DOI:
10.1039/C9SC01701A
(Edge Article)
Chem. Sci., 2019,
10, 6868-6875
Formation of an imidazoliumyl-substituted [(LC)4P4]4+ tetracation and transition metal mediated fragmentation and insertion reaction (LC = NHC)†
Received
7th April 2019
, Accepted 29th May 2019
First published on 30th May 2019
Abstract
Tetracationic cyclo-tetraphosphane [(LC)4P4]4+ as triflate salt (3[OTf]4) (LC = 4,5-dimethyl-1,3-diisopropyl-imidazol-2-yl) is obtained in high yield from the reduction of [LCPCl2]+ (4[OTf]) with 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine (6) and represents the first salt of the cationic cyclo-phosphane series with the general formula [LnPn]n+. Theoretical calculations reveal the electrophilic nature of the P atoms within the P4-ring due to the influence of the imidazoliumyl-substituents. Further reduction of 3[OTf]4 with 6 affords the unexpected formation of the notricyclane P7-type cation [(LC)3P7]3+ (9[OTf]3). Selective transition metal mediated [2 + 2]-fragmentation of 34+ is achieved when 3[OTf]4 is reacted with Fe2(CO)9, Pd(PPh3)4 and Pt(PPh3)4 leading to the formation of the dicationic diphosphene complexes [(η2-LCP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PLC)Fe(CO)4]2+ (12[OTf]2) and [(η2-LCPPLC)M(PPh3)2]2+ (13[OTf]2 for M = Pd; 14[OTf]2 for M = Pt). In contrast, the reaction of 3[OTf]4 with an excess of AuCl(tht) gives rise to the formation of the five-membered ring complex [((LC)4P4)AuCl2]3+ (15[OTf]3), where the Au(I) atom reductively inserts into a P–P bond of 34+.
PLC)Fe(CO)4]2+ (12[OTf]2) and [(η2-LCPPLC)M(PPh3)2]2+ (13[OTf]2 for M = Pd; 14[OTf]2 for M = Pt). In contrast, the reaction of 3[OTf]4 with an excess of AuCl(tht) gives rise to the formation of the five-membered ring complex [((LC)4P4)AuCl2]3+ (15[OTf]3), where the Au(I) atom reductively inserts into a P–P bond of 34+.
Introduction
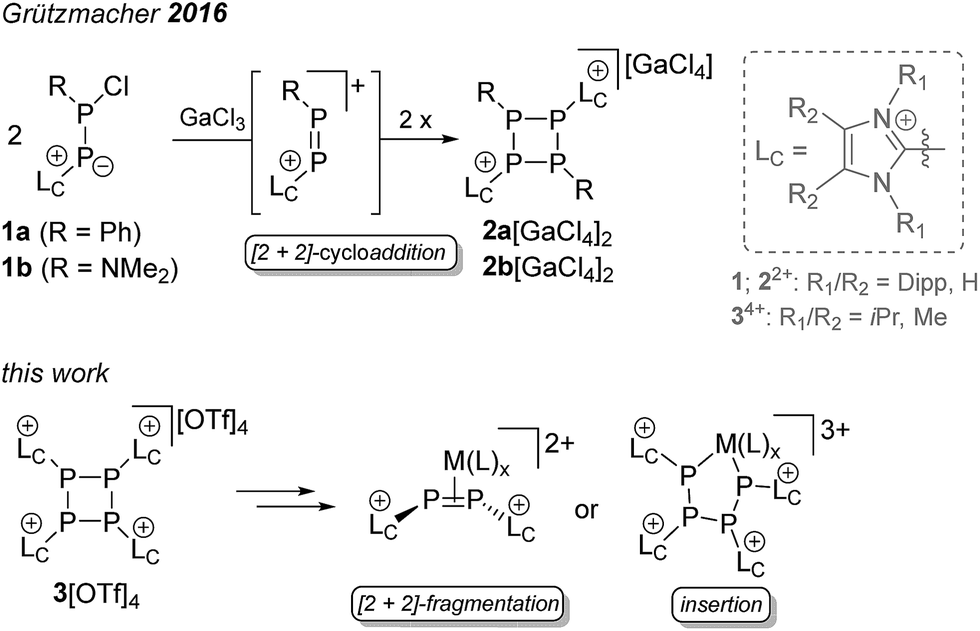
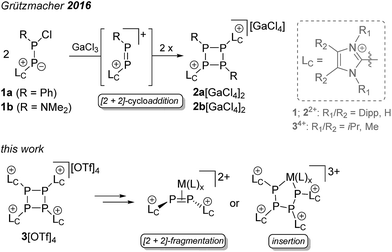
The fact that phosphorus tends to assemble into homo-nuclear cyclic and cage-like structural motifs is not only illustrated by the variety of phosphorus allotropes,1 but also by the multitude of known polyphosphanes.2 Particularly, neutral monocyclic polyphosphanes with the general formula PnRn (R = alkyl, aryl, n = 3–5) are already known for decades and their chemistry is well established.3–5 There are several synthetic protocols for this group of compounds, of which the reduction of a dichlorophosphane RPCl2 (R = alkyl, aryl) with various reducing agents represents the most common approach.6 The steric demand of the substituents at the P atom determines the favored ring-size.6,7 Thus, the reduction of CyPCl2 with Mg yields the thermodynamically favored cyclo-Cy4P4,8 whereas the reduction of PhPCl2 gives mainly cyclo-Ph5P5.9 A mixture of compounds with several ring sizes is often observed in the initial reaction, however, a scrambling to the thermodynamically favored ring-size typically occurs over time.6–9 When the steric demand of the substituent is too large, the formation of cyclo-phosphanes does not proceed, as illustrated by the reduction of Mes*PCl2 (Mes* = 2,4,6-tri(tert-butyl)phenyl) to diphosphene Mes*–PP–Mes*.10 Generally, mixed-substituted or cationic cyclo-phosphanes are very scarce and the only examples of dicationic cyclo-phosphanes of type [R2(LC)2P4]2+ (2a,b2+) (LC = 1,3-bis(2,6-diisopropylphenyl)-imidazol-2-yl, 1a: R = Ph, 1b: R = NMe2; Scheme 1) were recently introduced by Grützmacher and co-workers as a result of a GaCl3-induced dimerization reaction of 1a,b.11 Mechanistically, it has been shown that cyclo-phosphanes 2a,b2+ are formed via a [2 + 2]-cycloaddition of the generated diphosphene intermediates. We targeted the replacement of all substituents R in cyclo-PnRn by an imidazoliumyl-substituent LC, resulting in the new group of cationic cyclo-phosphanes of type [(LC)nPn]n+ (Scheme 1). These cationic substituents LC are frequently employed in phosphorus chemistry as they are known to have a significant influence on the reactivity of the directly bonded P atom.12–15 In this contribution we present the formation of a tetracationic cyclo-tetraphosphane [(LC)4P4]4+ as triflate salt (3[OTf]4) via the reduction of [LCPCl2]+ (4[OTf])12 and elucidate the mechanism for its formation. Furthermore, the reductive build-up reaction to a larger cationic phosphorus framework and selective transition metal mediated [2 + 2]-fragmentation and ring opening reactions were investigated.
