
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective C–H activation and regiospecific annulation using propargylic carbonates†
Qingquan
Lu‡
 ,
Shobhan
Mondal‡
,
Sara
Cembellín
,
Steffen
Greßies
and
Frank
Glorius
*
,
Shobhan
Mondal‡
,
Sara
Cembellín
,
Steffen
Greßies
and
Frank
Glorius
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
First published on 27th May 2019
Abstract
Herein we describe an unprecedented RuII-catalyzed site-selective and regiospecific annulation of benzoic acids with propargylic carbonates. The weakly coordinating carboxylic acid moiety outperformed other typically used directing groups in C–H activation, including ketone, nitrile, sulfonamide, amide and strongly coordinating nitrogen heterocycles. This is an important step towards the application of C–H activation reactions in complex (functional) real-world molecules.
Localization of a metal catalyst with a coordinating group is the vital step in directed C–H activation (Scheme 1a).1 To ensure positional selectivity, the intended directing group must overcome other coordinating groups present in the substrate. This has significantly limited the application of directed C–H activation to the preparation of valuable molecules typically containing prevalent amines, medicinally important N-heterocycles, as well as other strongly coordinating groups, which can interact with the metal catalyst and further deactivate the catalyst completely.2 Very recently, positionally-selective C–H activation that can elegantly tolerate strongly coordinating N-heterocycles has been proven feasible by the groups of Yu and Ackermann using an installed X-type N-directing group as the site selectivity-ensuring entities,2b,2c and the precise nature of the activation model still remains unclear. To avoid the time- and labor-intensive installation and removal (often under harsh conditions) of directing groups in C–H activation, the use of naturally abundant and bioactive carboxylic acids to achieve robust control of positional selectivity is therefore highly desirable,3 which can accelerate the discovery of new medicines. However, the low σ-donating ability of the carboxylate poses substantial challenges towards anchoring metal centers close to the target C–H bond and reduces the activity of the resulting metallacycle towards electrophiles.4 In this regard, Yu et al. have identified carboxylic acids to direct C–H activation through weak coordination, and they proposed that the alkali metal cation could coordinate with carboxylate group in a κ2 fashion and force Pd(II) to coordinate with the unhindered lone pair of electrons on the oxygen atom, thus enabling the palladacycle formation step.1i,4c
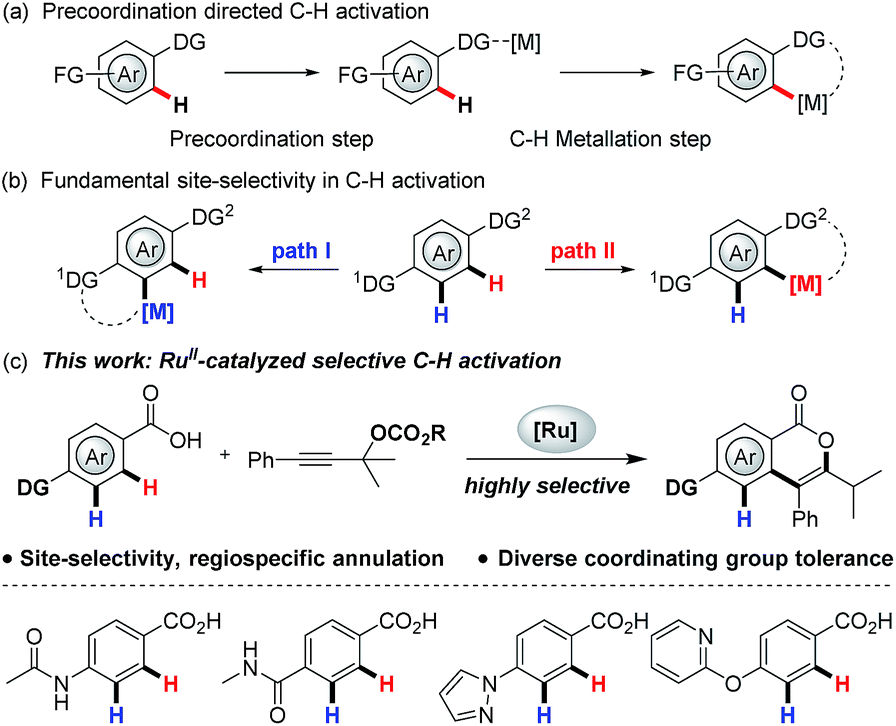 | ||
| Scheme 1 RuII-catalyzed site-selective C–H activation. | ||
Equally, annulation reactions with alkynes are one of the most popular methods to construct valuable cyclic structures, which have been extensively explored in C–H activation.3,5 However, almost all the methods reported to date suffer from the uncontrolled regioselectivity arises from alkyne insertion step when unsymmetrical alkynes are utilized, inevitably leading to regioisomeric product mixtures.3,5,6 To address this issue, installation of traceless directing groups (TDG) into an alkyne is an alternative pathway to discriminate the reactive sites, which could enable a regioselective alkyne insertion via precoordination to metal catalyst.7 Furthermore, this precoordination can also improve the activity of the resulting metallacycle with electrophilic alkyne, providing the possibility of access to site-selective and regiospecific annulation of a weakly O-coordinating carboxylate with propargylic compounds, which has not been explored yet.
