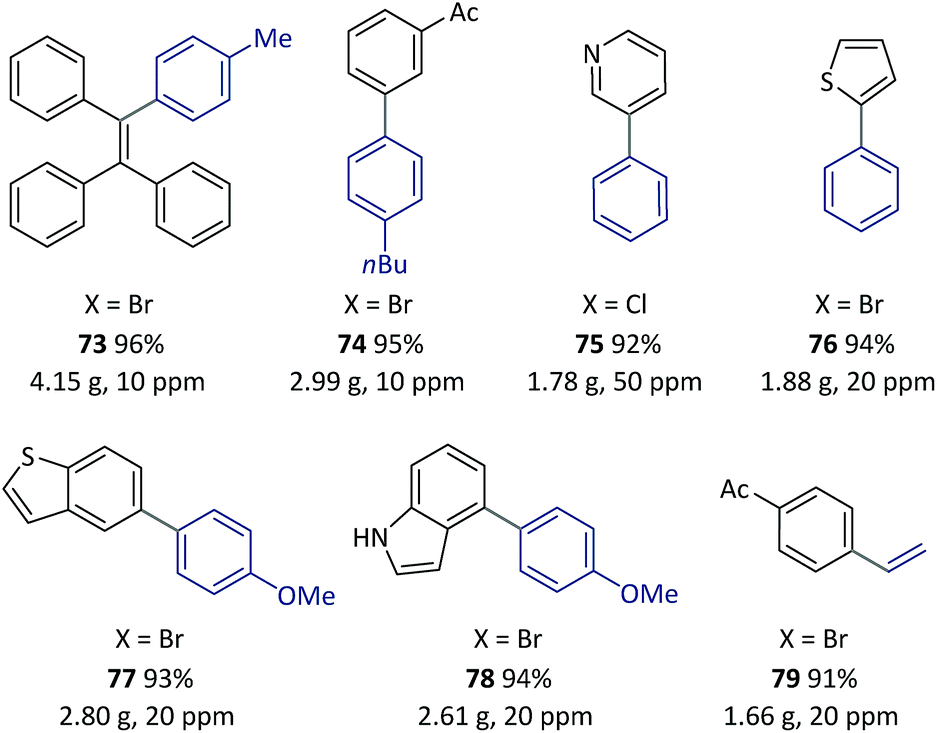 | ||
| Scheme 7 Examples of SM couplings with the continuous catalyst-recycling approach based on flow chemistry. Collections were based on 12.5 mmol aryl halides for all examples. The Pd loadings were calculated according to the molar ratio of [Pd]/[aryl halide]. See the ESI† for details of the flow set-up and operations. | ||
A solution of pre-catalyst and base in water was mixed with a solution of bromotriphenylethylene and 4-methylphenylboronic acid pinacol ester in toluene, and delivered into a packed bed reactor (packed with stainless steel beads)51 submerged in a preheated bath at 110 °C with a 30 min residence time. After the reaction, the resulting mixture was collected in a flask under a nitrogen atmosphere at RT. The water layer containing the catalyst was continuously extracted using pump 1 and then reinjected into the flow line. In this way, after 8.3 h of catalyst recycling, 4.15 g of 73 was isolated from the collected organic layer using only 1.3 mg of pre-catalyst 6a (13.8 μg Pd).
Once this flow technique was available, other bi(hetero)aryl compounds (74–78) and substituted styrene (79) were also successfully synthesized at the gram scale with 10–50 ppm of Pd. Based on the excellent behavior of the continuous catalyst-recycling synthesis using flow chemistry, it is believed that a larger scale preparation could be realized by extending the collection time without adding extra catalyst.
Conclusions
In conclusion, a novel thermoresponsive polymeric catalyst was developed, which can rapidly shuttle between water and organic phases, facilitating a highly efficient SM cross-coupling and tandem reaction with good to excellent isolated yields at ppm levels of catalyst usage. This method allows the preparation of a broad scope of bi(hetero)aryls, and can tolerate various functional groups. Furthermore, in combination with flow chemistry, the catalyst shuttling enables continuous catalyst-recycling, further promoting the scalability and efficiency of cross-coupling using ultralow loadings of palladium. Given the significant influence of transition-metal-catalyzed cross-coupling and increasing interest in sustainable chemistry, it is believed that, based on the strategy presented here, new response modes can be developed by tuning the structures of the ligands using different polymeric species. Also, the compatibility of this method with other metal-catalyzed reactions is under investigation, which would show promise for facilitating other diverse applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (NSFC, no. 21704016), start-up funding from Fudan University, and the National Program for Thousand Young Talents of China.Notes and references
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- P. Devendar, R.-Y. Qu, W.-M. Kang, B. He and G.-F. Yang, J. Agric. Food Chem., 2018, 66, 8914–8934 CrossRef CAS PubMed.
- M. J. Robb, S.-Y. Ku and C. J. Hawker, Adv. Mater., 2013, 25, 5686–5700 CrossRef CAS PubMed.
- A. Taheri Kal Koshvandi, M. M. Heravi and T. Momeni, Appl. Organomet. Chem., 2018, 32, e4210 CrossRef.
- M. Beller and A. Zapf, Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E.-i. Negishi, Wiley, New York, 2002, vol. 1, pp. 1209–1222 Search PubMed.
- B. H. Lipshutz, Chem, 2018, 4, 2004–2007 CAS.
- B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952–10958 CrossRef CAS PubMed.
- S. Handa, Y. Wang, F. Gallou and B. H. Lipshutz, Science, 2015, 349, 1087–1091 CrossRef CAS PubMed.
- S. Handa, M. P. Andersson, F. Gallou, J. Reilly and B. H. Lipshutz, Angew. Chem., Int. Ed., 2016, 55, 4914–4918 CrossRef CAS PubMed.
- L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392–6395 CrossRef CAS PubMed.
- S. M. Wong, C. M. So, K. H. Chung, C. P. Lau and F. Y. Kwong, Eur. J. Org. Chem., 2012, 2012, 4172–4177 CrossRef CAS.
- N. E. Leadbeater, V. A. Williams, T. M. Barnard and M. J. Collins, Org. Process Res. Dev., 2006, 10, 833–837 CrossRef CAS.
- R. K. Arvela, N. E. Leadbeater, M. S. Sangi, V. A. Williams, P. Granados and R. D. Singer, J. Org. Chem., 2005, 70, 161–168 CrossRef CAS PubMed.
- Á. Molnár, Chem. Rev., 2011, 111, 2251–2320 CrossRef PubMed.
- W. Wang, L. Cui, P. Sun, L. Shi, C. Yue and F. Li, Chem. Rev., 2018, 118, 9843–9929 CrossRef CAS PubMed.
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed.
- B. H. Lipshutz, Current Opinion in Green and Sustainable Chemistry, 2018, 11, 1–8 CrossRef.
- S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Green Chem., 2019, 21, 381–405 RSC.
- E. B. Landstrom, S. Handa, D. H. Aue, F. Gallou and B. H. Lipshutz, Green Chem., 2018, 20, 3436–3443 RSC.
- N. A. Isley, F. Gallou and B. H. Lipshutz, J. Am. Chem. Soc., 2013, 135, 17707–17710 CrossRef CAS PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- S. Harkal, F. Rataboul, A. Zapf, C. Fuhrmann, T. Riermeier, A. Monsees and M. Beller, Adv. Synth. Catal., 2004, 346, 1742–1748 CrossRef CAS.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS PubMed.
- M. Miura, Angew. Chem., Int. Ed., 2004, 43, 2201–2203 CrossRef CAS PubMed.
- U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366–374 CrossRef CAS PubMed.
- C. M. So, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 8059–8063 CrossRef CAS PubMed.
- C. Li, D. Chen and W. Tang, Synlett, 2016, 27, 2183–2200 CrossRef CAS.
- P. Slavik, D. W. Kurka and D. K. Smith, Chem. Sci., 2018, 9, 8673–8681 RSC.
- B. Li, Z. Guan, W. Wang, X. Yang, J. Hu, B. Tan and T. Li, Adv. Mater., 2012, 24, 3390–3395 CrossRef CAS PubMed.
- M. Chtchigrovsky, Y. Lin, K. Ouchaou, M. Chaumontet, M. Robitzer, F. Quignard and F. Taran, Chem. Mater., 2012, 24, 1505–1510 CrossRef CAS.
- S. Doherty, J. G. Knight, T. Backhouse, E. Abood, H. Alshaikh, A. R. Clemmet, J. R. Ellison, R. A. Bourne, T. W. Chamberlain, R. Stones, N. J. Warren, I. J. S. Fairlamb and K. R. J. Lovelock, Adv. Synth. Catal., 2018, 360, 3716–3731 CrossRef CAS.
- R. Shen, W. Zhu, X. Yan, T. Li, Y. Liu, Y. Li, S. Dai and Z.-G. Gu, Chem. Commun., 2019, 55, 822–825 RSC.
- Z. Wang, Y. Yu, Y. X. Zhang, S. Z. Li, H. Qian and Z. Y. Lin, Green Chem., 2015, 17, 413–420 RSC.
- E. Rangel Rangel, E. M. Maya, F. Sánchez, J. G. de la Campa and M. Iglesias, Green Chem., 2015, 17, 466–473 RSC.
- Y. M. A. Yamada, S. M. Sarkar and Y. Uozumi, J. Am. Chem. Soc., 2012, 134, 3190–3198 CrossRef CAS PubMed.
- S. Ogasawara and S. Kato, J. Am. Chem. Soc., 2010, 132, 4608–4613 CrossRef CAS PubMed.
- Z. Dong and Z. Ye, Adv. Synth. Catal., 2014, 356, 3401–3414 CrossRef CAS.
- C. Deraedt, L. Salmon and D. Astruc, Adv. Synth. Catal., 2014, 356, 2525–2538 CrossRef CAS.
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- C. P. Seath, J. W. B. Fyfe, J. J. Molloy and A. J. B. Watson, Angew. Chem., Int. Ed., 2015, 54, 9976–9979 CrossRef CAS PubMed.
- J. W. B. Fyfe, C. P. Seath and A. J. B. Watson, Angew. Chem., Int. Ed., 2014, 53, 12077–12080 CrossRef CAS PubMed.
- L. Xu, S. Ding and P. Li, Angew. Chem., Int. Ed., 2014, 53, 1822–1826 CrossRef CAS PubMed.
- N. Badi, Prog. Polym. Sci., 2017, 66, 54–79 CrossRef CAS.
- E. E. Dormidontova, Macromolecules, 2004, 37, 7747–7761 CrossRef CAS.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS PubMed.
- E. Bultz, M. Ouchi, K. Nishizawa, M. F. Cunningham and M. Sawamoto, ACS Macro Lett., 2015, 4, 628–631 CrossRef CAS.
- See the ESI for details.†.
- N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 RSC.
- C. S. Wei, G. H. M. Davies, O. Soltani, J. Albrecht, Q. Gao, C. Pathirana, Y. Hsiao, S. Tummala and M. D. Eastgate, Angew. Chem., Int. Ed., 2013, 52, 5822–5826 CrossRef CAS PubMed.
- Lower GC yields with other catalysts could due to: (1) insufficient catalyst concentration in toluene as caused by less efficient UCST behaviour of low-molecular-weight ligands (5b and 5c); (2) less efficient reaction caused by the large steric hindrance of the long polymeric chain (5d)..
- J. Alvarez-Builla, J. J. Vaquero and J. Barluenga, Modern Heterocyclic Chemistry, Wiley-VCH, Weinheim, 2011 Search PubMed.
- I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS PubMed.
- J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297–2307 CrossRef CAS PubMed.
- E. P. Gillis and M. D. Burke, Aldrichimica Acta, 2009, 42, 17–27 CAS.
- L. Xu, S. Zhang and P. Li, Chem. Soc. Rev., 2015, 44, 8848–8858 RSC.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- J. A. M. Lummiss, P. D. Morse, R. L. Beingessner and T. F. Jamison, Chem. Rec., 2017, 17, 667–680 CrossRef CAS PubMed.
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC.
- D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed.
- M. Chen and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 11628–11631 CrossRef CAS PubMed.
- M. Chen, S. Ichikawa and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 263–266 CrossRef CAS PubMed.
- X. Shen, H. Gong, Y. Zhou, Y. Zhao, J. Lin and M. Chen, Chem. Sci., 2018, 9, 1846–1853 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02171j |
| This journal is © The Royal Society of Chemistry 2019 |