 |
| | Scheme 1 Cationic cyclo-phosphanes of type [R2(LC)2P4]2+ (2a,b2+) and [(LC)4P4]4+ (34+) and subsequent transition metal mediated fragmentation and insertion reactions. | |
Results and discussion
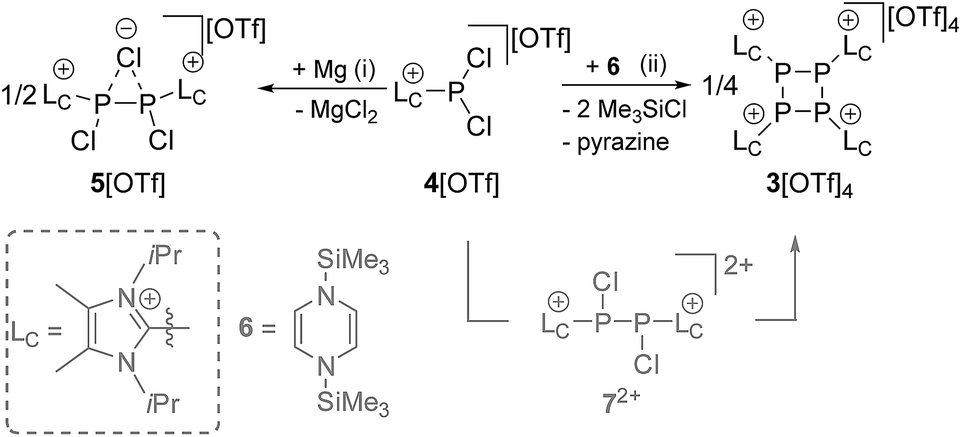
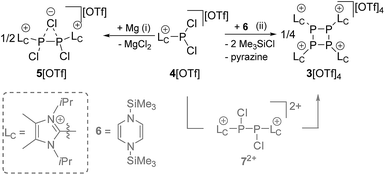
We first reduced compound 4[OTf] with Mg in THF and observed the formation of a yellow-colored reaction mixture (Scheme 2). After removing MgCl2 by filtration, the 31P NMR spectrum of the obtained clear solution reveals a singlet resonance at δ(P) = −24.0 ppm. This resonance can be attributed to the chloride bridged P2-compound 5[OTf] which was confirmed by X-ray analysis (see Fig. S2†).16 As compound 5[OTf] is similar to our previous findings, in which we employed a sterically more demanding imidazoliumyl-substituent (LC = 1,3-bis(2,6-diisopropylphenyl)-imidazol-2-yl), we abstain from a detailed discussion here.17,18 It is noteworthy to mention, however, that reduction of 4[OTf] with Na gives the same result. The reduction proceeds completely different when 4[OTf] is reacted with 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine (6) in fluorobenzene as solvent. Within 12 h the formation of a colorless precipitate is observed which after filtration and removal of all volatiles in vacuo gives the analytically pure triflate salt of the tetracationic cyclo-tetraphosphane 34+ in very good yield (86%, Scheme 2). Although compound 3[OTf]4 is well soluble in CH3CN it slightly decomposes in solution in the course of several days. The 31P NMR spectrum of 3[OTf]4 dissolved in CD3CN, shows a singlet resonance at δ(P) = −55.7 ppm, which is comparable to tBu4P4 (δ(P) = −58.1 ppm).19 Single crystals suitable for X-ray analysis are obtained by slow diffusion of Et2O into a saturated CH3CN solution of 3[OTf]4 at −35 °C. Fig. 1 shows the molecular structure of tetracation 34+ and reveals the typical butterfly-shaped P4 motif with an averaged P–P–P angle of 81.59°. All imidazoliumyl-substituents at the P4 ring system are arranged in equatorial and all-trans position. The P–P bond lengths in 34+ (av. 2.235 Å) are in the range for P–P single bonds (2.22 Å)20 and the P–C bond lengths (av. 1.822 Å) are similar to comparable imidazoliumyl-substituted phosphorus compounds.15 In order to elucidate whether the formation of tetracation 34+ proceeds through a diphosphene intermediate, we monitored the aforementioned reaction by means of 31P NMR spectroscopy (see Fig. S3†). Notably, Tamm and co-workers recently reported on the isolation of a diphosphene derivative obtained by the reduction of related dichlorophosphane with compound 6.21 The 31P NMR spectrum of the reaction mixture shows only one major prominent intermediate with a singlet resonance at δ(P) = −29.1 ppm, which is in the range for diphosphanes.22 We assign this intermediate to dication 72+ which is similar to findings of Goicoechea and co-workers who isolated a related dibromo-diphosphane compound featuring imidazoliumyl-substituents.18 To further proof our assumption we calculated the chemical shift of intermediate 72+ and found a theoretical 31P NMR shift of δ(P) = −33.4 ppm being in a reasonable range compared to the experimentally found shift.16,23 We confirmed the existence of dication 72+ by the addition of a chloride source, namely nBu4N[Cl] and observed the formation of chloride-bridged cation 5+ from 72+.16
 |
| | Scheme 2 Reduction of 4[OTf] with Mg (left) and 6 (right) yields either 5[OTf] or 3[OTf]4, respectively. Proposed reaction pathway with intermediate 72+ to 3[OTf]4 (grey); (i): +ex. Mg, 12 h, −2MgCl2, 99% (determined by 31P NMR spectroscopy); (ii): +6, C6H5F, 12 h, −pyrazine, −2Me3SiCl, 86%. | |
 |
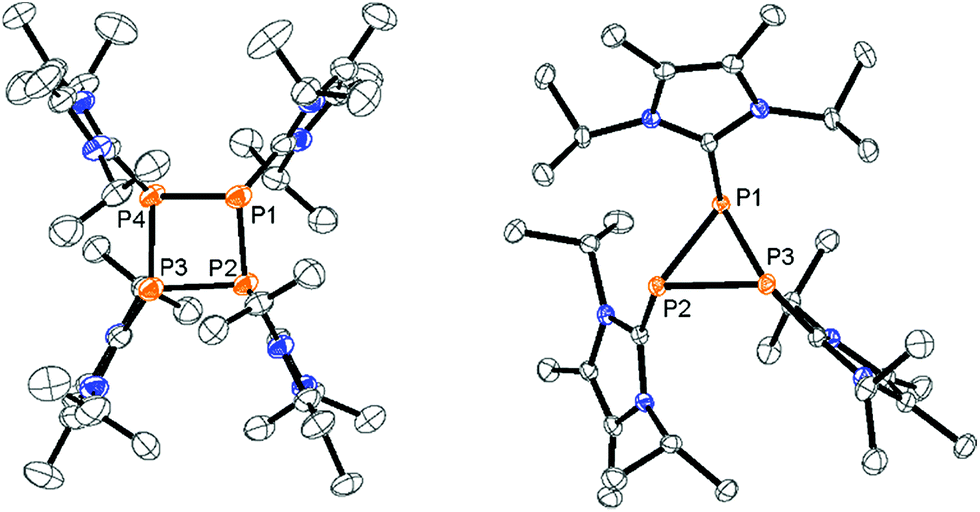
| | Fig. 1 Molecular structures of tetracation 34+ in 3[OTf]4·2CH3CN (left) and trication 83+ in 8[OTf]3 (right; hydrogen atoms, solvate molecules and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability); selected bond lengths (Å) and angles (°) for 34+: P1–P2 2.2382(8), P2–P3 2.2356(8), P3–P4 2.2400(8), P4–P1 2.2298(8), P1–C1 1.822(2), P2–C12 1.824(2), P3–C23 1.823(2), P4–C34 1.822(2); P4–P1–P2 81.69(3), P1–P2–P3 81.54(3), P2–P3–P4 81.53(3), P3–P4–P1 81.62(3); 83+: P1–P2 2.2459(6), P2–P3 2.2293(6), P1–P3 2.2139(6), P1–C1 1.8429(17), P2–C12 1.8373(17), P3–C23 1.8303(17); P1–P2–P3 59.300(18); P2–P3–P1 60.72(2), P3–P1–P2 59.975(18). | |
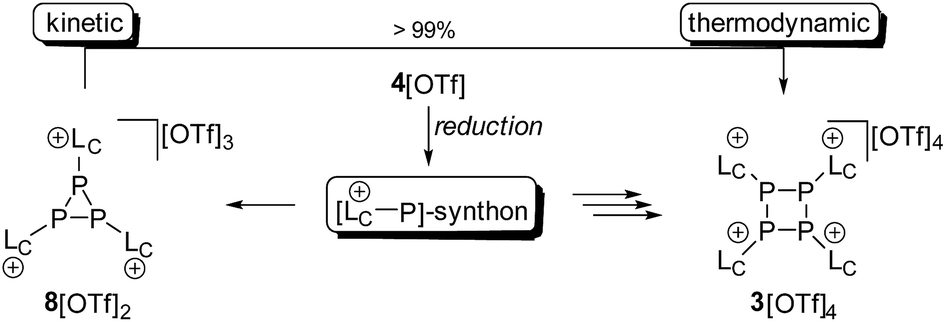
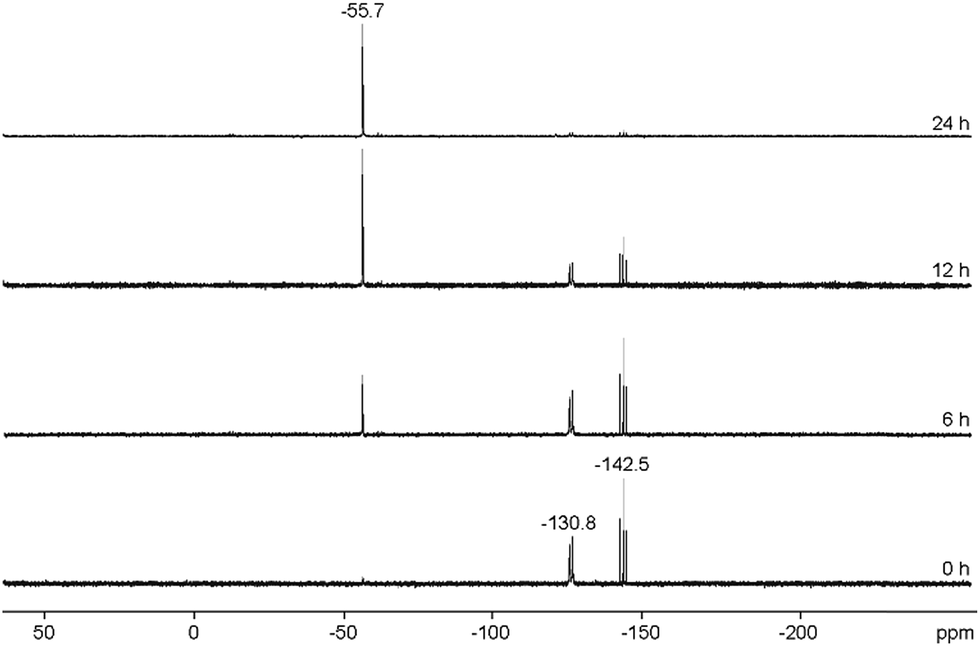