To deliver selectivity, there are several intricate challenges to be overcome:
(1) The initial C–H metalation step would have to overcome the interaction of the transition metal catalyst with the electrophile, while η1-and η3-propargylic/allenylic metal species might be generated through oxidative addition of the metal catalyst with propargylic compounds, which could directly react with nucleophilic benzoates.8
(2) As previously highlighted, C–H activation at the ortho position of the carboxyl group in the presence of other coordinating groups needs to occur in a highly selective fashion, and further second C–H activation might result after the formation of desired product.
(3) C–O reductive elimination competes with β-oxygen elimination from intermediate III (Scheme 7), and the relative reaction rate for the two processes will largely determine final product distribution.6
(4) The generation of an aryl-metal species in situ from decarboxylation of benzoates would result in an undesired decarboxylative coupling.9
In the present work, these processes were well controlled by selection of a suitable catalyst and propargylic compound, enabling a site-selective C–H activation and regiospecific annulation that fully tolerates strongly coordinating groups.
To probe the feasibility of our assumption, we began our investigation by reacting benzoic acid (1a) with tertiary propargylic alcohol under ruthenium catalysis (Scheme 2). To our delight, the desired isocoumarin 3aa was obtained in 14% yield. The low yield was supposed to be due to the less efficient precoordination of the hydroxyl group to the metal catalyst and the slow downstream β-oxygen elimination. On this basis, a series of propargylic compounds, which contain a coordinating carbonyl oxygen atom and a better leaving