
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA symbiotic hetero-nanocomposite that stabilizes unprecedented CaCl2-type TiO2 for enhanced solar-driven hydrogen evolution reaction†
Yuelan
Zhang
 a,
Liping
Li
*a,
Yan
Liu
a,
Tao
Feng
a,
Shibo
Xi
b,
Xiyang
Wang
a,
Chenglin
Xue
a,
Jingyu
Qian
c and
Guangshe
Li
*a
a,
Liping
Li
*a,
Yan
Liu
a,
Tao
Feng
a,
Shibo
Xi
b,
Xiyang
Wang
a,
Chenglin
Xue
a,
Jingyu
Qian
c and
Guangshe
Li
*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: lipingli@jlu.edu.cn
bInstitute of Chemical and Engineering Sciences, A*STAR, 1 Pesek Road, Jurong Island, Singapore 627833, Singapore
cState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, P. R. China
First published on 27th July 2019
Abstract
Symbiotic hetero-nanocomposites prevail in many classes of minerals, functional substances and/or devices. However, design and development of a symbiotic hetero-nanocomposite that contains unachievable phases remain a significant challenge owing to the tedious formation conditions and the need for precise control over atomic nucleation in synthetic chemistry. Herein, we report a solution chemistry approach for a symbiotic hetero-nanocomposite that contains an unprecedented CaCl2-type titania phase inter-grown with rutile TiO2. CaCl2 structured TiO2, usually occurring when bulk rutile-TiO2 is compressed at an extreme pressure of several GPa, is identified to be a distorted structure with a tilt of adjacent ribbons of the c-axis of rutile. The structural specificity of the symbiotic CaCl2/rutile TiO2 hetero-nanocomposite was confirmed by Rietveld refinement, HRTEM, EXAFS, and Raman spectra, and the formation region (TiCl4 concentration vs. reaction temperature) was obtained by mapping the phase diagram. Due to the symbiotic relationship, this CaCl2-type TiO2 maintained a high stability via tight connection by edge dislocations with rutile TiO2, thus forming a CaCl2/rutile TiO2 heterojunction with a higher reduction capacity and enhanced charge separation efficiency. These merits endow symbiotic CaCl2/rutile TiO2 with a water splitting activity far superior to that of the commercial benchmark photocatalyst, P25 under simulated sunlight without the assistance of a cocatalyst. Our findings reported here may offer several useful understandings of the mechanical intergrowth process in functional symbiotic hetero-nanocomposites for super interfacial charge separation, where interfacial dislocation appears to be a universal cause.
Introduction
Symbiotic hetero-nanocomposites prevail in many classes of minerals, functional substances and/or devices,1–3 providing opportunities to maximize interfacial/surface chemical reactions and synergistic effects between components essential for important applications. Important applications of these hetero-nanocomposites have been witnessed in a variety of fields, including catalysis, photovoltaic conversion, energy storage, decontamination, and clean energy production since these nanocomposites are merited by optimum interfacial/surface chemical reactions and synergistic effects of components.4–8 Depending on the interconnection of each component through van der Waals forces or covalent bonds and on the interplay of individual components, heterogeneous composites could always generate a superior performance. For example, strong interfacial interactions in between components enable charge transfer within heterogeneous composites, accounting for the effective separation of photo-generated electrons and holes through interfaces for excellent photocatalytic overall water splitting activities.9–14 When noble metals are strongly supported onto oxides in forming heterogeneous composites, excellent CO oxidation ability was achieved because of the simultaneous CO adsorption of noble metals and oxygen activation of MOx.15–17 Indeed, when growing nanocomposites or epitaxial oxide films, pronounced interfacial interactions could be possible because of the interfacial dislocations that directly influence the average stress and strain states in the composites,18 while upon cooling down from the growth temperature, tensile strain is stored in the composites or films (owing to the thermal expansion mismatch between component oxides) which could stabilize certain highly useful, but also barely accessible phases under ambient conditions.19 All these demonstrate a great possibility of using interfacial dislocations or strains in developing symbiotic hetero-nanocomposites that contain unachievable phases for many important applications, which however remains a significant challenge owing to the tedious formation conditions (like pre-synthesis and post-processing) and the need for precise control over atomic nucleation in synthetic chemistry. Consequently, it is highly necessary to explore one-step approaches for synthesizing symbiotic hetero-nanocomposites via precise control over the composition intergrowth during the nucleation and crystallization process.Depending on the synthetic chemistry, symbiotic hetero-nanocomposites could be achieved in many classes of oxide systems because of the intrinsic nature of polymorphs. For instance, TiO2 has many crystalline phases. Heterogeneous composites formed in between rutile, anatase, and/or brookite-type TiO2 have been evidenced (e.g., anatase/rutile, anatase/brookite, rutile/brookite or anatase/rutile/brookite) to show strong interfacial interactions because of the intergrowth of these phases during the synthesis of TiO2. Closely related to the specific combination of these constituent structures, heterogeneous composites have found a broad range of applications such as in photocatalysis, lithium-ion batteries, CO oxidation, and ammonia synthesis.17,20–22 P25 is a typical heterogeneous composite that consists of anatase and rutile at a molar ratio of 79 to 21, and has shown a preeminent performance in photocatalytic hydrogen production and sewage treatment. Indeed, the intergrowth compositions of TiO2 are rather limited to those of common phases, while heterogeneous composites that involve many other unconventional phases of TiO2 (like TiO2-H, CaCl2 phase, or α-PbO2 phase, etc.) are barely accessible. This is because the synthesis of these unconventional phases can only be achieved under harsh experimental conditions such as high pressure and high temperature, even though these phases have been predicted to have superior activity in batteries and photocatalysis.23,24 Consequently, an intergrowth of heterogeneous composites with those unconventional phases is highly important, but also fairly challenging.
We hold the opinion that the CaCl2 phase and rutile could most probably form a symbiotic hetero-nanocomposite with a broad impact if successful. We took CaCl2/rutile as the target to study based on the following considerations: (i) among all unconventional phases of TiO2, the orthorhombic CaCl2-type TiO2 polymorph with a space group of Pnnm is a distorted structure of tetragonal rutile with a relative tilt of adjacent ribbons about the c-axis, whose structure is very close but absolutely different from rutile. They are likely to connect with each other and grow together through orientation attachment, leading to dislocations in between them that could in turn stabilize this metastable phase; (ii) although the CaCl2 phase can only be synthesized under high pressure by assistance of foreign ions currently,25,26 solution chemistry especially the hydrothermal approach merited by a special subcritical reaction medium and abundant intermediate states could regulate the flexible reaction procedure to obtain several special phases with a metastable structure. Further, hydrothermal methods allow control over the nucleation and growth processes essential for synthesizing symbiotic hetero-nanocomposites with strong interactions.27 (iii) There are many other rutile counterparts like SnO2, SiO2, GeO2, and RuO2 that have been found to transform into the CaCl2 structure under high pressure.28–31 Thus, once such a solution methodology was established, one may expect more opportunities to develop other CaCl2/rutile symbiotic hetero-nanocomposites. As we all know, rutile is an excellent multi-functional material in a wide range of fields, and thus it is highly possible for symbiotic hetero-nanocomposites with a distorted rutile structure to show superior performances that have not yet been accessible.