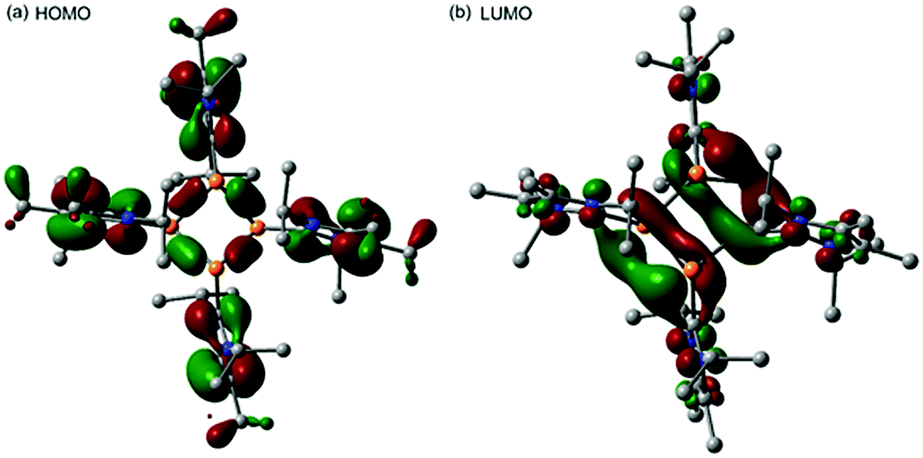
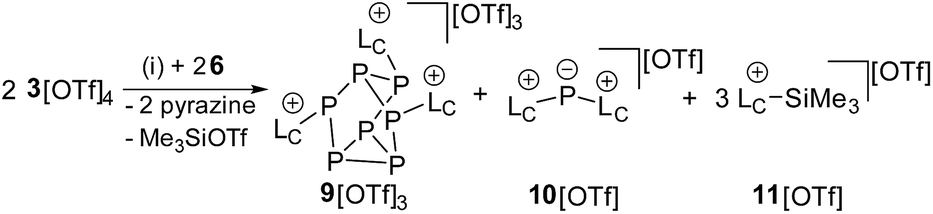
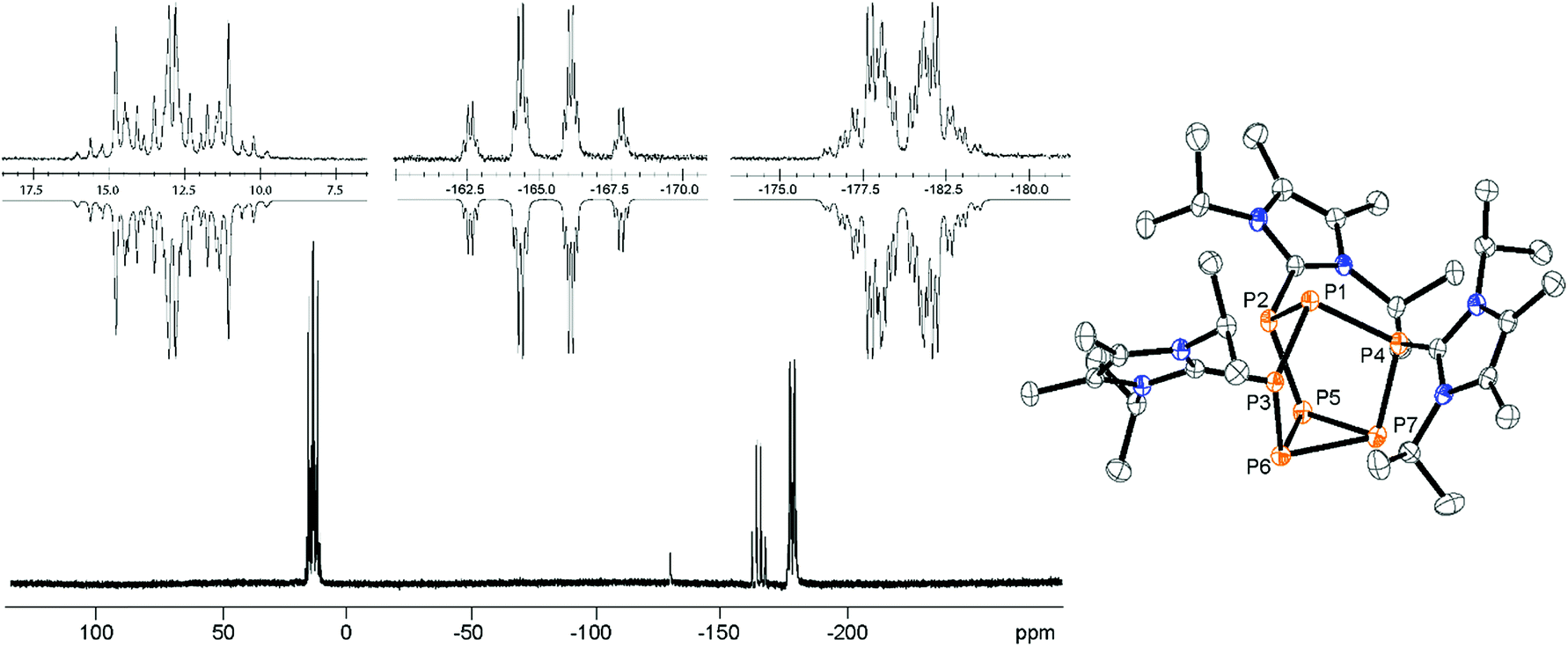
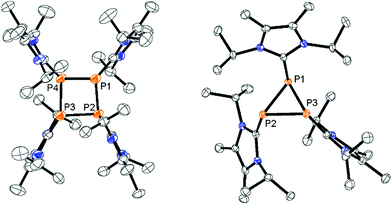
Mechanistically, we propose that 2 equivalents of 4+ react with one equivalent of 6 to give intermediate 72+ which subsequently reacts with another equivalent of 6 ultimately leading to tetracation 34+. The first reduction step might also lead to the formation of an [LCP]+-synthon which is supported by the formation of [(LC)3P3]+ (83+) in this reaction (Scheme 3). A few crystals of 8[OTf]3 could be isolated by overlaying the filtrate of the aforementioned reaction with n-hexane at −35 °C. The molecular structure is depicted in Fig. 1. The bonding parameters of trication 83+ are well in the expected range. Accordingly, one imidazoliumyl-substituent at the planar P3 ring is trans-arranged to the other two imidazoliumyl-substituents which results in the characteristic pattern in the 31P NMR spectrum.3,6 The 31P NMR spectrum of 83+ shows the AB2 spin system (δ(PA) = −142.5 ppm, δ(PB) = −130.8 ppm; 1J(PP) = −162 Hz) in the typical region for cyclo-triphosphanes.19 To our surprise, time dependent 31P NMR spectra reveal the rearrangement of 83+ into cyclo-tetraphosphane 34+ in a CD3CN solution in the course of 24 h (Fig. 2), suggesting that 83+ is the kinetic product of this reaction, while 34+ is thermodynamically favorable. Although such ring expansions are known for neutral cyclo-phosphanes, it seems, due to electrostatic repulsive effects, thermodynamically contradicting that a trication (83+) transforms into the corresponding tetracation (34+),24 rather than its fragmentation into charge separated species.24,25 We calculated the HOMO and the LUMO and optimized the structure by DFT methods using the PBE0/def2-TZVP level of theory (Fig. 3).16 The HOMO of cation 34+ is basically located at the imidazoliumyl-substituents, whereas the LUMO is mostly located at the P4 ring, suggesting an electrophilic nature of the P atoms within the P4 ring. The small degree of delocalization of the LUMO onto the imidazoliumyl-substituents also contributes to the stability of the tetracation 34+. We further reduced cyclo-tetraphosphane 3[OTf]4 again using the metal free reducing agent 6 (Scheme 4). Upon reaction of 3[OTf]4 with 6 in CH3CN, the reaction mixture turned from colorless to orange. The 31P NMR spectrum of this mixture showed after 8 hours a broad singlet resonance at δ(P) = −124.6 ppm and a dynamic AA′A′′BXX′X′′ spin system with resonances at δ(PA) = −178.5 ppm, δ(PB) = −164.9 ppm and δ(PX) = 13.2 ppm (see Fig. S5†).16 The singlet resonance is assigned to cation 10+ which was first reported by Macdonald and co-workers26 whereas the spin system of higher order is attributed to the P7 trication [(LC)3P7]3+ (93+). Imidazolium cation 11+ has been identified by its characteristic chemical shift in the 29Si NMR spectrum.129[OTf]3 can be isolated by washing the obtained crude oil with THF and CH2Cl2. After removal of all volatiles in vacuo, salt 9[OTf]3 is obtained as colorless solid in an overall yield of 61%. Fig. 3 shows the 31P{H} NMR spectrum of 93+ at 260 K which is iteratively fitted to an AA′A′′BXX′X′′ spin system and indicates the formation of a C3-symmetric stereoisomer (Fig. 4).27 The A part at δ(PA) = −178.5 ppm is assigned to the P atoms of the basal P3-ring, the B part at δ(PB) = −164.9 ppm to the apical P atom and the X part at δ(PX) = 13.2 ppm to the bridging P atoms carrying the imidazoliumyl-substituents. Details on coupling constants are included in the ESI in Table S1.† Single crystals suitable for X-ray analysis are obtained by slow diffusion of CH2Cl2 into a saturated CH3CN solution of 9[OTf]3 at −35 °C. The molecular structure confirms the C3-symmetric, heptaphosphanortricyclane cage with three imidazoliumyl-substituents at the bridging phosphorus atoms P2, P3 and P4 (Fig. 4). The P–C bond lengths (av. 1.834 Å) are comparable to those of tetracation 34+ (av. 1.822 Å) and other imidazoliumyl-substituted phosphorus derivatives.15 The bonding parameters within the P7-cage are comparable to those of our previously reported triarsonium-substituted P7 cage compound,27ai.e. the P–P bond lengths of the basal P atoms and the bridging P atoms (P2–P5, P3–P6 and P4–P7: av. 2.2238 Å) are slightly longer compared to those in the basal P3-ring (av. 2.2166 Å) and to those between the apical and the bridging P atoms (P1–P2, P1–P3 and P1–P4: av. 2.1882 Å).