group that might accelerate the catalyst–substrate interaction and β-oxygen elimination, were explored. It was found that propargylic carbonates exhibit high reactivity and the yield of 3aa was improved to 88%. Further experiments showed that the catalyst also plays a crucial role (for details, see the ESI†). State-of-the-art precatalysts widely employed in C–H activation, such as [Cp*Rh(MeCN)3](SbF6)2, [Cp*IrCl2]2, [Cp*Co(MeCN)3](SbF6)2 and [MnBr(CO)5] were inactive in this new challenging transformation.
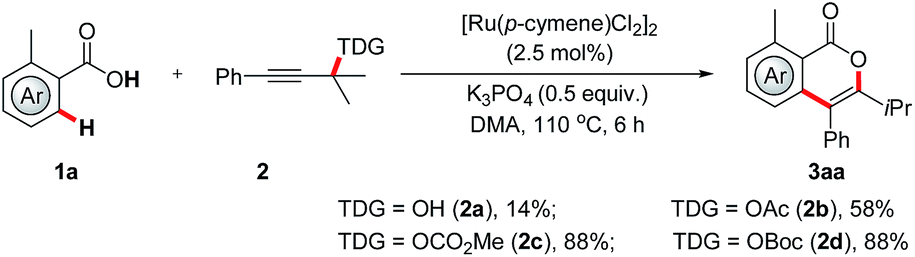 | ||
| Scheme 2 RuII-catalyzed regiospecific synthesis of isocoumarins. | ||
With the optimized reaction conditions in hand, we first examined the scope of carboxylic acids. As shown in Scheme 3, an extensive array of benzoic acids, bearing diverse neutral, electron-donating and electron-withdrawing groups, reacted smoothly with 2c to afford the corresponding isocoumarins 3aa–3oa in 42% to 92% yield. Significantly, a variety of functionalities, such as formyl, halogen, alkenyl, unprotected amino and hydroxy groups, were well tolerated. Typically, these functional groups offer vectors for further elaboration. Further application of this method to heteroaromatic carboxylic acids was also successful, thereby delivering valuable thiophene and indole derivatives 3la–3na in 64% to 92% yields. It is particularly noteworthy that this protocol provides direct access to late-stage derivatization of drug molecules (adapalene, 3oa) and (probenecid, 3sa in Scheme 4). Acrylic acid derivative, as exemplified by cyclohex-1-ene-1-carboxylic acid, could also serve as a suitable reaction partner in this protocol to give the desired pyrone 3pa in moderate yield. Next, the scope of the reaction with respect to propargylic carbonates was explored. The