Herein, we successfully synthesized a symbiotic hetero-nanocomposite that consists of metastable CaCl2 structured TiO2 and rutile TiO2via a mild solution chemistry approach. The composite structure of symbiotic CaCl2/rutile TiO2 was confirmed by XRD data refinement, EXAFS, and Raman spectra. Then, the phase diagram for the symbiotic heteronanocomposite was depicted based on the reactant concentration and reaction temperature, in which the Ti(OH)2(OH2)42+ saturation ion played a crucial role. Despite the presence of metastable CaCl2 structured TiO2, the as-prepared hetero-nanocomposite showed a high stability and a superior performance in photocatalytic H2 evolution reactions through forming a type II heterojunction. This work provides insights into developing new methods for synthesizing high-performance symbiotic hetero-nanocomposites that contain metastable compounds.
Results and discussion
Titania polymorphs in this work were synthesized through a high-acidity hydrothermal process utilizing TiCl4 as the titanium source, and no other reagents were needed except for the aqueous solution. After reaction at 120 °C for 2 h, the product was examined by powder X-ray diffraction (XRD) to identify the structure. As shown in Fig. 1c, the XRD data of the product were readily indexed to a rutile phase of TiO2 in tetragonal structure (JCPDS no. 21-1276). However, unlike the traditional rutile TiO2, the diffraction line (110) around two theta of 28° is clearly asymmetric (Fig. 1a). This diffraction peak could be fitted well using two Lorentz fitting peaks: one shifted towards a higher diffraction angle, while the other one shifted oppositely and became symmetric gradually after calcination at 200 °C and 400 °C (Fig. S1† and 1b). Typically, the origins of the doublet peaks are regarded as Kα1 and Kα2 rays of the Cu target during XRD measurements. However, the peak splitting caused by the Cu target couldn’t be so large.32 Thus, the influence of the instrument was excluded, and the peak splitting around two theta of 28° could be attributed to the change of the structure. To the best of our knowledge, the peak splitting of diffraction peaks could be the consequences of (i) strain; for instance, polymer capping induced strain may result in changes in the lattice constant, and crystal boundary strain in the twin crystal could give rise to doublet XRD peaks;33–35 (ii) surface species, such as water vapour on the surface of ice;36 and (iii) distortion of the structure.37 Asymmetry of diffraction peaks induced by strain and surface species is often composed of one strong intrinsic peak accompanied by weak peaks at the shoulder, differing from two comparative peaks. The sole possibility is appearance of the composition in a new phase. Firstly, we assumed that the peak splitting originates from two rutile TiO2 phases intergrown with a shrinking lattice and an expanded lattice. That is, two tetragonal TiO2 structures with space group of P42/mnm coexist in this material. Thus, according to eqn (S1),† two sets of lattice parameters could be obtained when calculated using d values of peaks (110) and (101), as shown in Table S1.† The lattice volume obtained from the left peak (peak 1) is well consistent with the experimental curve of V–1/D of rutile TiO2, while the lattice volume obtained from the right peak (peak 2) severely deviates from the curve (Fig. S2†), giving a hint that peak 2 does not originate from rutile TiO2 with a contracted lattice. This means that one has to look for other structured TiO2 similar to rutile. Coincidentally, a similar phenomenon has been found in rutile SnO2 under a pressure of 250 bar, and the author assigned the structure to a CaCl2 structure (space group Pnnm) that forms through a second-order phase transition from the rutile phase.38 Typically, the CaCl2 structure is an orthorhombic structure that occurs at a very high pressure, which has been found in many rutile structured compounds including SnO2, SiO2, GeO2, RuO2, TiO2, etc.25,28–31 It seems reasonable that there is a CaCl2 phase in the as-prepared TiO2, which explains the peak splitting around two theta of 28° in Fig. 1a. Thus, the XRD patterns were fitted in terms of rutile and CaCl2 structure. As one expected, the results agree well with the data (Fig. 1, Tables 1, S2 and S3†). The lattice parameters of the CaCl2 structure in CaCl2/rutile TiO2 were determined to be a = 4.57 Å, b = 4.48 Å, c = 2.99 Å. It is authentic to identify the existence of CaCl2-type structured TiO2 in CaCl2/rutile TiO2. | ||
| Fig. 1 Structural analyses by X-ray diffraction (XRD) refinement: (a) XRD pattern of the enlarged peak (110) for CaCl2/rutile TiO2. Inset illustrates the crystal structures for rutile TiO2 and CaCl2 phase TiO2. (b) XRD pattern of the calcined product CaCl2/rutile-400. (c) Rietveld structural data refinement results for CaCl2/rutile TiO2. Black, red, and blue curves represent the experimental diffraction data, calculated data, and deviation between the experimental and calculated data, respectively. The vertical bars below the pattern denote the standard diffraction data for internal standard Al (blue), rutile TiO2 (black) and CaCl2 phase TiO2 (rose red). (d) The structural refinement results for components rutile and CaCl2 in the composite. | ||
| Sample name (phase) | Lattice | Space group | a/Å | b/Å | c/Å | V/Å3 | d(110)/Å | χ 2 |
|---|---|---|---|---|---|---|---|---|
| CaCl2/rutile TiO2 (CaCl2 phase) | Orthorhombic | Pnnm | 4.569 | 4.484 | 2.988 | 61.21 | 3.200 | 1.575 |
| CaCl2/rutile TiO2 (rutile phase) | Tetragonal | P42/mnm | 4.642 | 4.642 | 2.947 | 63.51 | 3.283 | |
| CaCl2/rutile-400 (rutile phase) | Tetragonal | P42/mnm | 4.596 | 4.596 | 2.957 | 62.46 | 3.250 | 5.513 |
| Ref.TiO2 (rutile phase) | Tetragonal | P42/mnm | 4.593 | 4.593 | 2.958 | 62.42 | 3.248 | 2.542 |
In order to further verify our speculation, CaCl2/rutile TiO2 was further characterized by field-emission scanning electron microscopy (SEM) and transmission electron microscopy (TEM) which display a cone shape at a dimension of about 2–3 μm (Fig. 2b and S6†). The cone was composed of numerous nanorods with a width of about 5 nm grouped by small nanoparticles through orientation attachment growth. High resolution transmission electron microscopy (HRTEM) indicates that the lattice spacing is ordered locally, while the lattice parameters vary in different parts (Fig. 2c, f and g). The interplanar spacing (d) in different parts of Fig. 2c differ (such as d1 = 0.320 Å, d2 = 0.328 Å), which are in accordance with the plane (110) in rutile and CaCl2 structure from the refinement results of CaCl2/rutile TiO2 (Table 1). Besides that, there are a large number of dislocations in the joint point of both parts (Fig. 2c). Benefitting from the dislocation, the neighbouring particles attached to each other and tended to grow together, as illustrated in the schematic diagram in Fig. 2a. Thus, the formation of CaCl2-type structured TiO2 intergrown with rutile TiO2 was confirmed.