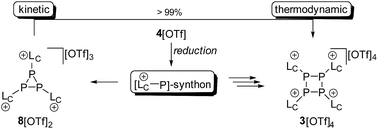 |
| | Scheme 3 Formation of 3[OTf]4 and 8[OTf]3 from the proposed [LC–P]+-synthon by reduction of 4[OTf]. Compound 8[OTf]3 readily rearranges in a CH3CN solution at room temperature within 24 h to 3[OTf]4. | |
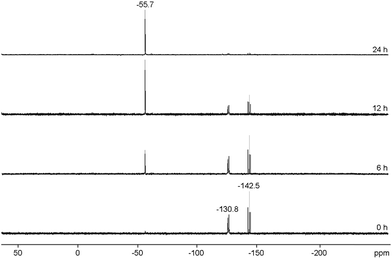 |
| | Fig. 2 Time dependent 31P NMR spectra of 83+, revealing a slow conversion of triphosphirane 83+ into cyclo-tetraphosphane 34+ over the course of 24 h (300 K, CD3CN). The resonances can be assigned as follows: δ(P) = −55.7 ppm to 34+; and AB2 spin system δ(PA) = −142.5 ppm, δ(PB) = −130.8 ppm to 83+. | |
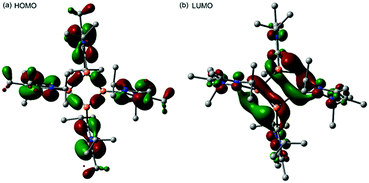 |
| | Fig. 3 HOMO (a) and LUMO (b) from the DFT optimized structure of 34+ using the PBE0/def2-TZVP functional. | |
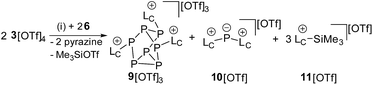 |
| | Scheme 4 Reaction of 3[OTf]4 with 6 to 9[OTf]3, 10[OTf] and 11[OTf]; (i) +2 6, CH3CN, 8 h, −2 pyrazine, –Me3SiOTf, 61%. | |
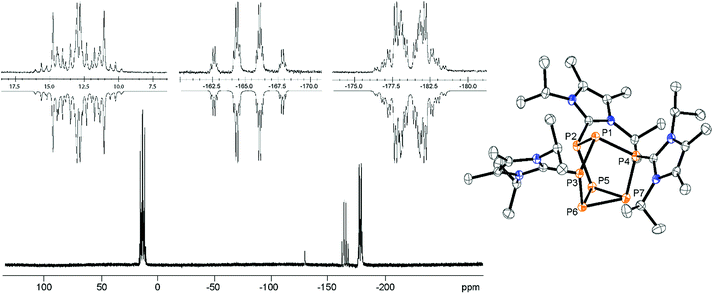 |
| | Fig. 4
31P{H} NMR spectrum of 93+ (CD3CN, 260 K). Insets show the extension of the experimental (upwards) and the iteratively fitted (downwards) AA′A′′BXX′X′′ spin system (left); molecular structure of trication 93+ in 9[OTf]3·0.8CH3CN·1.7CH2Cl2 (right); hydrogen atoms, solvate molecules and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability; selected bond lengths (Å) and angles (°) for: P1–P2 2.1810(6), P1–P3 2.1816(6), P1–P4 2.2022(6), P2–P5 2.2189(7), P3–P6 2.2282(7), P4–P7 2.2243(7), P5–P6 2.2250(7), P5–P7 2.2021(7), P6–P7 2.2227(7), P2–C1 1.8339(19), P3–C12 1.8291(19), P4–C23 1.8418(19). | |
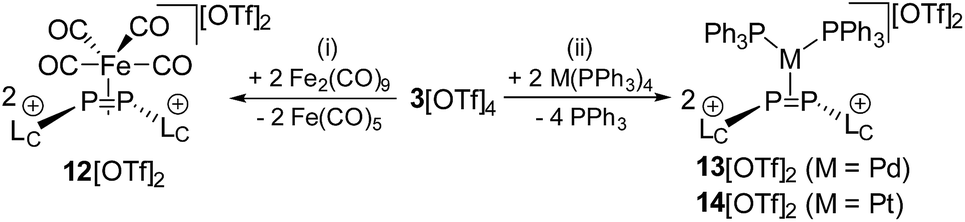
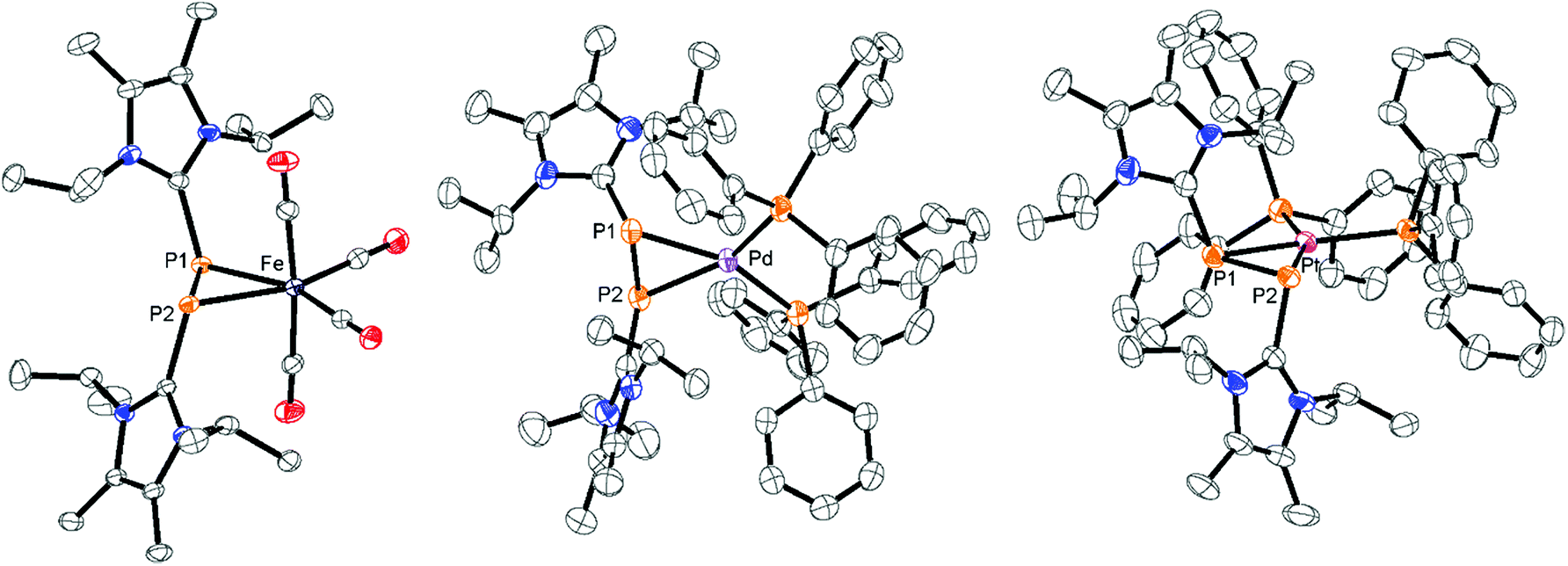
As the DFT calculations suggest rather electrophilic P atoms in 34+, we investigated its reactivity towards a series of low valent transition metal compounds in order to evaluate the ligand properties of 34+. 3[OTf]4 readily reacts with two equivalents of Fe2(CO)9 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of THF/CH3CN under the immediate formation of a brown-colored reaction mixture. The addition of Et2O gives a yellow-colored precipitate in a 98% yield which was identified as complex 12[OTf]2.16 In this complex, a Fe(CO)4 moiety is η2-coordinated by the disphosphene ligand [LCPPLC]2+, which is formed via the metal induced [2 + 2]-fragmentation (Scheme 5). Single crystals suitable for X-ray analysis are obtained by slow diffusion of n-hexane into a saturated CH2Cl2 solution of 12[OTf]2. The complex reveals a trigonal core including the P atoms of the [LCPPLC]2+ ligand and the Fe atom of the Fe(CO)4 moiety. The imidazoliumyl-substituents of the [LCPPLC]2+ ligand are arranged in an E-configuration (C1–P1–P2–C12 149.205(1)°) and the P1–Fe–P2 angle is relatively acute with a value of 54.490(19)°. The shortest distance between the diphosphene unit and the Fe atom is 2.105 Å as a result of the rather long Fe–P contacts (Fe–P1 2.3774(6) Å and Fe–P2 2.3590(6) Å). In contrast, typically Fe–P distances in η1-coordination complexes are usually in the region of 2.2 Å.28 The P–P bond length with a value of 2.1684(7) Å is significantly longer compared to those in a typical diphosphene such as Mes*–PP–Mes*: (2.034(2) Å),10 but slightly shorter compared to a typical P–P single bond (compare 34+: av. 2.235 Å). This also supports the η2-coordination of the P2 ligand. The IR stretching frequencies of the CO ligands are found at 2048 cm−1, 2074 cm−1 and 2116 cm−1 and are significantly shifted to higher wave numbers compared to related reported phosphorus Fe(CO)4 complexes (e.g. [(L2–P–L2)Fe(CO)4][BPh4]: 2029 cm−1, 1947 cm−1, 1907 cm−1).29 According to Tolman analysis,30 this suggests that the [LCPPLC]2+ ligand is a very strong π-acceptor which even surpasses PF3 ([(PF3)Fe(CO)4]: 2101 cm−1, 2022 cm−1, 2018 cm−1, 1999 cm−1)31 due to the electron withdrawing effect of the imidazoliumyl-substituents.16,32 We therefore computed the π-accepting properties of the [LCPPLC]2+ ligand which is related to the LUMO energy and shape of the ligand (see Fig. S12†).16 Our calculations show that the LUMO is the π*(PP) bond which is perfectly pre-organized to interact with the d-orbitals of the Fe atom. Furthermore, the calculations indicated that, due to the dicationic charge, the LUMO is very low in energy (−9.313 eV; compare [LC–PP–Dipp]+: −5.900 eV;16,33 LC = 4,5-dichloro-1,3-bis(Dipp)-imidazol-2-yl; Dipp = 2,6-diisopropylphenyl; see Fig. S12†) which is beneficial for π-bonding with the corresponding transition metal. In addition, the second order perturbation analysis of 12+ reveals a strong back-bonding from the d-orbitals of the Fe atom into the σ*(P–C)-orbital (E(2) = 35.77 kcal mol−1) indicating the stability of this complex.16 Similar findings have been recently reported by Macdonald and co-workers for a related diphosphene nickel complex featuring two phosphoniumyl-substituents.34