sterically hindered alkyl groups, such as ethyl and cyclopentyl, located on the tertiary carbon of the propargylic carbonate, were found to marginally affect the efficiency of these reactions, providing the products 3ab–3ad in 52% to 78% yields. The R1 group in the propargylic carbonates was tolerated when it was an aryl or heteroaryl group (3aa, 3ae). In addition, the regioselectivity of this protocol was confirmed by the X-ray crystallography of 3aa.10
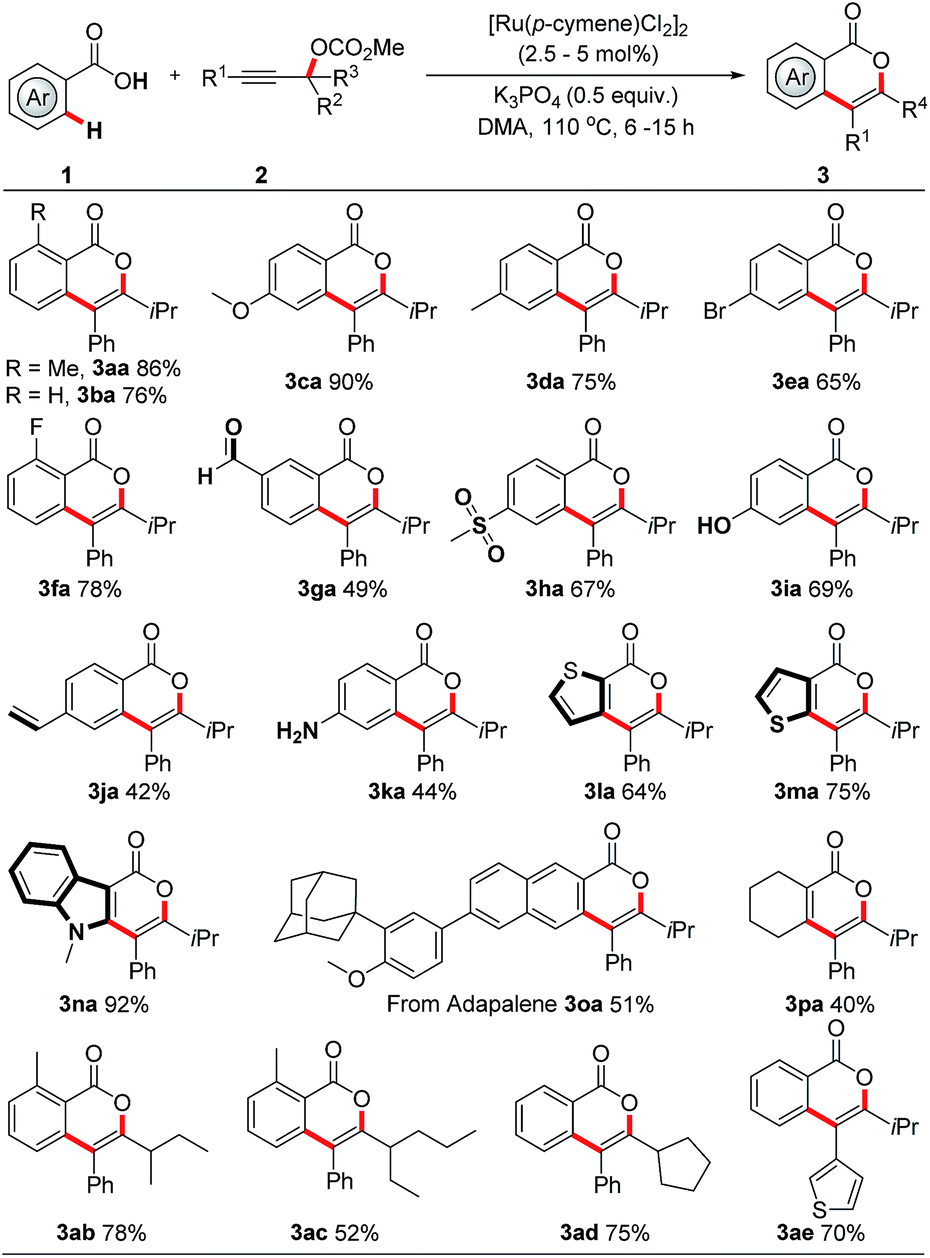 | ||
| Scheme 3 RuII-catalyzed regiospecific synthesis of isocoumarins. For details, see the ESI.† | ||
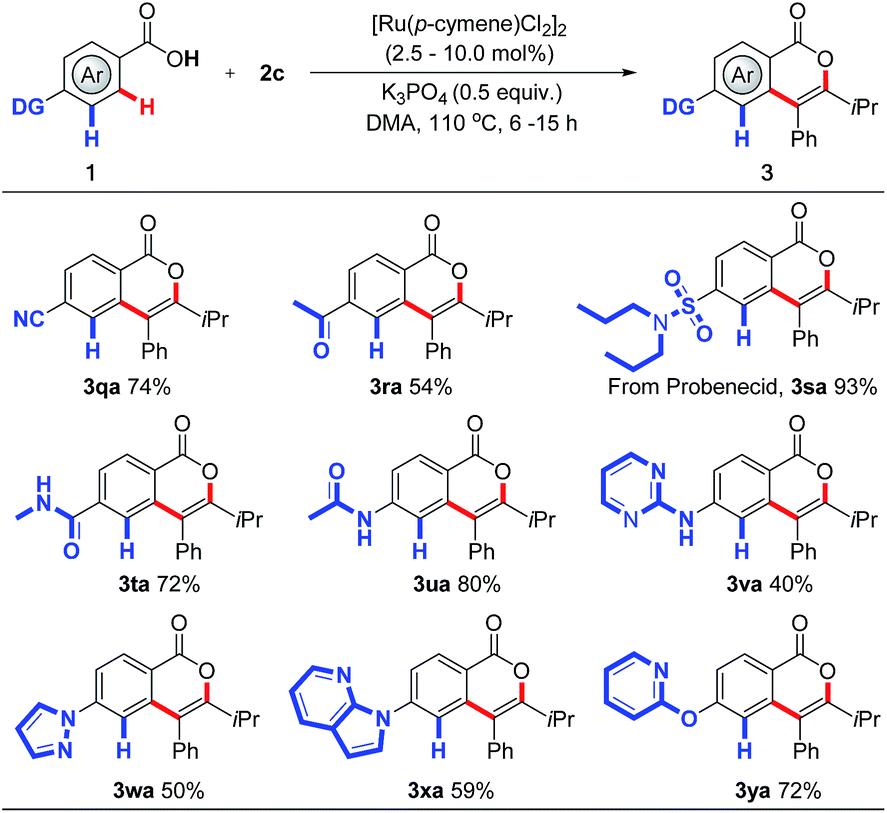 | ||
| Scheme 4 RuII-catalyzed site-selective C–H activation. For details, see the ESI.† | ||
Encouraged by the broad tolerance of functionality, we extended this approach to control the site-selectivity in the presence of multiple chelating groups (Scheme 4), which has remained a great challenge in C–H activation. Interestingly, compared with keto and cyano groups, which have proved to exhibit proximity effects in C–H activation,11 carboxylic group displays preferential reactivity, affording the corresponding products 3qa and 3ra in 74% and 54% yields respectively. Importantly, further experiments showed that yet more efficient directing groups can also be compatible in this protocol. The weakly coordinating carboxylic acid overrides conventionally efficient directing groups in C–H activation including sulfonamide, amide, pyrazole, pyrimidine, and even pyridine,11 delivering the desired products 3sa–3ya in moderate to excellent yields. Indeed, this site-selectivity pattern has been seldom seen in C–H activation,2b,2c reflecting the benefit of this new strategy. We believe this may prove instrumental in future drug-discovery applications.