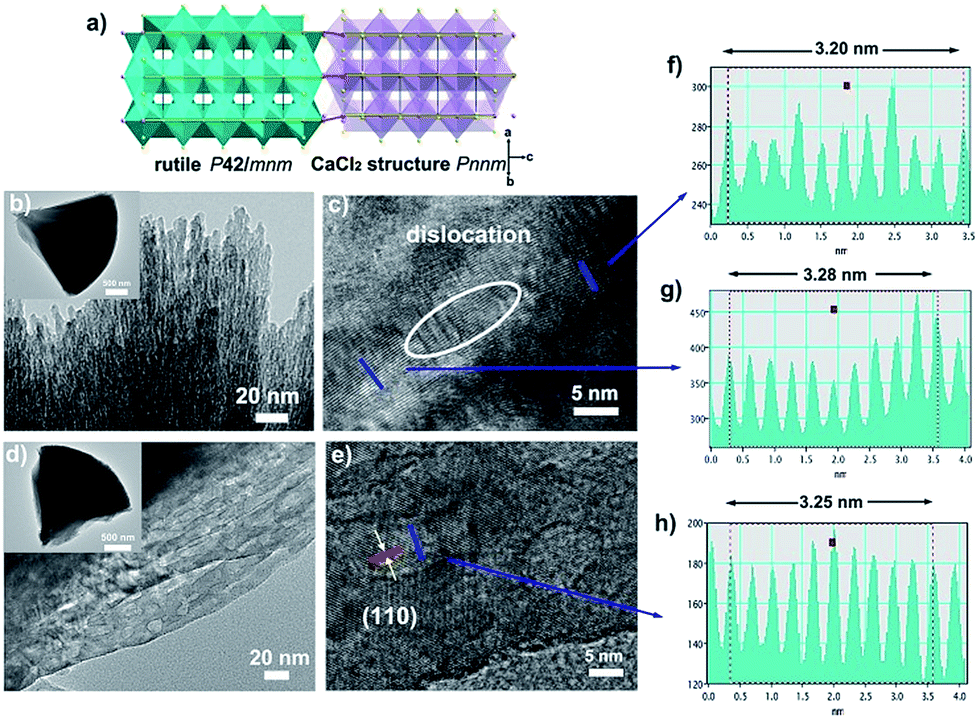 | ||
| Fig. 2 Morphology and assembly structure of the composite: (a) schematic diagram of the assembly structure, (b) TEM and (c) HRTEM images for CaCl2/rutile TiO2. (d) TEM and (e) HRTEM images for CaCl2/rutile-400. Profiles of lattice fringes for (f and g) CaCl2/rutile TiO2 in different areas; and (h) profiles of lattice fringe for CaCl2/rutile-400. Scale value in insets of (b) and (d) are 500 nm. | ||
When the intergrowth structure was calcined at high temperatures, the lattice tends to be regular and transformed entirely into a rutile TiO2 phase with a little shrinkage relative to the rutile phase in CaCl2/rutile TiO2. This observation is likely due to the phase transition of the metastable CaCl2 structure to the rutile structure and the growth of crystals. The d spacing could be identified to decrease to a value of 0.325 Å from HRTEM, which is also consistent with XRD refinement results of CaCl2/rutile-400 and commercial rutile (Fig. S3 and S4, Tables S4 and S5†). It should be mentioned that uniform mesopores (∼8 nm) appeared with the cone-shape maintained after calcination (Fig. 2d and S6b†), in line with the pore distributions obtained by N2 adsorption/desorption isotherm curves (Fig. S5†). Lattice rearrangement should be responsible for the appearance of mesopores. As the calcination temperature increased, the specific areas decreased (Table S6†) due to the grain growth.
The Raman spectrum is often used as a fingerprint to analyze the symmetry and structural order. As shown in Fig. 3a, three signals located at about 140, 400, and 600 cm−1 are associated with the modes B1g, Eg, and A1g of the rutile TiO2 lattice, respectively, while that at 200 cm−1 can be assigned to the multi-phonon modes of rutile TiO2.39,40 It should be pointed out that in CaCl2/rutile TiO2, a weak signal of the B1g mode for rutile TiO2 at 140 cm−1 became inconspicuous and overlapped by a new peak at about 110 cm−1. In Raman active modes of rutile, the central cation Ti is silent, and the Raman modes are purely O vibrations.39 As shown in the schematic diagram in Fig. 3a, the oxygen ions vibrate perpendicular to the Ti–O bond in the B1g mode. Typically, the B1g mode is highly sensitive to the long-range order of TiO2 crystals and softens significantly more than other modes under high pressures.41 The Raman shift in rutile often has a relationship with the strain, O/Ti ratio, size effect, O isotope, and volume effect, which usually give rise to several wavenumber shifts.39,40,42,43 The most significant factor could be the pressure-induced-volume effect as represented by a shift of about 20 cm−1 softened at 6 GPa.40 Several viewpoints have attributed the origin of anomalistic softening in the B1g mode to a thermal/pressure-induced-lattice contraction or an incipient structural phase transition, which explains well the softening of mode B1g. Thus, the new emerging peak at 110 cm−1 most likely originates from CaCl2 structured TiO2 in CaCl2/rutile TiO2. Due to the intense signal of this vibration, the intrinsic signal for rutile is covered by that of CaCl2 phase TiO2.
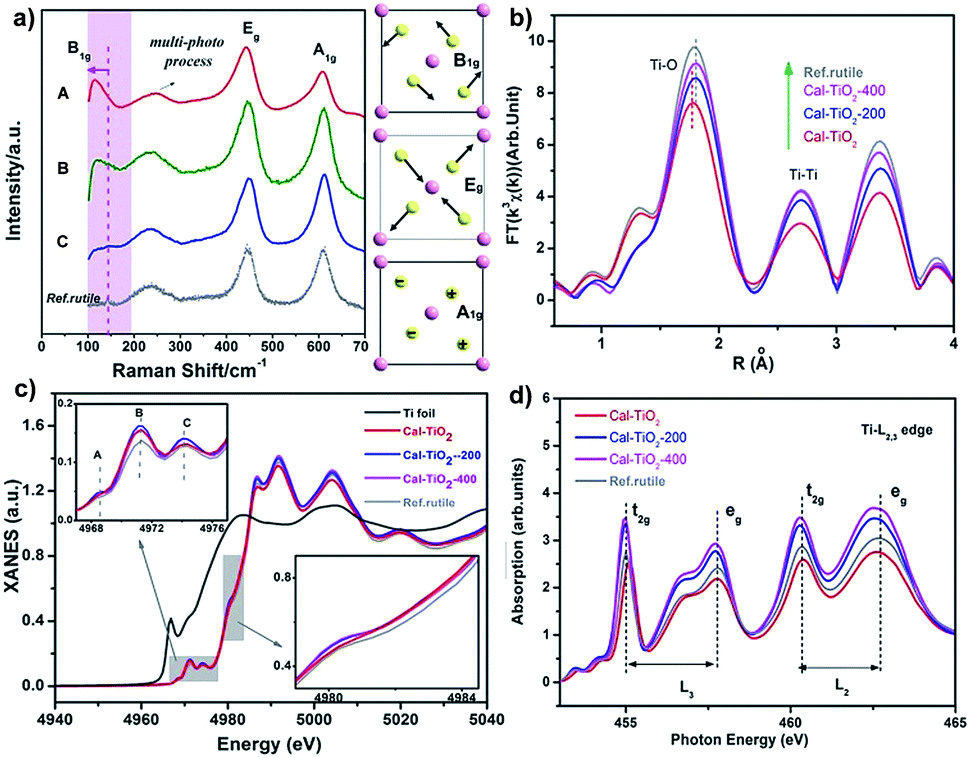 | ||
| Fig. 3 Structural information from Raman and EXAFS. (a) Raman spectra, (b) R-space Fourier-transformed FT (k3χ(k)) of Ti K-edge EXAFS, (c) XANES spectra of the Ti K-edge and (d) Ti L2,3-edge of the samples CaCl2/rutile TiO2 (A), CaCl2/rutile-200 (B), CaCl2/rutile-400 (C) and commercial rutile TiO2. The gray area in (c) highlights the pre-edge region. | ||
In order to investigate the coordination environment of CaCl2/rutile TiO2, Ti K-edge X-ray adsorption spectra of CaCl2/rutile TiO2 and its calcined samples were measured and compared. As shown in Fig. 3b, Ti K-edge EXAFS of CaCl2/rutile TiO2, CaCl2/rutile-200, CaCl2/rutile-400, and commercial rutile TiO2 (Ref.rutile) are transformed into the radial distribution function via R-space Fourier-transformed FT (k3χ(k)). The first peak at about 1.8 Å is attributed to the six Ti–O bonds in the first coordination sphere (TiO6 octahedron) and the second peak at about 2.7 Å could be ascribed to Ti–Ti distances of these samples. The third peak at 3.4 Å corresponds to Ti–O bonds at about 3.5 Å and Ti–Ti bonds at 3.57 Å. Obviously, the Ti–O distance in CaCl2/rutile TiO2 is shorter than those of other samples, which could be caused by CaCl2-type structured