:1 mixture of THF/CH3CN under the immediate formation of a brown-colored reaction mixture. The addition of Et2O gives a yellow-colored precipitate in a 98% yield which was identified as complex 12[OTf]2.16 In this complex, a Fe(CO)4 moiety is η2-coordinated by the disphosphene ligand [LCPPLC]2+, which is formed via the metal induced [2 + 2]-fragmentation (Scheme 5). Single crystals suitable for X-ray analysis are obtained by slow diffusion of n-hexane into a saturated CH2Cl2 solution of 12[OTf]2. The complex reveals a trigonal core including the P atoms of the [LCPPLC]2+ ligand and the Fe atom of the Fe(CO)4 moiety. The imidazoliumyl-substituents of the [LCPPLC]2+ ligand are arranged in an E-configuration (C1–P1–P2–C12 149.205(1)°) and the P1–Fe–P2 angle is relatively acute with a value of 54.490(19)°. The shortest distance between the diphosphene unit and the Fe atom is 2.105 Å as a result of the rather long Fe–P contacts (Fe–P1 2.3774(6) Å and Fe–P2 2.3590(6) Å). In contrast, typically Fe–P distances in η1-coordination complexes are usually in the region of 2.2 Å.28 The P–P bond length with a value of 2.1684(7) Å is significantly longer compared to those in a typical diphosphene such as Mes*–PP–Mes*: (2.034(2) Å),10 but slightly shorter compared to a typical P–P single bond (compare 34+: av. 2.235 Å). This also supports the η2-coordination of the P2 ligand. The IR stretching frequencies of the CO ligands are found at 2048 cm−1, 2074 cm−1 and 2116 cm−1 and are significantly shifted to higher wave numbers compared to related reported phosphorus Fe(CO)4 complexes (e.g. [(L2–P–L2)Fe(CO)4][BPh4]: 2029 cm−1, 1947 cm−1, 1907 cm−1).29 According to Tolman analysis,30 this suggests that the [LCPPLC]2+ ligand is a very strong π-acceptor which even surpasses PF3 ([(PF3)Fe(CO)4]: 2101 cm−1, 2022 cm−1, 2018 cm−1, 1999 cm−1)31 due to the electron withdrawing effect of the imidazoliumyl-substituents.16,32 We therefore computed the π-accepting properties of the [LCPPLC]2+ ligand which is related to the LUMO energy and shape of the ligand (see Fig. S12†).16 Our calculations show that the LUMO is the π*(PP) bond which is perfectly pre-organized to interact with the d-orbitals of the Fe atom. Furthermore, the calculations indicated that, due to the dicationic charge, the LUMO is very low in energy (−9.313 eV; compare [LC–PP–Dipp]+: −5.900 eV;16,33 LC = 4,5-dichloro-1,3-bis(Dipp)-imidazol-2-yl; Dipp = 2,6-diisopropylphenyl; see Fig. S12†) which is beneficial for π-bonding with the corresponding transition metal. In addition, the second order perturbation analysis of 12+ reveals a strong back-bonding from the d-orbitals of the Fe atom into the σ*(P–C)-orbital (E(2) = 35.77 kcal mol−1) indicating the stability of this complex.16 Similar findings have been recently reported by Macdonald and co-workers for a related diphosphene nickel complex featuring two phosphoniumyl-substituents.34
 |
| | Scheme 5 Transition metal induced [2 + 2]-fragmentation of 3[OTf]4 to give diphosphene complexes 12[OTf]2, 13[OTf]2 and 14[OTf]2; (i) +2 Fe2(CO)9, CH3CN/THF, 12 h, −2 Fe(CO)5, 98%; (ii) +2 M(PPh3)4 (M = Pd, Pt), CH3CN/THF, 12 h, −4PPh3, 93% for 13[OTf]2 and 88% for 14[OTf]2. | |
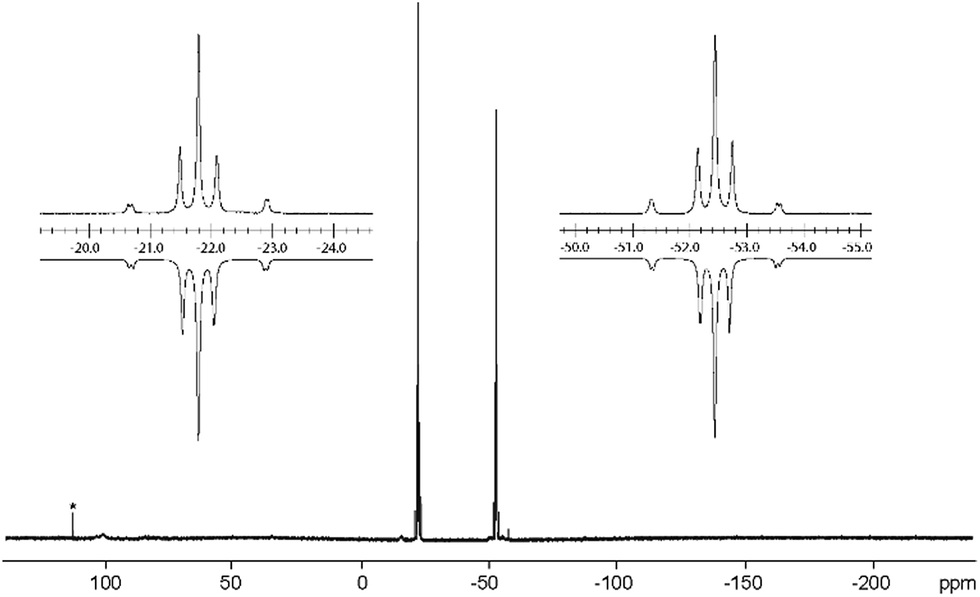
Similar [2 + 2]-fragmentation reactions are observed when 3[OTf]4 is reacted with 2 equivalents of M(PPh3)4 (M = Pd, Pt) in a 1:1 mixture of THF/CH3CN which leads to the formation of diphosphene complexes 13[OTf]2 (Pd(PPh3)4) and 14[OTf]2 (Pt(PPh3)4), respectively, under concomitant release of 4 equivalents PPh3 (Scheme 5). After workup, complexes 13[OTf]2 and 14[OTf]2 are obtained as analytically pure red-colored (13[OTf]2, 93%) or yellow-colored (14[OTf]2, 88%) crystalline powders. The 31P NMR spectrum of 13[OTf]2 at 300 K shows a broad singlet resonance at δ(P) = 10.2 ppm for the phosphorus atoms of the diphosphene moiety which upon cooling to 270 K appears as pseudo triplet with a 2J(PP) coupling constant of 31 Hz. The two PPh3 ligands give rise to a pseudo-triplet resonance at δ(P) = 23.4 ppm (2J(PP) = 31 Hz). The chemical shift is comparable to the related diphosphene complex [(η2-F3C–PP–CF3)Pd(PPh3)2].35 In contrast to the trans-2J(PP) coupling, the magnitude of the cis2J(PP) coupling is not resolved, resulting in the observed pseudo-triplet splitting of this resonance. The 31P NMR spectrum of 14[OTf]2 shows dynamic behavior at 300 K which upon cooling resolves to an AA′BB′X spin system. The high field shifted A part at δ(PA) = – 42.9 ppm is assigned to the diphosphene moiety and shows a 2J(PP) coupling constant of 29 Hz similar to that of 13[OTf]2. The 1J(PPt) coupling with a value of 235 Hz is significantly smaller compared to typical 1J(PPt) coupling constants,35 indicating the η2-coordination of the disphosphene ligand and the Pt atom.35,36 The B part at δ(PB) = 25.1 ppm (2J(PP) = 29 Hz) is