This procedure can be readily scaled to gram quantities with high efficiency. For example, 1.42 g of 3aa was isolated, corresponding to 85% yield (Scheme 6a). The synthetic utility of this method was further illustrated by other high-value molecule synthesis (Scheme 5). For example, isoquinolines, which are common motifs within pharmaceuticals and natural products,12 were delivered in moderate yields and exclusive regioselectivity using this unique ruthenium catalysis.
 | ||
| Scheme 5 RuII-catalyzed regiospecific synthesis of isoquinolines. | ||
To gain insight into the reaction mechanism, a series of experiments were conducted (Scheme 6). The reaction of isotopically-labeled [D5]-benzoic acid ([D]-1b) with 2c was first carried out. A 32% deuterium loss on the C-6 site in [D]-3ba was observed (Scheme 6b), revealing that the C–H ruthenation step is reversible. Moreover, an apparent kinetic isotope effect (kH/kD = 3.34) was observed from two parallel reactions of 1b or [D]-1b with 2c (for details, see the ESI†), which suggests that cleavage of the C–H bond was involved in the rate-determining step. Furthermore, intermolecular competition experiments showed that electron-deficient benzoic acids were more reactive (for details, see the ESI†), which indicates a carboxylate-assisted, concerted metalation-deprotonation type C–H ruthenation mechanism. Additionally, a side product 3aa′ was isolated from the gram-scale synthesis (Scheme 6a), which can be quantitatively transformed into the desired product 3aa under standard condition.13 Next, no chirality transfer was observed when chiral carbonate (S)-2e (98% ee) was applied.
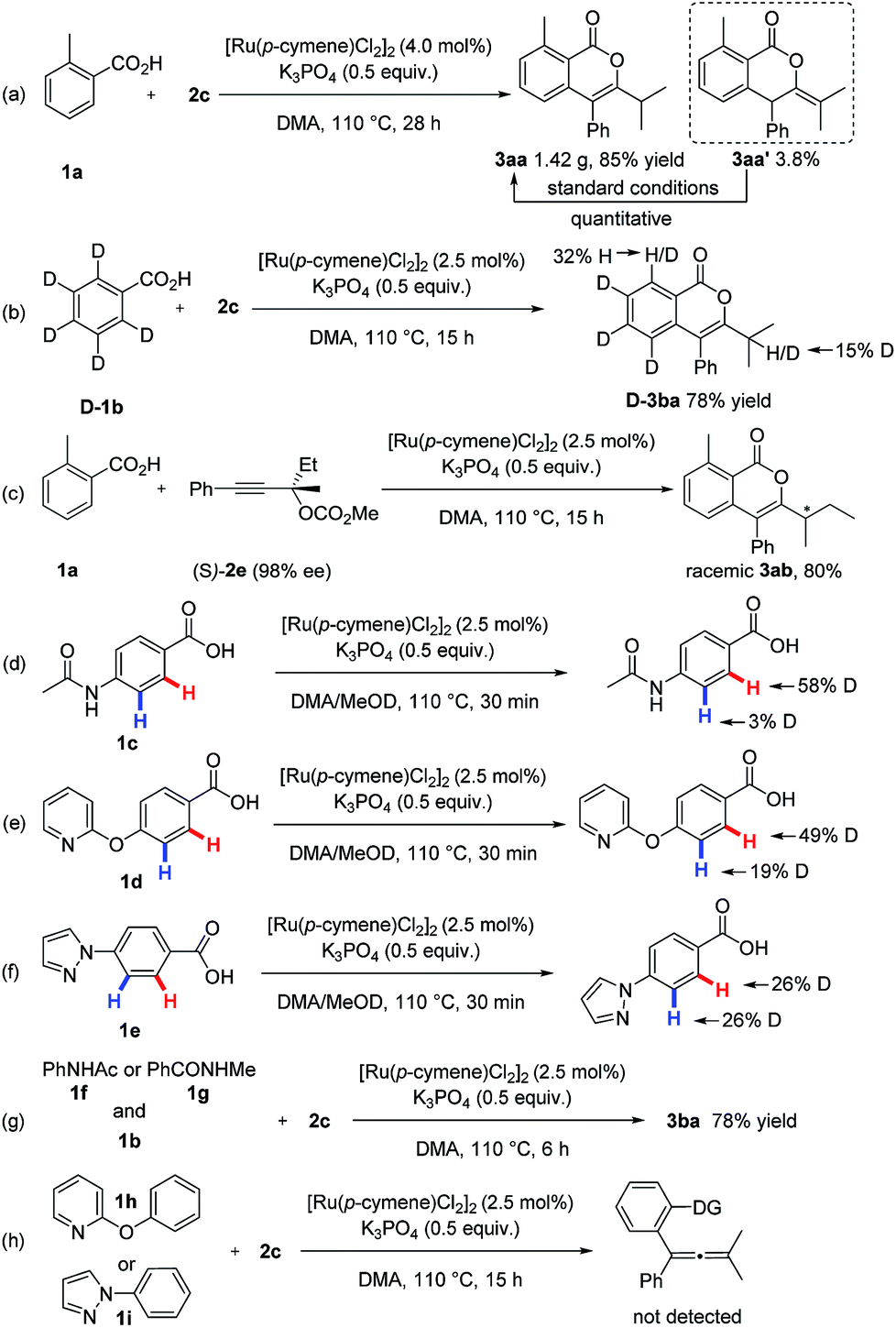 | ||
| Scheme 6 Gram synthesis and mechanistic studies. | ||
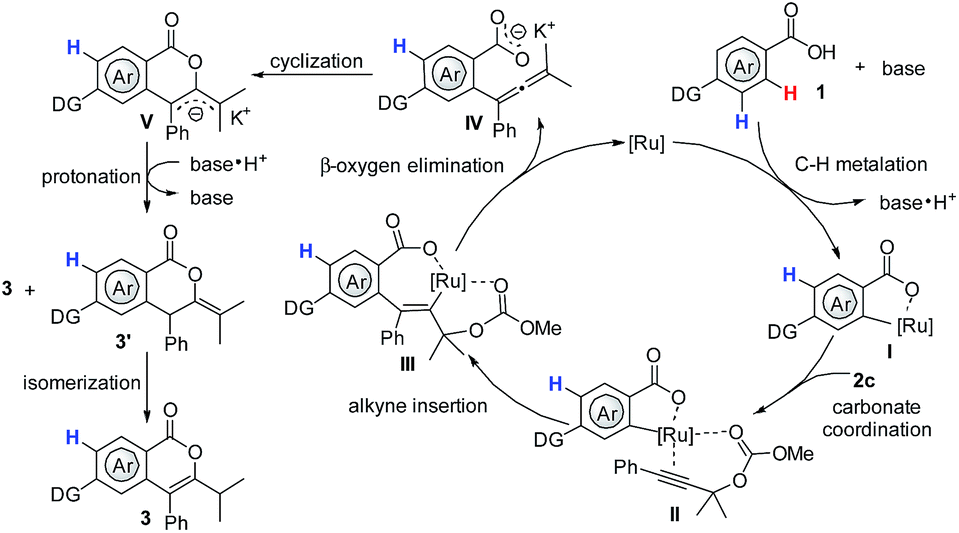 | ||
| Scheme 7 Proposed mechanism. | ||