TiO2 with a contracted unit cell. Normalized Ti K-edge XANES spectra are shown in Fig. 3c. The line shape of CaCl2/rutile TiO2, CaCl2/rutile-200 and CaCl2/rutile-400 is similar to that of commercial rutile TiO2 except for a subtle change in the relative intensities of peaks and the absorption edge. The three major features in the pre-peaks of the Ti K-pre-edge region are labelled A, B, and C, which are identified to be associated with hybridized Ti 3d–4p, and 1s → 2t2g and 1s → 3eg transitions, respectively.44,45 The intensity of pre-peak B in CaCl2/rutile TiO2, CaCl2/rutile-200, and CaCl2/rutile-400 is stronger than that of commercial rutile TiO2. It is well established that the intensity of pre-peak B enhanced with increasing concentration of defects.45,46 Oxygen vacancy signals were then detected by ESR at g = 2.00 (Fig. S7†),47 the calcined samples showed obvious signals of oxygen vacancies, whereas, CaCl2/rutile TiO2 did not show any single electron signal (Ti3+ or single electron trapped in oxygen vacancies), indicating the absence of defect sites in CaCl2/rutile TiO2. Even so, there are dislocations (line defects) in CaCl2/rutile TiO2, which should be responsible for the enhanced pre-peak B. Another change in normalized Ti K-edge XANES spectra is the absorption edge. It is easy to understand why the lower absorption edge energy of calcined samples stems from the existence of Ti3+, while, the inconformity of the absorption edge of CaCl2/rutile TiO2 cannot be explained in terms of valence characteristics of Ti ions. Instead, the covalency in CaCl2/rutile TiO2 was enhanced due to the shortened Ti–O distance.
In addition, Ti L2,3-edge absorption spectra are also measured to study the electronic structure, as shown in Fig. 3d. Two sets of local maxima over this energy range could be assigned to Ti 2p3/2 (L3: peaks t2g and eg) and Ti 2p1/2 (L2: peaks t2g and eg) core levels into empty Ti 3d states.48,49 It should be noted that the difference value (ΔE) of peaks t2g (L3) to eg (L3) of CaCl2/rutile TiO2 decreases compared with other samples, which is caused by the lattice distortion of rutile TiO2.
It is well known that the formation of TiO2 in different phases (like rutile, anatase, and brookite) proceeds under specific conditions, and these phases often transform into each other through external energy, as previously reported.50,51 However, CaCl2 structured TiO2 has not been obtained under mild conditions and the formation inducement has not been uncovered. Herein, we initiated experiments under a broad range of conditions with varying the amount of reactants and reaction temperature with an aim to obtain the phase diagram for given phases and nanostructures (Fig. 4a, S8 and S9†). It seems that solution reaction with a high concentration of TiCl4 leads to the occurrence of CaCl2-type structured TiO2, while a low concentration of TiCl4 causes an appearance of the anatase phase. In the boundary of the CaCl2 phase and anatase phase, pure rutile formed. With increasing the amount of TiCl4, no products could be obtained except for a solution. Through this mapping, we didn’t find an indication that pure CaCl2 phase TiO2 is formed through the hydrolytic process of TiCl4. This may be due to the metastable structure of CaCl2 phase TiO2, which makes it hard to stabilize by itself. Further, the symbiotic structure is vital in stabilizing the metastable CaCl2 phase TiO2. Based on the synthetic maps, the possible formation process of CaCl2/rutile TiO2 was proposed in terms of the schematic diagram in Fig. 4b: when the starting reactant TiCl4 was dissolved in water, water-soluble precursor Ti(OH)2(OH2)4Cl2 was formed initially, and the solution became highly acidic. Then, the precursor was dissociated and condensed to crystallize TiO2 through an oxolation. At a high concentration of TiCl4, Ti(OH)2(OH2)4Cl2 could not be completely disintegrated to create a crystalline TiO2 phase,52 leaving lots of redundant saturation Ti(OH)2(OH2)42+ ions and Cl− ions scattered around the crystals that affect the crystallization and the subsequent grain growth process. Even after the reaction, there remained a great number of Ti(OH)2(OH2)42+ ions in the solution. In order to verify the existence of soluble titanium compounds, the filter liquor was collected. At first, ultra-pure H2O was added, the solution showed a phenomenon of liquid miscibility with visible flow traces, and no precipitates appeared, which is due to the large viscosity and mobility difference between H2O and the filter liquor with a high concentration of Ti(OH)2(OH2)42+. Then, excessive NaOH was put in and plenty of white precipitates appeared which indicates the presence of titanium compounds in this filter liquor. This observation shows that a high concentration of Ti(OH)2(OH2)42+ ions plays an important role in the formation of symbiotic CaCl2/rutile TiO2. Similar cases have been reported elsewhere, in which ions can regulate the morphology and structure of final products.53,54