assigned to the PPh3 ligands and reveals a typical 1J(PPt) coupling constant of 3279 Hz. The 195Pt NMR spectrum shows a triplet of triplet resonance at δ(Pt) = −5015.7 ppm (1J(PtP) = 3279 Hz; 1J(PtP) = 235 Hz; see Fig. S8†). For both complexes a set of resonances with low intensities indicates the presence of rotational isomers in the NMR spectra which, however, only appears at temperatures below 270 K (see Fig. S6 and S7†). Single crystals suitable for X-ray analysis are obtained for both complexes by slow diffusion of Et2O into a saturated CH3CN solutions (Fig. 5). Similar to complex 12[OTf]2, the η2-coordination of the corresponding metal by the diphosphene ligand is observed with the metal atoms being in a distorted square planar bonding environment. The averaged distance between the diphosphene unit and metals are in the same range (132+: Pd⋯P–P 2.124 Å, Pd–P1 2.3543(6) Å, Pd–P2 2.4003(8) Å; 142+: Pt⋯P–P 2.114 Å, Pt–P1 2.3530(7) Å, Pt–P2 2.3930(6) Å). The P–P bond lengths are only marginally shorter (132+ 2.1340(12) Å and 142+ 2.1562(9) Å) compared to that of complex 122+ (2.1684(7) Å). Similar to the Fe complex the [LCPPLC]2+ ligand is in both cases also in the E-configuration (132+: C1–P1–P2–C12 148.393°; 142+: C1–P1–P2–C12 147.088°).
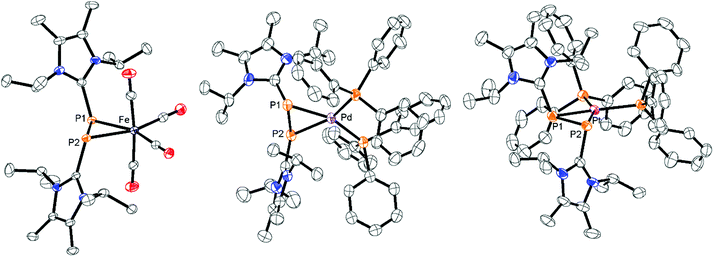 |
| | Fig. 5 Molecular structures of diphosphene complexes 122+ in 12[OTf]2, 132+ in 13[OTf]2·3C6H5F and 142+ in 14[OTf]2·3C6H5F; hydrogen atoms, solvate molecules and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability; selected bond lengths (Å) and angles (°) for 122+(Fe): P1–P2 2.1684(7), Fe⋯P–P 2.105, Fe–P1 2.3774(6), Fe–P2 2.3590(6); C1–P1–P2–C12 149.205(1), P1–Fe–P2 54.490(19); 132+(Pd): P1–P2 2.1340(12), Pd⋯P–P 2.124, Pd–P1 2.3543(6), Pd–P2 2.4003(8); C1–P1–P2–C12 148.393; 142+ (Pt): 2.1562(9), Pt⋯P–P 2.114, Pt–P1 2.3530(7), Pt–P2 2.3930(6); C1–P1–P2–C12 147.088. | |
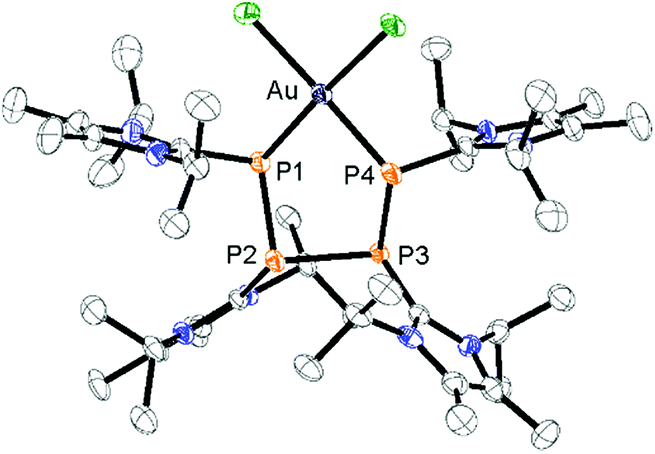
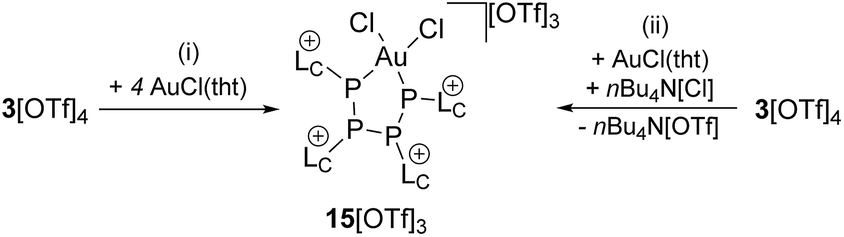
Notably, the known diphosphene complexes [(η2-F3C–PP–CF3)M(PPh3)2] (M = Pd, Pt)35,37 are also synthesized from the reaction of the tetraphosphetane (CF3)4P4 and the corresponding transition metal precursor M(PPh3)4 (M = Pd, Pt) suggesting the necessity of electron withdrawing substituents at the P atoms for the observed [2 + 2]-fragmentation. However, the formation of the dicationic diphosphene complexes (122+, 132+ and 142+), featuring imidazoliumyl-substituents at the P atom, illustrate the potential use of cyclo-tetraphosphane 34+ to synthesize a variety of novel cationic transition metal complexes. When 3[OTf]4 is reacted with one equivalent AuCl(tht) in THF/CH3CN (1:1) the formation of a small set of two coupled broad resonances at δ(P) = −49.9 ppm and δ(P) = −24.7 ppm along with a singlet resonance for 1[OTf] (δ(P) = 107.8 ppm) can be observed in the 31P NMR spectrum of the reaction mixture (see Fig. S9†). A complete consumption of 3[OTf]4 is achieved with 4 equivalents of AuCl(tht) (see Fig. S10†). Filtration of the reaction mixture after 12 h and recrystallization from THF, complex 15[OTf]3 can be isolated as a yellow powder. The yield can be increased by further recrystallization steps up to 57%. The 31P NMR spectrum of 15[OTf]3 dissolved in CD2Cl2 shows at 300 K two broad resonances which resolve to an AA′BB′ spin system at 260 K (Fig. 6). The A part at δ(P) = −49.9 ppm can be assigned to the backbone P atoms and the B part at δ(P) = −24.7 ppm to the Au-coordinating P atoms. Details on coupling constants are included in the ESI in Table S2.† Single crystals suitable for X-ray analysis are obtained by overlaying a saturated CH2Cl2 solution of 15[OTf]3 with pentane at −35 °C (Fig. 7). The Au atom is coordinated by two P atoms resulting in a five-membered P4Au-core structure which reveals an envelope conformation. Hey-Hawkins and co-workers reported on a similar structural motif where a catena-tetraphophosphane-1,4-diide coordinates Ni(0) and Pd(0), respectively.38 The gold atom in 153+ reveals a square-planar geometry where the P4-chain chelates the gold atom via P1 and P4. The phosphorus gold bond lengths (P1–Au 2.3053(11) Å and P4–Au 2.3103(10) Å) are well in the range for related P–AuIII bonds (av. 2.314).39 The four P atoms exhibit a pyramidal bonding environment with P–P bond lengths (av. 2.237 Å) being in the range of typical P–P single bonds (2.22 Å).20 As expected, the bond angles around the phosphorus atoms P1 and P4 (Au–P1–P2 107.79(5) ° and P3–P4–Au 107.90(5) Å) are wider compared to the other angles within the P4Au-core structure (P1–Au–P4 92.63(4) Å, P1–P2–P3 93.22(5) Å, P2–P3–P4 90.66(5) Å). Different from the above mentioned reactions, the reaction of 3[OTf]4 with an excess of AuCl leads to a reductive insertion of the gold atom into the P4-ring of 3[OTf]4 leading to the formation of a five-membered P4Au-core structure (Scheme 6).
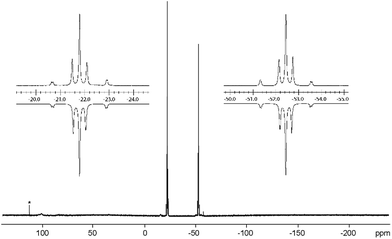 |
| | Fig. 6
31P{H} NMR spectrum of 153+ (CD2Cl2, 260 K). Insets show the extension of the experimental (upwards) and the iteratively fitted (downwards) AA′XX′ spin system; trace amounts of unidentified side product is marked with an asterisk. | |
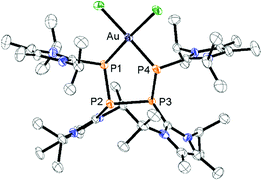 |
| | Fig. 7 Molecular structure of gold complex 153+ in 15[OTf]3·3CH2Cl2; hydrogen atoms, solvate molecules and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability; selected bond lengths (Å) and angles (°): P1–P2 2.2364(14), P2–P3 2.2304(13), P3–P4 2.2467(15), P1–Au 2.3053(11), P4–Au 2.3103(10); P1–Au–P4 92.63(4), Au–P1–P2 107.79(5), P1–P2–P3 93.22(5), P2–P3–P4 90.66(5), P3–P4–Au 107.90(5). | |
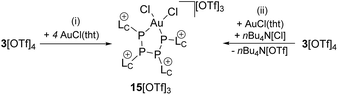 |
| | Scheme 6 Formation of the 5-membered P4Au-complex 15[OTf]3; (i) +4 AuCl(tht), THF/CH3CN, 8 h 57%; (ii) +AuCl(tht), +nBu4N[Cl], THF/CH3CN, −nBu4N[OTf], 76% determined by 31P NMR spectroscopy. | |
In this reaction, the Au(I) atom reductively inserts into a P–P bond of 34+ and is oxidized to Au(III). The free coordination site at the Au atom is saturated by a further chloride anion, leading to the square planar coordination environment at the Au(III) atom. Thus, it appears that the excess of the AuCl(tht) merely serves as chloride source in this reaction. We found that the reaction can be carried out with one equivalent AuCl(tht) and one equivalent nBu4N[Cl] as chloride source which leads to the consumption of 3[OTf]4 up to 90% and to the formation of the P4Au-complex 15[OTf]3 in 70% yield as judged by 31P NMR spectroscopy (see Fig. S11†).
Conclusions
In summary, we successfully synthesized the first example of a cationic cyclo-phosphane with the general formula [(LC)nPn]n+ (3[OTf]4) via the reduction of 4[OTf] using 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine (6). Due to the electrophilic nature of the P atoms within the P4 ring, which was shown by theoretical calculations, we further reduced compound 3[OTf]4 to give the notricyclane P7 compound 9[OTf]3. The reaction of 3[OTf]4 with low oxidation state transition metal complexes Fe2(CO9), Pd(PPh3)4 and Pt(PPh3)4 leads to the formation of dicationic diphosphene complexes 12[OTf]2, 13[OTf]2 and 14[OTf]2, respectively. The transition metal mediated [2 + 2]-fragmentation reaction of 34+ is attributed to the cationic imidazoliumyl-substituents causing the electrophilic nature of the P atoms in 34+. Due to the dicationic charge, the P2 ligand in the aforementioned complexes is a very good π-acceptor with an exceptionally low lying LUMO (π*-PP bond) rendering this ligand optimal for the complexation of a π-basic metal center. In contrast, the reaction of 3[OTf]4 with an excess of AuCl(tht) gives rise to the formation of complex 15[OTf]3 featuring a five-membered P4Au-core structure. In this reaction, the Au(I) atom reductively inserts into a P–P bond of 34+ and is oxidized to Au(III). As the DFT calculations of the optimized structure of 34+ reveal rather Lewis acidic P atoms, the reactivity towards nucleophiles promises a variety of novel phosphorus compounds featuring interesting bonding motifs and properties.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the German Science Foundation (DFG Grant number WE 4621/3-1). A. F. and A. B. thank MINECO/AEI of Spain (project CTQ2017-85821-R FEDER funds) for financial support. We also thank Philipp Lange for experimental assistance and EA measurements.
Notes and references
-
(a)
D. E. C. Corbridge, Phosphorus Chemistry, Biochemistry and Technology, Elsevier, Amsterdam, 2000 Search PubMed;
(b)
N. Wiberg, Lehrbuch der Anorganischen Chemie, de Gruyter, Berlin, 102 edn, 2007, pp. 758–775 Search PubMed.
-
(a) A. H. Cowley, Chem. Rev., 1965, 65, 617 CrossRef CAS;
(b) L. Lamandé, K. Dillon and R. Wolf, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 103, 1 CrossRef;
(c) M. Donath, F. Hennersdorf and J. J. Weigand, Chem. Soc. Rev., 2016, 45, 1145 RSC.
- Selected examples for the chemistry of neutral triphosphiranes:
(a) M. Bentner and W. Uhl, Dalton Trans., 2000, 3133 Search PubMed;
(b) C. A. Dyker, N. Burford, G. Menard, M. D. Lumsden and A. Decken, Inorg. Chem., 2007, 46, 4277 CrossRef CAS PubMed;
(c) M. H. Holtausen, D. Knackstedt, N. Burford and J. J. Weigand, Aust. J. Chem., 2013, 66, 1155 CrossRef;
(d) A. P. M. Robertson, C. A. Dyker, P. Gray, B. O. Patrick, A. Decken and N. Burford, J. Am. Chem. Soc., 2014, 136, 14941 CrossRef CAS PubMed;
(e) S. S. Chitnis, H. A. Sparkes, V. T. Annibale, N. E. Pridmore, E. Natalie, A. M. Oliver and I. Manners, Angew. Chem., Int. Ed., 2017, 56, 9536 (
Angew. Chem.
, 2017
, 129
, 9664
) CrossRef CAS PubMed.
- Selected examples for the chemistry of neutral tetraphosphetanes:
(a) S. Parveen, P. Kilian, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2006, 2586 RSC;
(b) C. A. Dyker, S. D. Riegel, N. Burford, M. D. Lumsden and A. Decken, J. Am. Chem. Soc., 2007, 129, 7464 CrossRef CAS PubMed;
(c) J. J. Weigand, N. Burford, R. J. Davidson, T. S. Cameron and P. Seelheim, J. Am. Chem. Soc., 2009, 131, 17953 CrossRef PubMed;
(d) E. Conrad, N. Burford, U. Werner-Zwanziger, R. McDonald and M. J. Ferguson, Chem. Commun., 2010, 46, 2465 RSC.
- Selected examples for the chemistry of neutral pentaphospholanes:
(a) A. J. Arduengo III, H. V. R. Dias and J. C. Calabrese, Chem. Lett., 1997, 143 CrossRef;
(b) I. P. Gray, P. Bhattacharyya, A. M. Z. Slawin and J. D. Woollins, Chem.–Eur. J., 2005, 11, 6221 CrossRef CAS PubMed;
(c) R. B. Baker, C. Jones, D. P. Mills, D. M. Murphy, E. Hey-Hawkins and R. Wolf, Dalton Trans., 2006, 64 RSC;
(d) R. Wolf, M. Finger, C. Limburg, C. A. Willis, S. B. Wild and E. Hey-Hawkins, Dalton Trans., 2006, 831 RSC;
(e) S. Geier and D. W. Stephan, Chem. Commun., 2010, 46, 1026 RSC;
(f) J. H. Barnard, P. A. Brown, K. L. Shuford and C. D. Martin, Angew. Chem., Int. Ed., 2015, 54, 12083 (
Angew. Chem.
, 2015
, 127
, 12251
) CrossRef CAS PubMed.
- For reviews see:
(a) M. Baudler and K. Glinka, Chem. Rev., 1993, 93, 1623 CrossRef CAS;
(b) G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815 CrossRef CAS PubMed.
-
(a) L. Weber, Chem. Rev., 1992, 92, 1839 CrossRef CAS;
(b) A.-M. Caminade, J.-P. Majoral and R. Mathieu, Chem. Rev., 1991, 91, 575 CrossRef CAS.
- L. R. Smith and J. L. Mills, J. Am. Chem. Soc., 1976, 98, 3852 CrossRef CAS.
-
(a) M. Baudler and K. Hammerstroem, Z. Naturforsch., B: J. Chem. Sci., 1965, 20, 810 Search PubMed;
(b) A. H. Cowley and R. P. Pinnell, Inorg. Chem., 1966, 5, 459 Search PubMed.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587 CrossRef CAS.
- A. Beil, R. J. Gilliard Jr and H. Grützmacher, Dalton Trans., 2016, 45, 2044 RSC.
-
(a) J. J. Weigand, K.-O. Feldmann and F. D. Henne, J. Am. Chem. Soc., 2010, 132, 16321 CrossRef CAS PubMed;
(b) F. D. Henne, A. T. Dickschat, F. Hennersdorf, K.-O. Feldmann and J. J. Weigand, Inorg. Chem., 2015, 54, 6849 CrossRef CAS PubMed.
- F. D. Henne, F. A. Watt, K. Schwedtmann, F. Hennersdorf, M. Kokoschka and J. J. Weigand, Chem. Commun., 2016, 52, 2023 RSC.
- K. Schwedtmann, M. H. Holthausen, K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 14204 (
Angew. Chem.
, 2013
, 125
, 14454
) CrossRef CAS PubMed.
- For reviews see:
(a) K. Schwedtmann, G. Zanoni and J. J. Weigand, Chem.–Asian J., 2018, 13, 1388 CrossRef CAS PubMed;
(b) T. Krachko and C. Slootweg, Eur. J. Inorg. Chem., 2018, 2734 CrossRef CAS.
- For details see ESI.†.
- F. D. Henne, E.-M. Schnoeckelborg, K.-O. Feldmann, J. Grunenberg, R. Wolf and J. J. Weigand, Organometallics, 2013, 32, 6674 CrossRef CAS.
- For the bromide-bridged analogue see: J. B. Waters, T. A. Everitt, W. K. Myers and J. M. Goicoechea, Chem. Sci., 2016, 4, 77 Search PubMed.
- M. Baudler, J. Hahn, H. Dietsch and G. Fürstenberg, Z. Naturforsch., B: J. Chem. Sci., 1976, 31, 1305 Search PubMed.
-
(a)
M. Nieger, E. Niecke and J. Tirree, Private Communication, 2002, CCD, 178050;
(b) L. Heurer, D. Schromburg and R. Schmutzler, Phosphorus, Sulfur Silicon Relat. Elem., 1989, 45, 217 CrossRef.
-
(a) L. P. Ho, A. Nasr, P. G. Jones, A. Altun, F. Neese, G. Bistoni and M. Tamm, Chem.–Eur. J., 2018, 24, 18922 CAS;
(b) Y.-y. Carpenter, N. Burford, M. D. Lumsden and R. McDonald, Inorg. Chem., 2011, 50, 3342 CrossRef CAS PubMed.
- S. Aime, R. K. Harris, E. M. McVicker and M. Fild, J. Chem. Soc., Dalton Trans., 1976, 2144 RSC.
-
(a) V. Robert, S. Petit, S. A. Borshch and B. Bigot, J. Phys. Chem. A, 2000, 104, 4586 CrossRef CAS;
(b) S. K. Latypov, F. M. Polyancev, D. G. Yakhvarov and O. G. Sinyashin, Phys. Chem. Chem. Phys., 2015, 17, 6976 RSC.
-
(a) D. A. Klumpp, Chem.–Eur. J., 2008, 14, 2004 CrossRef CAS PubMed;
(b) D. Schröder and H. Schwarz, J. Phys. Chem. A, 1999, 103, 7385 CrossRef.
-
(a) G. A. Olah, G. Rasul, A. K. Yudin, A. Burrichter, G. K. S. Prakash, A. L. Chistyakov, I. V. Stankevich, I. S. Akhrem, N. P. Gambaryan and M. E. Vol'pin, J. Am. Chem. Soc., 1996, 118, 1446 CrossRef CAS;
(b) G. K. S. Prakash, T. Mathew, D. Hoole, P. M. Esteves, Q. Wang, G. Rasul and G. A. Olah, J. Am. Chem. Soc., 2004, 126, 15770 CrossRef CAS PubMed;
(c) R. Weiss and S. Engel, Angew. Chem., Int. Ed., 1992, 31, 216 (
Angew. Chem.
, 1992
, 104
, 239
) CrossRef;
(d) F. G. Pühlhofer and R. Weiss, Eur. J. Inorg. Chem., 2004, 1002 CrossRef.
- B. D. Ellis, C. A. Dyker, A. Decken and C. L. B. Macdonald, Chem. Commun., 2005, 1965 RSC.
- For related P7 compounds see:
(a) M. Donath, M. Bodensteiner and J. J. Weigand, Chem.–Eur. J., 2014, 20, 17306 CrossRef CAS PubMed;
(b) J. J. Weigand, M. H. Holthausen and R. Fröhlich, Angew. Chem., Int. Ed., 2009, 48, 295 (
Angew. Chem.
, 2009
, 121
, 301
) CrossRef CAS PubMed;
(c) W. Hönle, H. G. von Schnering, A. Schmidpeter and G. Burget, Angew. Chem., Int. Ed. Engl., 1984, 23, 817 (
Angew. Chem.
, 1984
, 96
, 796
) CrossRef;
(d) C. Mujica, D. Weber and H.-G. von Schnering, Z. Naturforsch., B: J. Chem. Sci., 1986, 41b, 991 CAS.
-
(a) S. Roy, S. K. S. Mazinani, T. L. Groy, L. Gan, P. Tarakeshwar, V. Mujica and A. K. Jones, Inorg. Chem., 2014, 53, 8919 CrossRef CAS PubMed;
(b) L. D. Field, R. W. Guest and P. Turner, Inorg. Chem., 2010, 49, 9086 CrossRef CAS PubMed;
(c) C. Federsel, A. Boddien, R. Jackstell, R. Jennerhahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777 (
Angew. Chem.
, 2010
, 122
, 9971
) CrossRef CAS PubMed.
- J. F. Binder, S. C. Kosnik and C. L. B. Macdonald, Chem.–Eur. J., 2018, 24, 3556 CrossRef CAS PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS.
- H. Mahnke, R. J. Clark, R. Rosanske and R. K. Sheline, J. Chem. Phys., 1974, 60, 2997 CrossRef CAS.
-
(a) M. Alcarazo, Chem.–Eur. J., 2014, 20, 7968 CrossRef PubMed;
(b) M. Alcarazo, Acc. Chem. Res., 2016, 49, 1797 CrossRef CAS PubMed.
- K. Schwedtmann, M. H. Holthausen, C. H. Sala, F. Hennersdorf, R. Fröhlich and J. J. Weigand, Chem. Commun., 2016, 52, 1409 RSC.
- S. C. Kosnik, J. F. Binder, M. C. Nascimento, A. Swidan and C. L. B. Macdonald, Chem.–Eur. J., 2019, 25, 1208 CAS.
-
(a) J. Chatt, P. B. Hitchcock, A. Pidcock, C. P. Warrens and K. R. Dixon, J. Chem. Soc., Dalton Trans., 1984, 2237 RSC;
(b) J. Chatt, P. B. Hitchcock, A. Pidcock, C. P. Warrens and K. R. Dixon, J. Chem. Soc., Dalton Trans., 1982, 932 CAS.
-
(a)
O. Kühl, Phosphorus-31 NMR Spectroscopy, Springer, Heidelberg, 2008 Search PubMed;
(b) A. Pidcock, R. E. Richards and L. M. Venanzi, J. Chem. Soc. A, 1966, 1707 RSC.
- I. G. Phillips, R. G. Ball and R. G. Cavell, Inorg. Chem., 1992, 31, 1633 CrossRef CAS.
-
(a) S. Gómez-Ruiz and E. Hey-Hawkins, Dalton Trans., 2007, 5678 RSC;
(b) S. Gómez-Ruiz and E. Hey-Hawkins, Coord. Chem. Rev., 2011, 255, 1360 CrossRef.
-
(a) H. Kawai, W. J. Wolf, A. G. DiPasquale, M. S. Winston and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 587 CrossRef CAS PubMed;
(b) O. Schuster and H. Schmidbaur, Organometallics, 2005, 24, 2289 CrossRef CAS;
(c) O. Schuster, R.-Y. Liau, A. Schier and H. Schmidbaur, Inorg. Chim. Acta, 2005, 358, 1429 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1884156–1884162 and 1904570. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01701a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Antonio
Frontera
b,
Antonio
Frontera