To better understand the origin of the observed site-selectivity, competitive H/D scrambling experiments were conducted. It was found that H/D exchanges happened in the both ortho-positions of the different coordinating groups (Scheme 6d–f), clearly illustrating that the C–H metalation step does not determine the site-selectivity. Subsequently, intermolecular competition reactions between N-phenylacetamide 1f or N-methylbenzamide 1g with 1b were performed. Carboxylate-directed product 3ba was exclusively formed (Scheme 6g), showing the carboxylic acid outcompeting strongly coordinating groups in this protocol. Furthermore, 2-phenoxypyridine 1h and 1-phenyl-1H-pyrazole 1i did not furnish the potential C–H activation products with 2c under standard conditions (Scheme 6h). These findings indicate that the site-selectivity is determined in the alkyne insertion step. Since the β-oxygen elimination was proven to be a facile process in this ruthenium catalysis, thus the feasibility of propargylic carbonate insertion into the C–Ru bond within the rutheniumcycle plays a crucial role in determining the site-selectivity. The importance of steric “match/mismatch” effect between transition metal species generated in situ and external coupling partner in regard to site-selectivity has been underutilized in C–H activation.14
Based on the aforementioned results and previous reports,3,6,7,15 a plausible mechanism was proposed. The reaction commences with base-assisted cycloruthenation of benzoic acid 1, forming a five-membered ruthenacycle I. Afterwards, coordination of the carbonyl oxygen of the carbonate 2c to I forms ruthenacycle II, which is followed by a regioselective alkyne insertion and subsequent β-oxygen elimination to deliver the active allene intermediate IV and regenerate the active Ru(II) catalyst. Finally, a highly selective intramolecular cyclization of IV would furnish the desired isocoumarins 3 and 3′, the later could be further transformed into 3in situ.