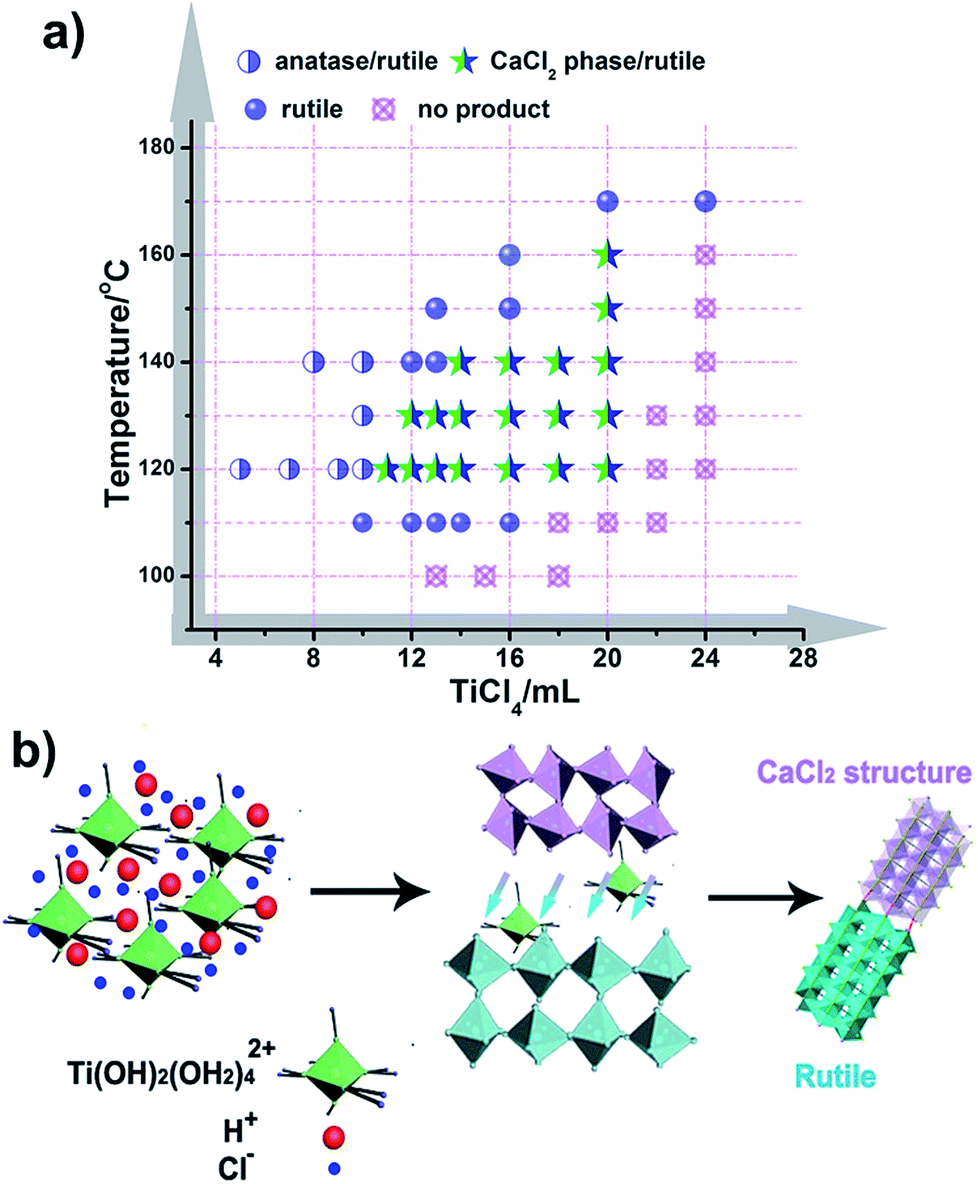 | ||
| Fig. 4 Chemical synthesis routes. (a) Maps for the synthesis of TiO2 at various hydrothermal temperatures and addition amounts of TiCl4. (b) The synthetic schematic diagram for the formation of CaCl2/rutile TiO2. | ||
The stability of CaCl2/rutile TiO2 was measured using in situ XRD through increasing the temperature gradually. The double peak feature of plane (110) is still clear when calcined at temperatures <200 °C, and then fades away as the calcination temperature increases (Fig. 5a). Meanwhile, such a feature remains unchanged if the samples are kept at room temperature for as long as three years (Fig. 5c), suggesting a good stability. Clearly, in the phase transition process, the double peaks located at two theta of about 27.0° and 27.9° gradually change into a single peak at two theta of about 27.5° (Fig. 5b). This reflects the lattice change process more intuitively: the CaCl2 phase fades away little by little, and the rutile phase contracts in the lattice with grain growth.
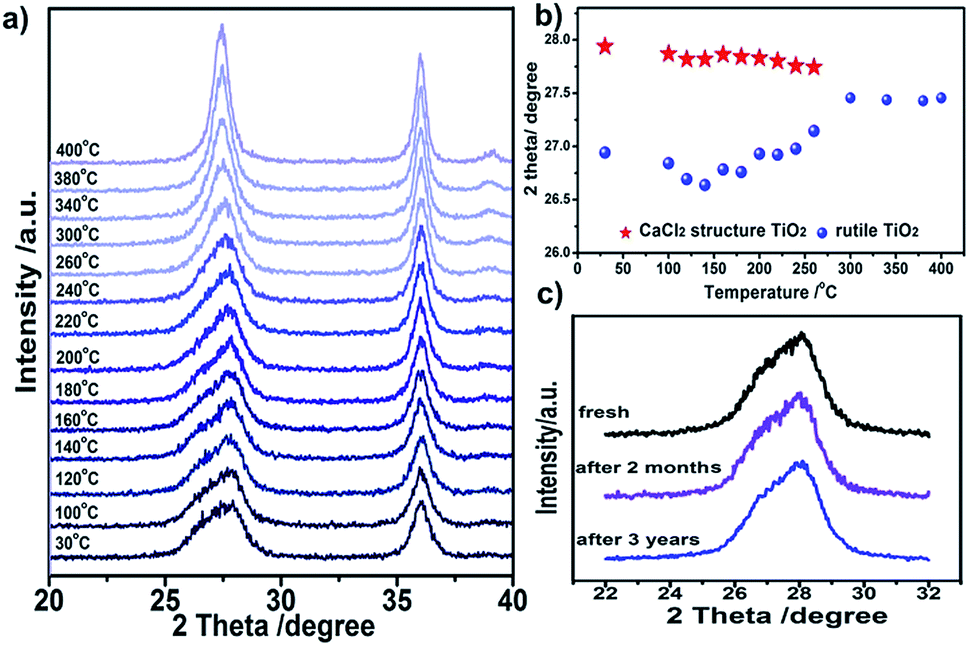 | ||
| Fig. 5 Stability of symbiotic CaCl2/rutile TiO2. (a) In situ XRD patterns for CaCl2/rutile TiO2 with increasing temperature from 30 to 400 °C. (b) Peak position variation as temperature increases. (c) XRD patterns for fresh CaCl2/rutile TiO2 and the samples after being stored in air for two months and three years as well. | ||
It is well known that hetero-nanocomposites of TiO2 like P25 are excellent photocatalysts, however, no one knows what the performance is for symbiotic hetero-nanocomposite CaCl2/rutile TiO2. Since the band structure of CaCl2-type TiO2 directly determines its photocatalytic activity, we calculated the density of states (DOS) of CaCl2 phase TiO2 and rutile TiO2 in CaCl2/rutile TiO2 (Fig. 6a). The conduction band minimum of both materials originates from the Ti 3d orbital, and the valence band maximum consists mainly of the O 2p orbital. Nevertheless, the band gap energy of CaCl2 structured TiO2 (Eg ≈ 3.3 eV) is obviously larger than that of rutile TiO2 (Eg ≈ 3.1 eV). The origin of the difference in band structure lies in the unique features of CaCl2 phase TiO2, which owns an entirely different octahedral configuration and Ti–O bond length when compared to rutile TiO2. The change in bond length could affect the interaction between orbitals, leading to variations in the band position.55 Experimentally, we measured the diffuse reflectance ultraviolet-visible spectra and XPS valence band spectra. The absorbance range of CaCl2/rutile TiO2 is similar to that of rutile TiO2 (Eg ≈ 3.1 eV) except for an enhanced absorption intensity around 360 nm (Fig. S10†). Combined with the DOS analysis results in which the band gap of CaCl2 phase TiO2 is wider than that of rutile TiO2, we could attribute the enhanced light absorption near 360 nm to the electron excitation from the O 2p orbital to the Ti 3d orbital of the CaCl2 phase TiO2. The edge absorption around 400 nm primarily originates from electron excitation from the O 2p orbital to the Ti 3d orbital of rutile TiO2. From XPS spectra in Fig. S11,† the valence band of CaCl2/rutile TiO2 is determined to be about 0.2 eV lower than that of rutile TiO2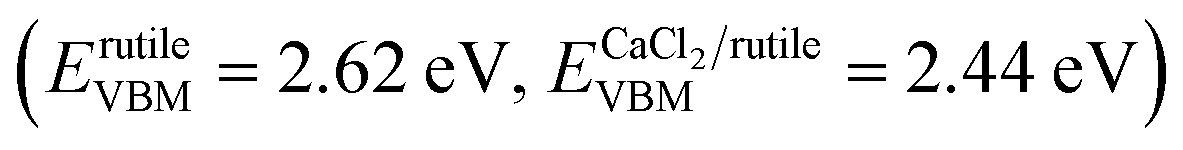 . Thus, CaCl2/rutile TiO2 forms a type II heterojunction similar to that of P25, as illustrated in the schematic diagram (Fig. 6f). Photoexcited charges would transport and accumulate in semiconductors.56,57 That is, photo-generated electrons in CaCl2 structured TiO2 could transport to rutile TiO2 preferentially to conduct reduction reactions. Meanwhile, the holes left in the valence of rutile TiO2 would transport to CaCl2 structured TiO2 preferentially to react with the sacrificial agent.
. Thus, CaCl2/rutile TiO2 forms a type II heterojunction similar to that of P25, as illustrated in the schematic diagram (Fig. 6f). Photoexcited charges would transport and accumulate in semiconductors.56,57 That is, photo-generated electrons in CaCl2 structured TiO2 could transport to rutile TiO2 preferentially to conduct reduction reactions. Meanwhile, the holes left in the valence of rutile TiO2 would transport to CaCl2 structured TiO2 preferentially to react with the sacrificial agent.
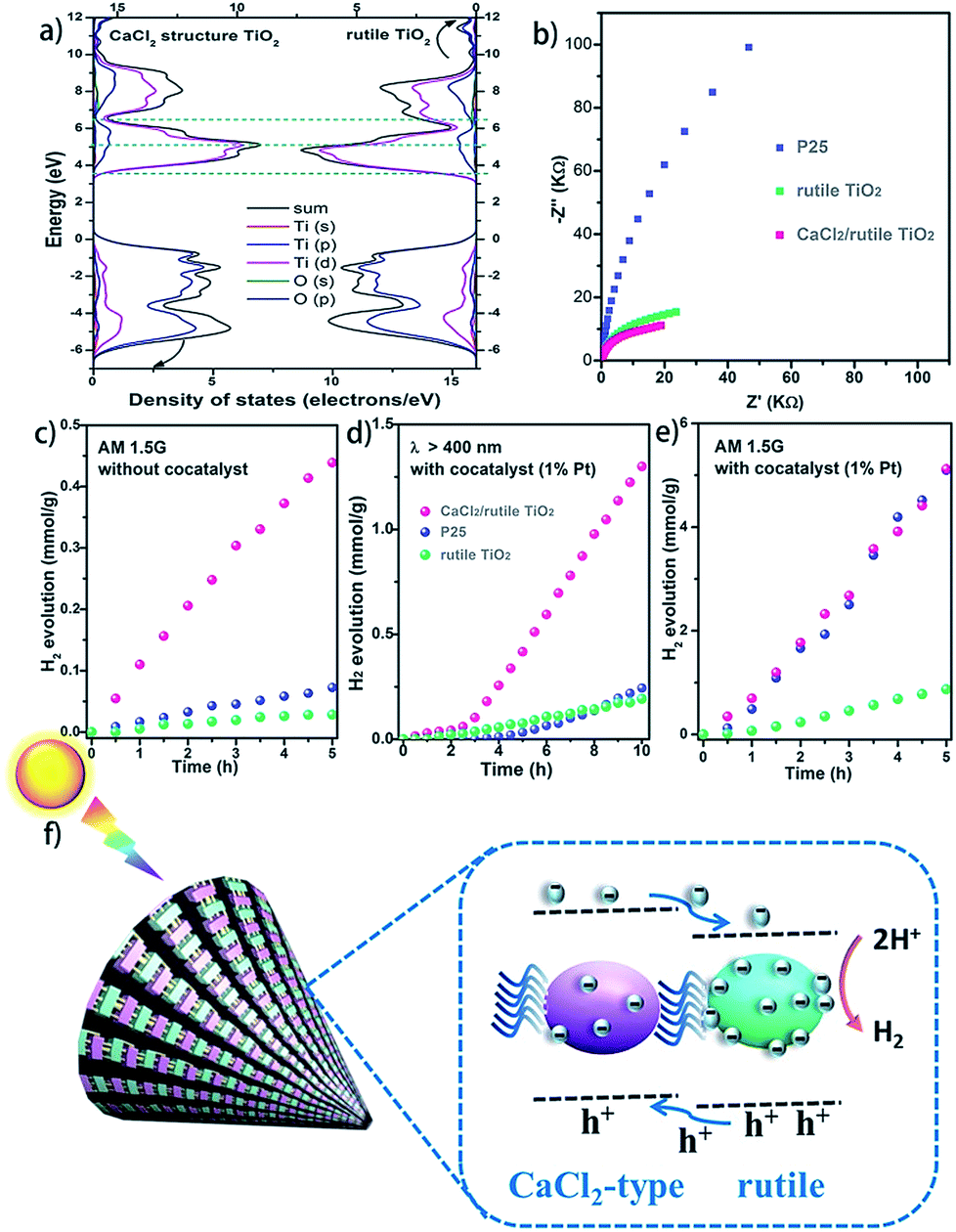 | ||
| Fig. 6 Photocatalytic performance of CaCl2/rutile TiO2. (a) Calculated density of states (DOS) of CaCl2 structured TiO2 and rutile TiO2; (b) Nyquist plots of the EIS for CaCl2 structured TiO2, P25, and commercial rutile TiO2 (Ref.rutile); Water splitting hydrogen evolution activities for CaCl2/rutile TiO2, commercial rutile TiO2 (Ref.rutile), and P25: (c) without any cocatalyst under simulated sunlight (with an AM 1.5G filter), (d) with the assistance of 1% Pt as the cocatalyst under simulated sunlight (with an AM 1.5G filter) and (e) visible light (wavenumber > 400 nm) illumination. (f) Schematic diagram for the electron transfer process under the illumination of light for CaCl2/rutile TiO2. | ||
The charge separation and transport efficiency were evaluated through electrochemical impedance measurements (EIS). As shown in Fig. 6b, the arc in the low-frequency range originates from electron transfer resistance (Rt). Obviously, the arc of CaCl2/rutile TiO2 is smaller than those of Ref.TiO2 and P25, indicating a more rapid electron transfer process in CaCl2/rutile TiO2. As expected, the as-prepared CaCl2/rutile TiO2 exhibits a superior performance in mass-specific H2 evolution (1024 μmol g−1 h−1) to rutile TiO2 (174 μmol g−1 h−1), and a comparable performance with P25 (1019 μmol g−1 h−1) under AM 1.5G simulated sunlight with the assistance of 1% Pt cocatalyst (Fig. 6e). It is well known that P25 is an excellent ultraviolet light absorber, while its visible light absorbance ability is low. Comparatively, the absorption edge of CaCl2/rutile TiO2 showed an obvious red shift compared with P25, which means that CaCl2/rutile TiO2 owns a wider range of light absorption (Fig. S10†). Besides this, dislocations that involve defects and/or local disorder may provide a mid-gap state in CaCl2/rutile TiO2 to broaden the light absorption range.58 Thus, under visible light illumination (λ > 400 nm), CaCl2/rutile TiO2 exhibited the highest H2 evolution activity (177 μmol g−1 h−1), much higher than that of commercial rutile TiO2 (23 μmol g−1 h−1) and P25 (42 μmol g−1 h−1) (Fig. 6d). Surprisingly, without the assistance of a cocatalyst, CaCl2/rutile TiO2 still showed the most outstanding performance under the illumination of simulated sunlight (Fig. 6c), which also indicates the excellent charge separation ability of symbiotic CaCl2/rutile TiO2. The cycle tests of CaCl2/rutile TiO2 for photocatalytic H2 generation proved a high stability in performance (Fig. S12†), which is further confirmed by the maintained structural feature after photocatalytic tests (Fig. S13†). Such an excellent performance can be primarily attributed to (i) the special structure of CaCl2-phase TiO2 which provides the CaCl2/rutile TiO2 hetero-nanocomposite with a higher reduction ability, and (ii) the symbiotic relationship that allows the CaCl2/rutile TiO2 hetero-nanocomposite to show a strong interaction, which could separate and transport carriers efficiently.
TiO2 is a multipurpose material, and many efforts have been made investigating the microwave absorption and terahertz absorption performance of hydrogenated TiO2.59,60 Thus, we examined the microwave absorption performance (1–18 GHz) of CaCl2/rutile TiO2, Ref.TiO2 and P25 (Fig. S14 and S15†). Compared with Ref.TiO2 and P25, CaCl2/rutile TiO2 improved the reflection loss or absorption of the microwave irradiation (see details in the ESI†). This is likely from the existence of CaCl2 phase TiO2. There are also many other potential applications worth studying, and the symbiotic CaCl2/rutile TiO2 hetero-nanocomposite is believed to be a promising candidate in other areas.
Conclusions
We report the synthesis of a symbiotic CaCl2/rutile TiO2 hetero-nanocomposite through a facile hydrothermal approach. The successful synthesis relies on the existence of high concentration Ti(OH)2(OH2)42+, which affects nucleation and crystallization behaviour, resulting in the formation of CaCl2 structured TiO2 intergrown with rutile TiO2. In this composite, orthorhombic CaCl2 phase TiO2 with a space group of Pnnm was stabilized by tetragonal phase rutile nanocrystals through dislocation connection in between both structures. Such a symbiotic relationship leads to a type II heterojunction, which enhances the charge separation and transport ability. The specific structure of CaCl2/rutile TiO2 elevated the reduction capacity, enabling a superior photocatalytic water splitting activity higher than that of the most widely used photocatalyst P25. We believe that the synthesis of the symbiotic CaCl2/rutile TiO2 hetero-nanocomposite reported here would facilitate its extensive research in those fields that have not yet been found and would provide guidance for the synthesis of other advanced hetero-nanocomposite materials.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the technical assistance of the Beijing Synchrotron Radiation Facility (BSRF) and National Synchrotron Radiation Laboratory (NSRL), Hefei, China. We also thank Pro. Xiaolong Wang (Jilin University), Pro. Minqiang Wang (Xi'an Jiaotong University), Dr Song Xia (Xi'an Jiaotong University), and Dr Xiangli Che (Shanghai Institute of Silicate, Chinese Academy of Sciences) for their assistance with the measurement and analysis of microwave absorption. This research is supported by the financial assistance of NSFC (21671077, 21871106, 21571176, and 21771075) and Interdisciplinary Research Grant for PhDs of Jilin University (10183201816).Notes and references
- C. Xia, Y. X. Cai, Y. Ma, B. Y. Wang, W. Zhang, M. Karlsson, Y. Wu and B. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 20748–20755 CrossRef CAS PubMed.
- C. L. Tan and H. Zhang, J. Am. Chem. Soc., 2015, 137, 12162–12174 CrossRef CAS PubMed.
- H. Y. Wang, H. S. Yin, H. Huang, K. L. Li, Y. L. Zhou, G. I. N. Waterhouse, H. Lin and S. Y. Ai, Biosens. Bioelectron., 2018, 108, 89–96 CrossRef CAS PubMed.
- G. Chen, Y. Zhao, G. Fu, P. N. Duchesne, L. Gu, Y. Zheng, X. Weng, M. Chen, P. Zhang, C. W. Pao, J. F. Lee and N. Zheng, Science, 2014, 344, 495–499 CrossRef CAS PubMed.
- B. Kong, J. Tang, Y. Zhang, T. Jiang, X. Gong, C. Peng, J. Wei, J. Yang, Y. Wang, X. Wang, G. Zheng, C. Selomulya and D. Zhao, Nat. Chem., 2016, 8, 171–178 CrossRef CAS PubMed.
- J. Cai, M. Wu, Y. Wang, H. Zhang, M. Meng, Y. Tian, X. Li, J. Zhang, L. Zheng and J. Gong, Chem, 2017, 2, 877–892 CAS.
- X. Y. Zheng, C. K. Yang, X. H. Chang, T. Wang, M. Ye, J. Lu, H. H. Zhou, J. Zheng and X. G. Li, Adv. Mater., 2018, 30, 1704353 CrossRef PubMed.
- D. Jung, L. M. A. Saleh, Z. J. Berkson, M. F. El-Kady, J. Y. Hwang, N. Mohamed, A. I. Wixtrom, E. Titarenko, Y. Shao, K. McCarthy, J. Guo, I. B. Martini, S. Kraemer, E. C. Wegener, P. Saint-Cricq, B. Ruehle, R. R. Langeslay, M. Delferro, J. L. Brosmer, C. H. Hendon, M. Gallagher-Jones, J. Rodriguez, K. W. Chapman, J. T. Miller, X. Duan, R. B. Kaner, J. I. Zink, B. F. Chmelka and A. M. Spokoyny, Nat. Mater., 2018, 17, 341–348 CrossRef CAS PubMed.
- M. Zhu, Z. Sun, M. Fujitsuka and T. Majima, Angew. Chem., Int. Ed., 2018, 57, 2160–2164 CrossRef CAS PubMed.
- Y. P. Zang, L. P. Li, Y. S. Xu, Y. Zuo and G. S. Li, J. Mater. Chem. A, 2014, 2, 15774–15780 RSC.
- B.-R. Wulan, S.-S. Yi, S.-J. Li, Y.-X. Duan, J.-M. Yan and Q. Jiang, Appl. Catal., B, 2018, 231, 43–50 CrossRef CAS.
- Q. Li, F. Zhao, C. Qu, Q. Shang, Z. Xu, L. Yu, J. R. McBride and T. Lian, J. Am. Chem. Soc., 2018, 140, 11726–11734 CrossRef CAS PubMed.
- B. Dong, J. Cui, Y. Gao, Y. Qi, F. Zhang and C. Li, Adv. Mater., 2019, e1808185 CrossRef PubMed.
- X. Chen, L. Li, Y. Xu, Y. Zhang and G. Li, RSC Adv., 2016, 6, 995–1003 RSC.
- Y. Suchorski, S. M. Kozlov, I. Bespalov, M. Datler, D. Vogel, Z. Budinska, K. M. Neyman and G. Rupprechter, Nat. Mater., 2018, 17, 519–522 CrossRef CAS PubMed.
- H. Jeong, J. Bae, J. W. Han and H. Lee, ACS Catal., 2017, 7, 7097–7105 CrossRef CAS.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates Jr, Science, 2011, 333, 736–739 CrossRef CAS PubMed.
- M. Shahidi, B. Pichler and C. Hellmich, Acta Mech., 2016, 227, 229–252 CrossRef.
- V. Thery, A. Boulle, A. Crunteanu, J. C. Orlianges, A. Beaumont, R. Mayet, A. Mennai, F. Cosset, A. Bessaudou and M. Fabert, Phys. Rev. B, 2016, 93, 184106 CrossRef.
- H. Liang, Q. Meng, X. Wang, H. Zhang and J. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 14145–14152 CrossRef CAS PubMed.
- H. Liu, W. Li, D. Shen, D. Zhao and G. Wang, J. Am. Chem. Soc., 2015, 137, 13161–13166 CrossRef CAS.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- J. C. Perez-Flores, C. Baehtz, A. Kuhn and F. Garcia-Alvarado, J. Mater. Chem. A, 2014, 2, 1825–1833 RSC.
- H. Razavi-Khosroshahi, K. Edalati, M. Hirayama, H. Emami, M. Arita, M. Yamauchi, H. Hagiwara, S. Ida, T. Ishihara, E. Akiba, Z. Horita and M. Fuji, ACS Catal., 2016, 6, 5103–5107 CrossRef CAS.
- A. Escudero, L. Delevoye and F. Langenhorst, J. Phys. Chem. C, 2011, 115, 12196–12201 CrossRef CAS.
- A. Escudero and F. Langenhorst, J. Solid State Chem., 2012, 190, 61–67 CrossRef CAS.
- J. Zhang, L. P. Li, X. S. Huang and G. S. Li, J. Mater. Chem., 2012, 22, 10480–10487 RSC.
- K. Suito, N. Kawai and Y. Masuda, Mater. Res. Bull., 1975, 10, 677–680 CrossRef CAS.
- Y. Tsuchida and T. Yagi, nature, 1989, 340, 217–220 CrossRef CAS.
- J. Haines, J. M. Leger and C. Chateau, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 8701–8706 CrossRef CAS.
- S. Ono and K. Mibe, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 054114 CrossRef.
- N. Stojilovic, J. Chem. Educ., 2018, 95, 598–600 CrossRef CAS.
- S. Pereira, M. R. Correia, E. Pereira, K. P. O'Donnell, R. W. Martin, M. E. White, E. Alves, A. D. Sequeira and N. Franco, Mater. Sci. Eng., B, 2002, 93, 163–167 CrossRef.
- M. J. Shearer, L. Samad, Y. Zhang, Y. Zhao, A. Puretzky, K. W. Eliceiri, J. C. Wright, R. J. Hamers and S. Jin, J. Am. Chem. Soc., 2017, 139, 3496–3504 CrossRef CAS PubMed.
- H. Huang, H. Jia, Z. Liu, P. Gao, J. Zhao, Z. Luo, J. Yang and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 3594–3598 CrossRef CAS PubMed.
- D. B. Varshney, J. A. Elliott, L. A. Gatlin, S. Kumar, R. Suryanarayanan and E. Y. Shalaev, J. Phys. Chem. B, 2009, 113, 6177–6182 CrossRef CAS PubMed.
- J. Hafizovic, M. Bjorgen, U. Olsbye, P. D. C. Dietzel, S. Bordiga, C. Prestipino, C. Lamberti and K. P. Lillerud, J. Am. Chem. Soc., 2007, 129, 3612–3620 CrossRef CAS PubMed.
- K. M. Jensen, M. Christensen, P. Juhas, C. Tyrsted, E. D. Bojesen, N. Lock, S. J. Billinge and B. B. Iversen, J. Am. Chem. Soc., 2012, 134, 6785–6792 CrossRef CAS PubMed.
- O. Frank, M. Zukalova, B. Laskova, J. Kurti, J. Koltai and L. Kavan, Phys. Chem. Chem. Phys., 2012, 14, 14567–14572 RSC.
- T. Lan, X. Tang and B. Fultz, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 094305 CrossRef.
- S. Zhou, E. Čižmár, K. Potzger, M. Krause, G. Talut, M. Helm, J. Fassbender, S. A. Zvyagin, J. Wosnitza and H. Schmidt, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 113201 CrossRef.
- J. C. Parker and R. W. Siegel, Appl. Phys. Lett., 1990, 57, 943–945 CrossRef CAS.
- V. Swamy, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 195414 CrossRef.
- C. Ribeiro, C. Vila, D. B. Stroppa, V. R. Mastelaro, J. Bettini, E. Longo and E. R. Leite, J. Phys. Chem. C, 2007, 111, 5871–5875 CrossRef CAS.
- C. X. Kronawitter, J. R. Bakke, D. A. Wheeler, W. C. Wang, C. Chang, B. R. Antoun, J. Z. Zhang, J. Guo, S. F. Bent, S. S. Mao and L. Vayssieres, Nano Lett., 2011, 11, 3855–3861 CrossRef CAS PubMed.
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1705369 CrossRef PubMed.
- L. Li, J. Yan, T. Wang, Z. J. Zhao, J. Zhang, J. Gong and N. Guan, Nat. Commun., 2015, 6, 5881 CrossRef PubMed.
- G. C. Vásquez, S. Z. Karazhanov, D. Maestre, A. Cremades, J. Piqueras and S. E. Foss, Phys. Rev. B, 2016, 94, 235209 CrossRef.
- S. O. Kucheyev, T. van Buuren, T. F. Baumann, J. H. Satcher, T. M. Willey, R. W. Meulenberg, T. E. Felter, J. F. Poco, S. A. Gammon and L. J. Terminello, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 245102 CrossRef.
- W. Hu, L. Li, G. Li, Y. Liu and R. L. Withers, Sci. Rep., 2014, 4, 6582 CrossRef CAS PubMed.
- W. Hu, L. Li, W. Tong and G. Li, Chem. Commun., 2010, 46, 3113–3115 RSC.
- H. Lin, L. Li, M. Zhao, X. Huang, X. Chen, G. Li and R. Yu, J. Am. Chem. Soc., 2012, 134, 8328–8331 CrossRef CAS PubMed.
- Y. Liu, J. Wei, Y. Tian and S. Yan, J. Mater. Chem. A, 2015, 3, 19000–19010 RSC.
- J. Ning, Q. Dai, T. Jiang, K. Men, D. Liu, N. Xiao, C. Li, D. Li, B. Liu, B. Zou, G. Zou and W. W. Yu, Langmuir, 2009, 25, 1818–1821 CrossRef CAS PubMed.
- R. Kuriki, T. Ichibha, K. Hongo, D. Lu, R. Maezono, H. Kageyama, O. Ishitani, K. Oka and K. Maeda, J. Am. Chem. Soc., 2018, 140, 6648–6655 CrossRef CAS PubMed.
- T. Xia, N. Li, Y. Zhang, M. B. Kruger, J. Murowchick, A. Selloni and X. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 9883–9890 CrossRef CAS PubMed.
- L. Guan and X. Chen, ACS Appl. Energy Mater., 2018, 1, 4313–4320 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- M. A. Green, J. Xu, H. Liu, J. Zhao, K. Li, L. Liu, H. Qin, Y. Zhu, D. Shen and X. Chen, Mater. Today Phys., 2018, 4, 64–69 CrossRef.
- T. Xia, C. Zhang, N. A. Oyler and X. Chen, Adv. Mater., 2013, 25, 6905–6910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01216h |
| This journal is © The Royal Society of Chemistry 2019 |