Conclusions
We developed an unprecedented redox-neutral, ruthenium-catalyzed regiospecific annulation of benzoic acids with propargylic carbonates. The newly developed protocol is site-selective towards the respective C–H bonds in the presence of other chelating groups. An array of isocoumarins, with a wide range of functionalities, were prepared regioselectively, which have not been successfully reported as single isomers via previously reported C–H activation methodology. Mechanistic studies indicate that a steric “match/mismatch” effect plays a crucial role in the obtained site-selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Alexander von Humboldt Foundation (Q. L. and S. C.), the WWU Münster (Graduate School of Molecules and Interfaces, S. M.) and the Deutsche Forschungsgemeinschaft (Leibniz Award). We also thank Dr Michael J. James and Toryn Dalton for helpful discussions.Notes and references
- (a) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (e) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (f) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (g) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (h) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (i) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (j) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (k) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (l) P. Gandeepan and C.-H. Cheng, Chem.–Asian J., 2016, 11, 448 CrossRef CAS PubMed; (m) W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743 CrossRef CAS.
- (a) R. Mandal and B. Sundararaju, Org. Lett., 2017, 19, 2544 CrossRef CAS PubMed; (b) Y.-J. Liu, H. Xu, W.-J. Kong, M. Shang, H.-X. Dai and J.-Q. Yu, Nature, 2014, 515, 389 CrossRef CAS PubMed; (c) H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 10386 CrossRef CAS PubMed; (d) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383 CrossRef CAS PubMed.
- L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed.
- (a) A. Biafora, T. Krause, D. Hackenberger, F. Belitz and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 14752 CrossRef CAS PubMed; (b) H. Liangbin, B. Agostino, Z. Guodong, B. Valentina and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 6933 CrossRef PubMed; (c) J. He, M. Wasa, K. S. L. Chan, Q. Shao and Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed.
- (a) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (b) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (c) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466 CrossRef CAS; (d) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155 RSC; (e) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed.
- (a) L. Ackermann, J. Pospech, K. Graczyk and K. Rauch, Org. Lett., 2012, 14, 930 CrossRef CAS PubMed; (b) R. K. Chinnagolla and M. Jeganmohan, Chem. Commun., 2012, 48, 2030 RSC; (c) R. Prakash, K. Shekarrao and S. Gogoi, Org. Lett., 2015, 17, 5264 CrossRef CAS PubMed; (d) S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepötter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513 CrossRef CAS PubMed; (e) K. S. Singh, S. G. Sawant and P. H. Dixneuf, ChemCatChem, 2016, 8, 1046 CrossRef CAS; (f) Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5818 CrossRef CAS PubMed.
- (a) Q. Lu, S. Greßies, S. Cembellín, F. J. R. Klauck, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12778 CrossRef CAS PubMed; (b) S. Wu, X. Huang, W. Wu, P. Li, C. Fu and S. Ma, Nat. Commun., 2015, 6, 7946 CrossRef PubMed.
- (a) S. Ma, Eur. J. Org. Chem., 2004, 2004, 1175 CrossRef; (b) L.-N. Guo, X.-H. Duan and Y.-M. Liang, Acc. Chem. Res., 2011, 44, 111 CrossRef CAS PubMed; (c) M. Yoshida, Chem. Pharm. Bull., 2012, 60, 285 CrossRef CAS PubMed.
- (a) A. G. Myers, D. Tanaka and M. R. Mannion, J. Am. Chem. Soc., 2002, 124, 11250 CrossRef CAS; (b) L. J. Gooßen, G. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef PubMed; (c) L. J. Goossen, N. Rodríguez, B. Melzer, C. Linder, G. Deng and L. M. Levy, J. Am. Chem. Soc., 2007, 129, 4824 CrossRef CAS PubMed.
- CCDC 1866187 (3aa) contain the supplementary crystallographic data for this paper.†.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- V. A. Glushkov and Y. V. Shklyaev, Chem. Heterocycl. Compd., 2001, 37, 663 CrossRef CAS.
- There are two possibilities for the cyclization: carboxylic acid addition to the exo double bond of the allene, or carboxylic acid addition to the endo double bond of the allene followed by isomerization of the remaining exo double bond.
- M. P. Doyle, J. P. Morgan, J. C. Fettinger, P. Y. Zavalij, J. T. Colyer, D. J. Timmons and M. D. Carducci, J. Org. Chem., 2005, 70, 5291 CrossRef CAS PubMed.
- (a) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (b) P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1866187. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01703h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |